Whole-Genome Signatures of Selection in Sport Horses Revealed Selection Footprints Related to Musculoskeletal System Development Processes

 ,
,  , ,
, , 
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Whole-Genome Mapping and Variant Calling
2.3. Population Genetic Structure
2.4. Genome-Wide Selective Signals Scan and Gene Ontology (GO)
3. Results and Discussion
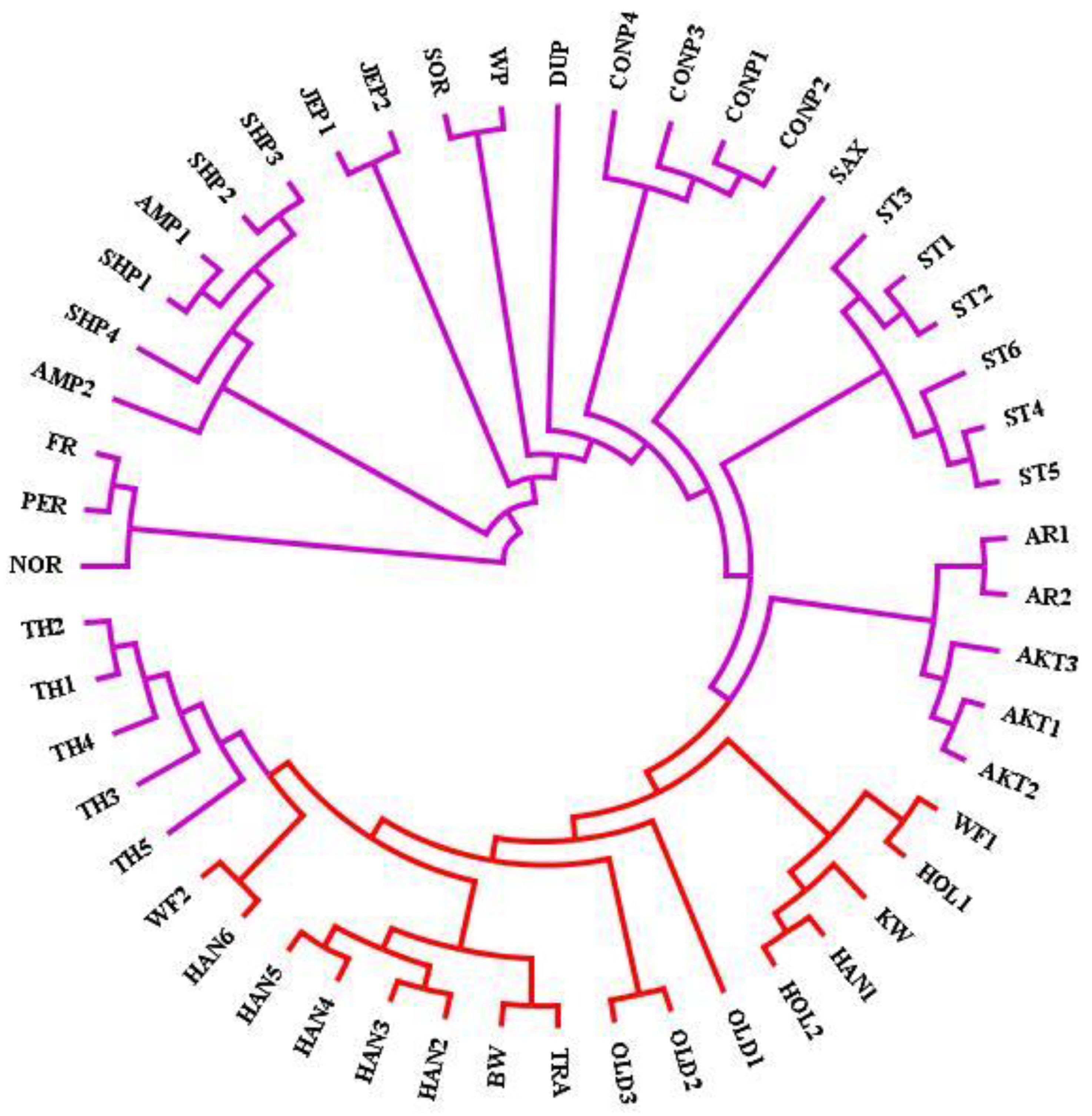
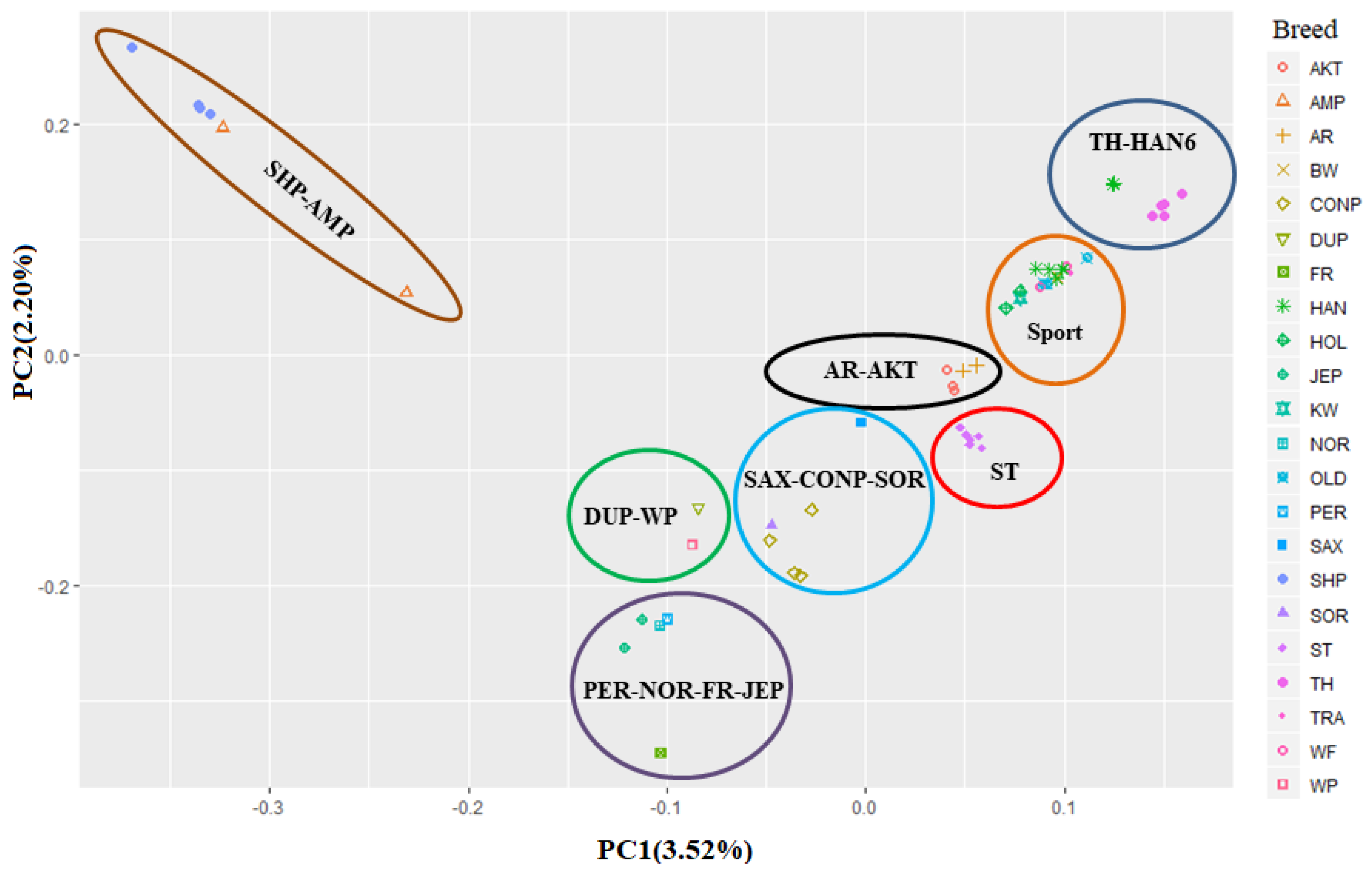
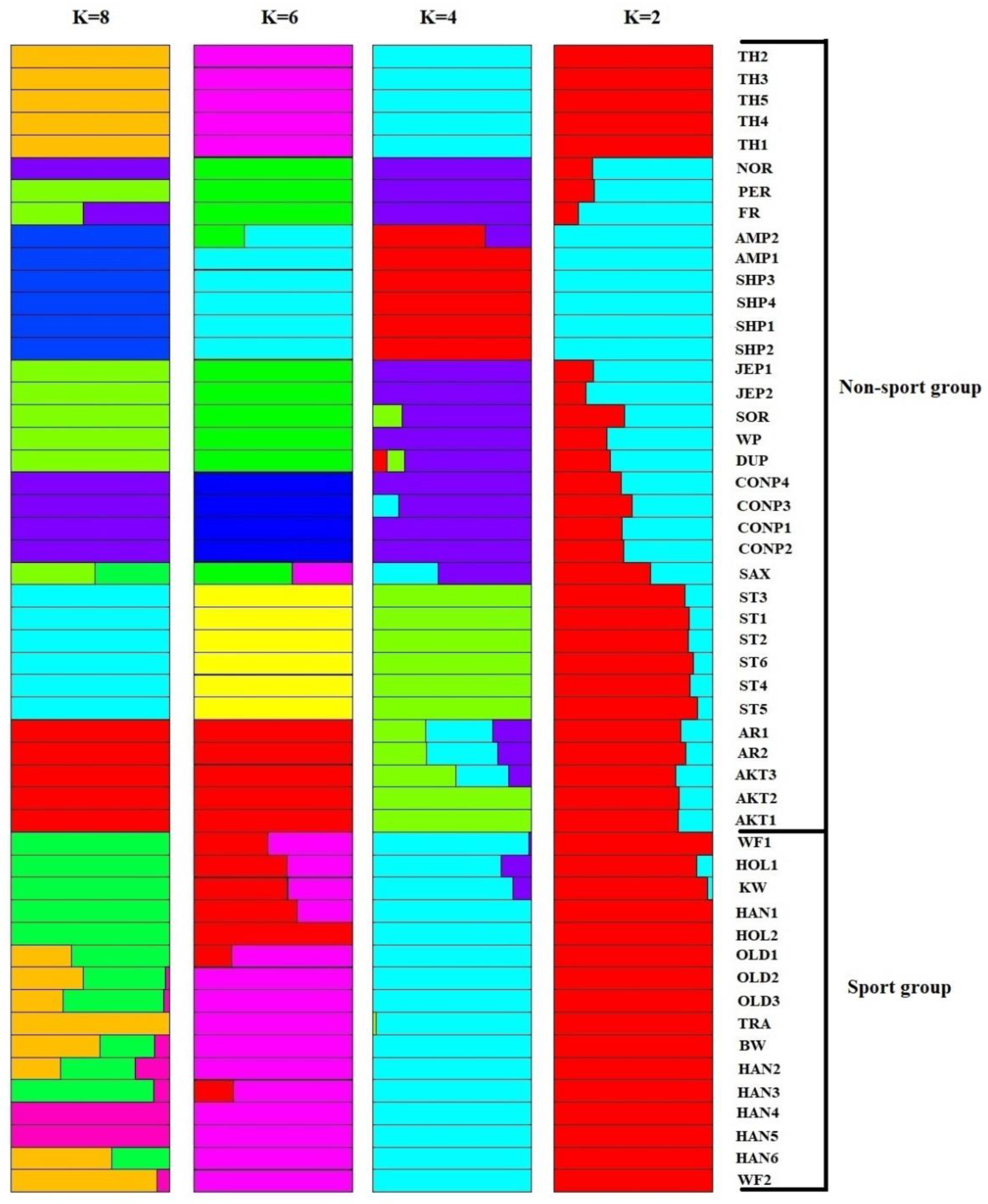
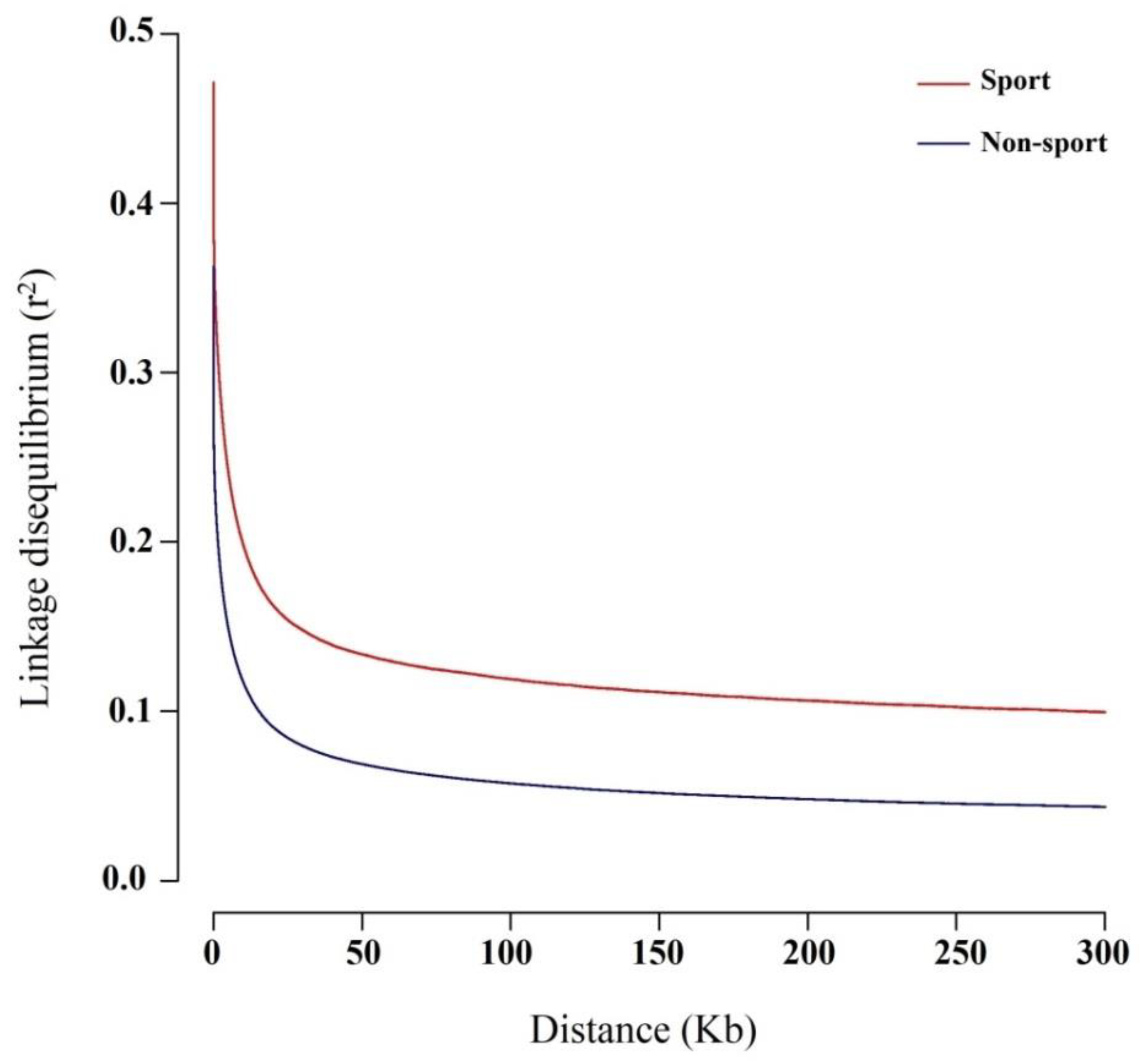
3.1. Genomic Variants and Population Genetic Structure
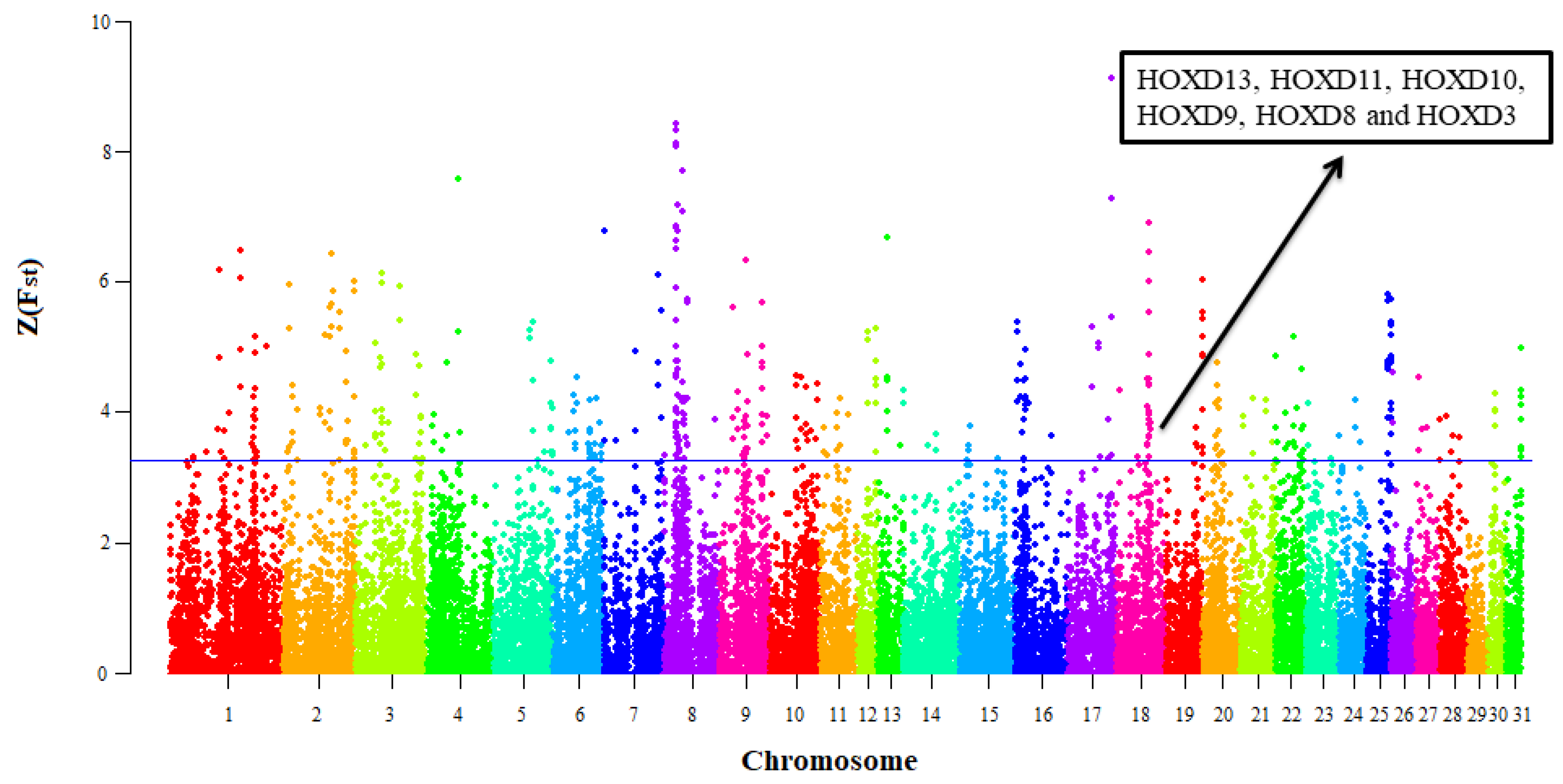
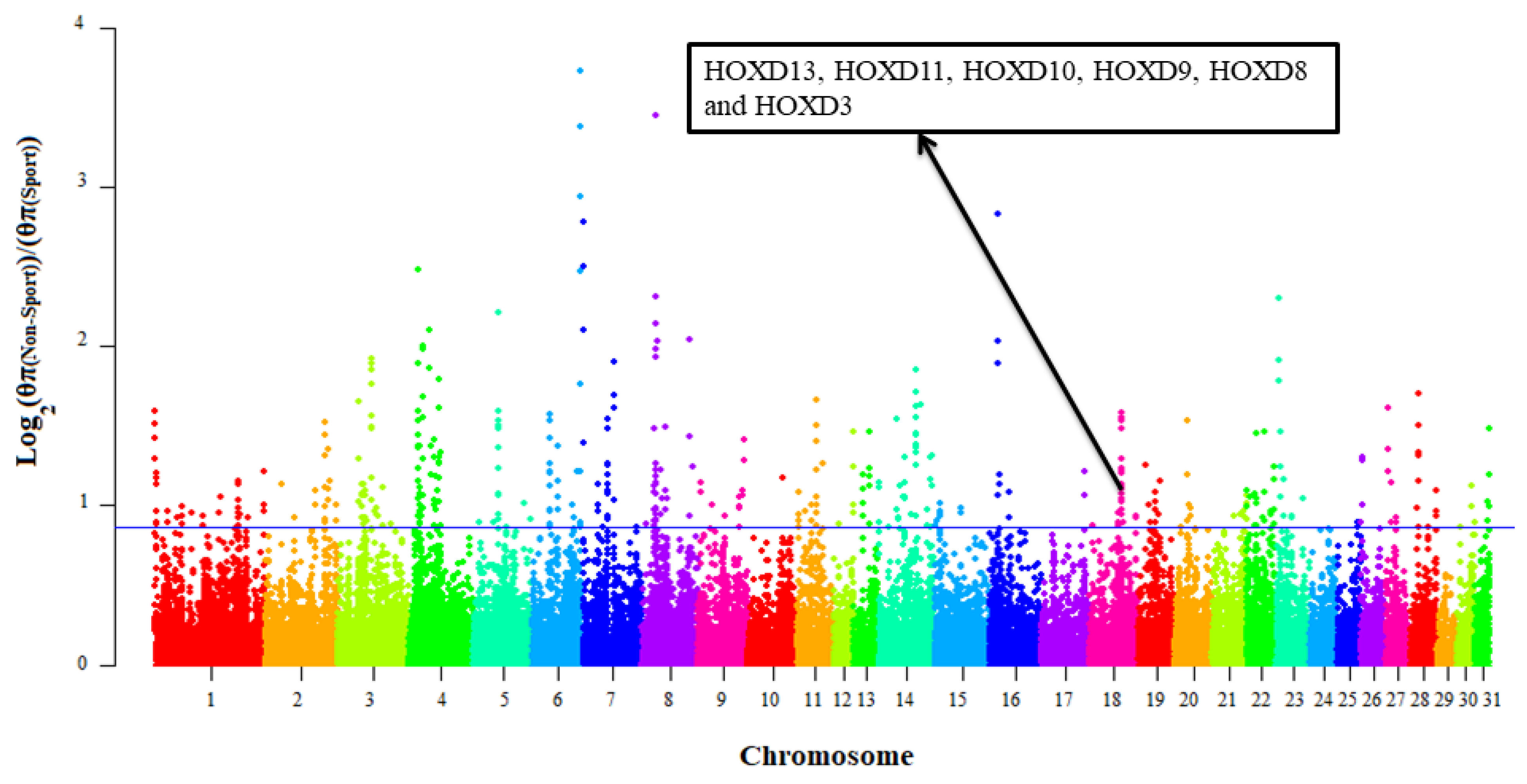
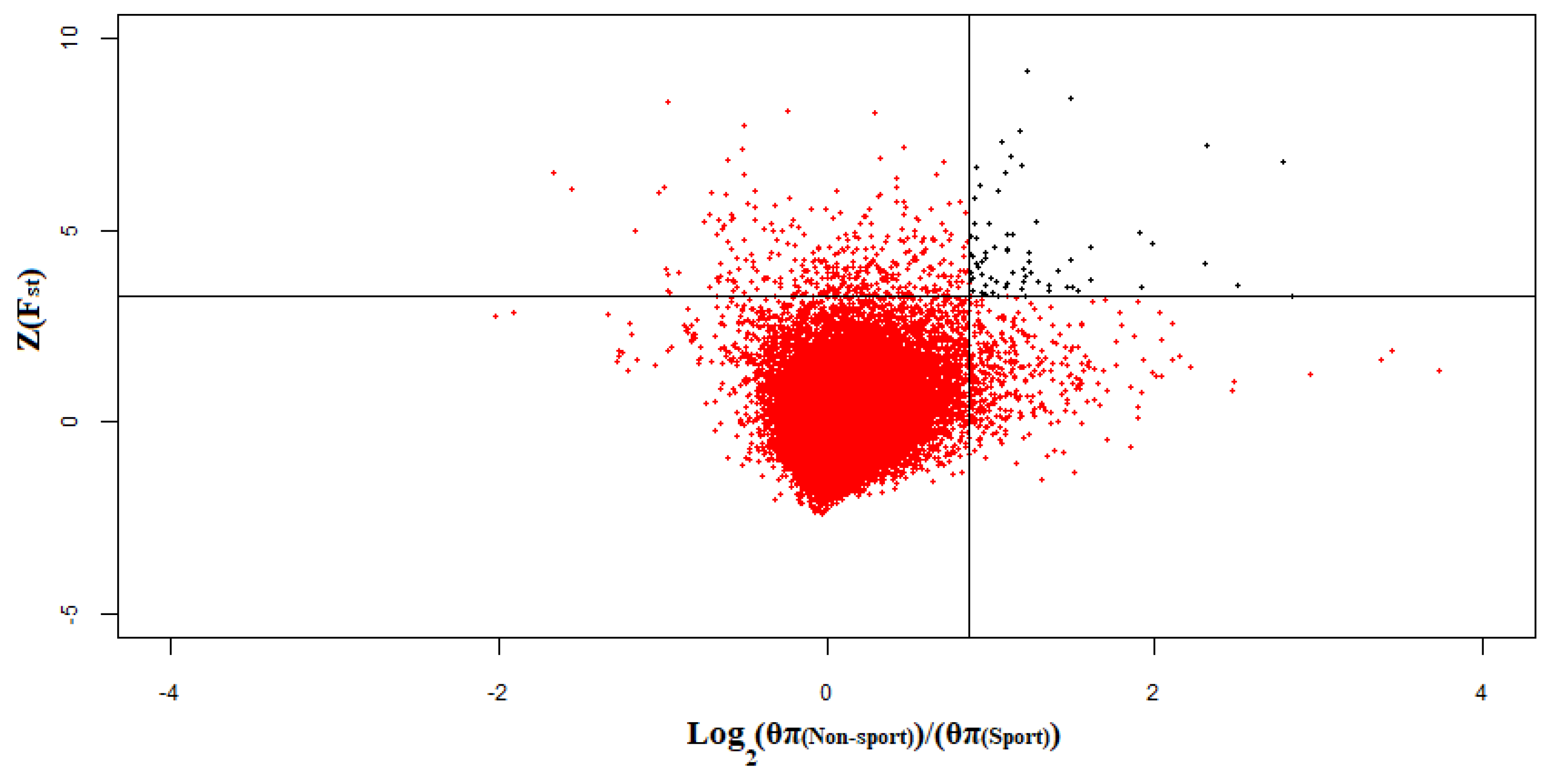
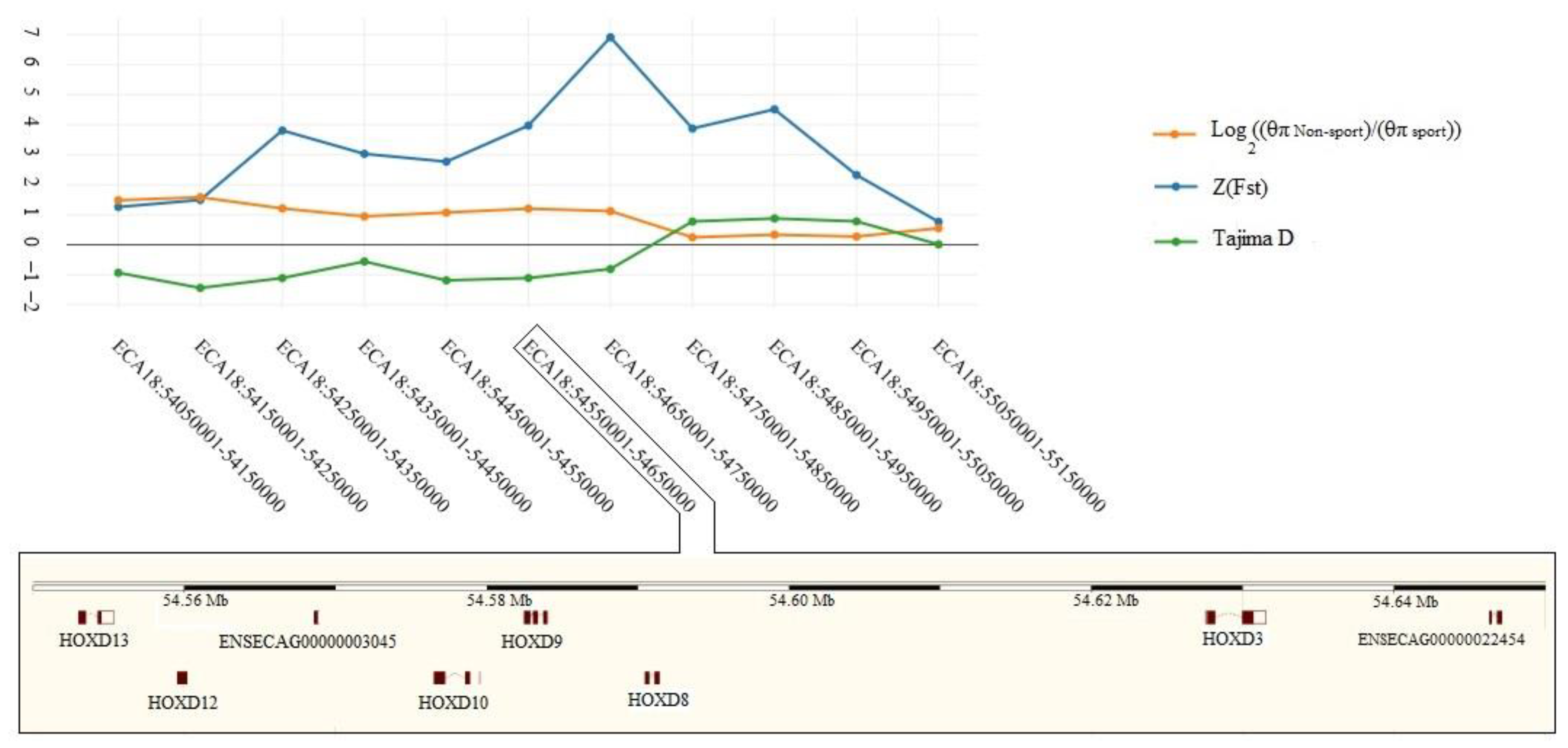
3.2. Selective Signals Detection
4. Conclusions
Supplementary Materials
Author Contributions
Data Availability
Conflicts of Interest
References
- Bowling, A.T.; Ruvinsky, A. The Genetics of the Horse; CABI: Cambridge, MA, USA, 2000. [Google Scholar]
- Koenen, E.; Aldridge, L.; Philipsson, J. An overview of breeding objectives for warmblood sport horses. Livest. Prod. Sci. 2004, 88, 77–84. [Google Scholar] [CrossRef]
- Nolte, W.; Thaller, G.; Kuehn, C. Selection signatures in four German warmblood horse breeds: Tracing breeding history in the modern sport horse. PLoS ONE 2019, 14, e0215913. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, W.R.; Lv, F.H.; He, S.G.; Tian, S.L.; Peng, W.F.; Sun, Y.W.; Zhao, Y.X.; Tu, X.L.; Zhang, M. Whole-genome sequencing of native sheep provides insights into rapid adaptations to extreme environments. Mol. Biol. Evol. 2016, 33, 2576–2592. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, J.; Zhou, G.; Guo, J.; Yan, H.; Niu, Y.; Li, Y.; Yuan, C.; Geng, R.; Lan, X. Whole-genome sequencing of eight goat populations for the detection of selection signatures underlying production and adaptive traits. Sci. Rep. 2016, 6, 38932. [Google Scholar] [CrossRef] [PubMed]
- Asadollahpour Nanaei, H.; Ayatollahi Mehrgardi, A.; Esmailizadeh, A. Comparative population genomics unveils candidate genes for athletic performance in Hanoverians. Genome 2019, 62, 279–285. [Google Scholar] [CrossRef]
- Stock, K.F.; Jönsson, L.; Ricard, A.; Mark, T. Genomic applications in horse breeding. Anim. Front. 2016, 6, 45–52. [Google Scholar] [CrossRef]
- Makvandi-Nejad, S.; Hoffman, G.E.; Allen, J.J.; Chu, E.; Gu, E.; Chandler, A.M.; Loredo, A.I.; Bellone, R.R.; Mezey, J.G.; Brooks, S.A. Four loci explain 83% of size variation in the horse. PLoS ONE 2012, 7, e39929. [Google Scholar] [CrossRef]
- Metzger, J.; Tonda, R.; Beltran, S.; Águeda, L.; Gut, M.; Distl, O. Next generation sequencing gives an insight into the characteristics of highly selected breeds versus non-breed horses in the course of domestication. BMC Genom. 2014, 15, 562. [Google Scholar] [CrossRef]
- Kader, A.; Li, Y.; Dong, K.; Irwin, D.M.; Zhao, Q.; He, X.; Liu, J.; Pu, Y.; Gorkhali, N.A.; Liu, X. Population variation reveals independent selection toward small body size in Chinese Debao pony. Genome Biol. Evol. 2015, 8, 42–50. [Google Scholar] [CrossRef]
- Metzger, J.; Rau, J.; Naccache, F.; Conn, L.B.; Lindgren, G.; Distl, O. Genome data uncover four synergistic key regulators for extremely small body size in horses. BMC Genom. 2018, 19, 492. [Google Scholar] [CrossRef]
- Rubin, C.J.; Megens, H.J.; Barrio, A.M.; Maqbool, K.; Sayyab, S.; Schwochow, D.; Wang, C.; Carlborg, Ö.; Jern, P.; Jørgensen, C.B. Strong signatures of selection in the domestic pig genome. Proc. Natl. Acad. Sci. USA 2012, 109, 19529–19536. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Su, R.; Wan, W.; Zhang, W.; Jiang, H.; Qiao, X.; Fan, Y.; Zhang, Y.; Wang, R.; Liu, Z. Identification of selection signals by large-scale whole-genome resequencing of cashmere goats. Sci. Rep. 2017, 7, 15142. [Google Scholar] [CrossRef] [PubMed]
- Metzger, J.; Karwath, M.; Tonda, R.; Beltran, S.; Águeda, L.; Gut, M.; Gut, I.G.; Distl, O. Runs of homozygosity reveal signatures of positive selection for reproduction traits in breed and non-breed horses. BMC Genom. 2015, 16, 764. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.; Lee, J.W.; Shin, D.; Shin, K.Y.; Kim, J.; Choi, I.Y.; Kim, J.; Kim, H. A genome-wide scan for selective sweeps in racing horses. Asian Australas. J. Anim. Sci. 2015, 28, 1525. [Google Scholar] [CrossRef]
- Petersen, J.L.; Mickelson, J.R.; Rendahl, A.K.; Valberg, S.J.; Andersson, L.S.; Axelsson, J.; Bailey, E.; Bannasch, D.; Binns, M.M.; Borges, A.S. Genome-wide analysis reveals selection for important traits in domestic horse breeds. PLoS Genet. 2013, 9, e1003211. [Google Scholar] [CrossRef]
- Frischknecht, M.; Flury, C.; Leeb, T.; Rieder, S.; Neuditschko, M. Selection signatures in Shetland ponies. Anim. Genet. 2016, 47, 370–372. [Google Scholar] [CrossRef]
- Gurgul, A.; Jasielczuk, I.; Semik-Gurgul, E.; Pawlina-Tyszko, K.; Stefaniuk-Szmukier, M.; Szmatoła, T.; Polak, G.; Tomczyk-Wrona, I.; Bugno-Poniewierska, M. A genome-wide scan for diversifying selection signatures in selected horse breeds. PLoS ONE 2019, 14, e0210751. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Cook, D.E.; Andersen, E.C. VCF-kit: Assorted utilities for the variant call format. Bioinformatics 2017, 33, 1581–1582. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.; Daly, M.J. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Dong, S.-S.; Xu, J.Y.; He, W.M.; Yang, T.L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2018, 35, 1786–1788. [Google Scholar] [CrossRef]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar]
- Nei, M.; Li, W.H. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. USA 1979, 76, 5269–5273. [Google Scholar] [CrossRef]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Andam, C.; Challagundla, L.; Azarian, T.; Hanage, W.; Robinson, D. Population Structure of Pathogenic Bacteria. In Genetics and Evolution of Infectious Diseases; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- McCue, M.E.; Bannasch, D.L.; Petersen, J.L.; Gurr, J.; Bailey, E.; Binns, M.M.; Distl, O.; Guérin, G.; Hasegawa, T.; Hill, E.W. A high density SNP array for the domestic horse and extant Perissodactyla: Utility for association mapping, genetic diversity, and phylogeny studies. PLoS Genet. 2012, 8, e1002451. [Google Scholar] [CrossRef]
- Petersen, J.L.; Mickelson, J.R.; Cothran, E.G.; Andersson, L.S.; Axelsson, J.; Bailey, E.; Bannasch, D.; Binns, M.M.; Borges, A.S.; Brama, P. Genetic diversity in the modern horse illustrated from genome-wide SNP data. PLoS ONE 2013, 8, e54997. [Google Scholar] [CrossRef]
- Zhang, C.; Ni, P.; Ahmad, H.I.; Gemingguli, M.; Baizilaitibei, A.; Gulibaheti, D.; Fang, Y.; Wang, H.; Asif, A.R.; Xiao, C. Detecting the population structure and scanning for signatures of selection in horses (Equus caballus) from whole-genome sequencing data. Evol. Bioinform. 2018, 14, 1176934318775106. [Google Scholar] [CrossRef] [PubMed]
- Dutson, J. Storey’s Illustrated Guide to 96 Horse Breeds of North America; Storey Publishing: North Adams, MA, USA, 2012. [Google Scholar]
- Georgescu, S.; Manea, M.; Dudu, A.; Costache, M. Phylogenetic relationships of the Hucul horse from Romania inferred from mitochondrial D-loop variation. Genet. Mol. Res. 2011, 10, 4104–4113. [Google Scholar] [CrossRef] [PubMed]
- Ardlie, K.G.; Kruglyak, L.; Seielstad, M. Patterns of linkage disequilibrium in the human genome. Nat. Rev. Genet. 2002, 3, 299. [Google Scholar] [CrossRef] [PubMed]
- Nalls, M.; Guerreiro, R.; Simon-Sanchez, J.; Bras, J.; Traynor, B.; Gibbs, J.; Launer, L.; Hardy, J.; Singleton, A. Extended tracts of homozygosity identify novel candidate genes associated with late-onset Alzheimer’s disease. Neurogenetics 2009, 10, 183–190. [Google Scholar] [CrossRef]
- Purfield, D.C.; McParland, S.; Wall, E.; Berry, D.P. The distribution of runs of homozygosity and selection signatures in six commercial meat sheep breeds. PLoS ONE 2017, 12, e0176780. [Google Scholar] [CrossRef]
- Rietbroek, N.J.; Dingboom, E.G.; Joosten, B.J.; Eizema, K.; Everts, M.E. Effect of show jumping training on the development of locomotory muscle in young horses. Am. J. Vet. Res. 2007, 68, 1232–1238. [Google Scholar] [CrossRef]
- Gouveia, J.J.d.S.; Silva, M.V.G.B.d.; Paiva, S.R.; Oliveira, S.M.P.d. Identification of selection signatures in livestock species. Genet. Mol. Biol. 2014, 37, 330–342. [Google Scholar] [CrossRef]
- Frischknecht, M.; Jagannathan, V.; Plattet, P.; Neuditschko, M.; Signer-Hasler, H.; Bachmann, I.; Pacholewska, A.; Drögemüller, C.; Dietschi, E.; Flury, C. A non-synonymous HMGA2 variant decreases height in Shetland ponies and other small horses. PLoS ONE 2015, 10, e0140749. [Google Scholar] [CrossRef]
- Ablondi, M.; Viklund, Å.; Lindgren, G.; Eriksson, S.; Mikko, S. Signatures of selection in the genome of Swedish warmblood horses selected for sport performance. BMC Genom. 2019, 20, 717. [Google Scholar] [CrossRef]
- Bordbari, M.; Penedo, M.; Aleman, M.; Valberg, S.; Mickelson, J.; Finno, C.J. Deletion of 2.7 kb near HOXD 3 in an Arabian horse with occipitoatlantoaxial malformation. Anim. Genet. 2017, 48, 287–294. [Google Scholar] [CrossRef]
- Gmel, A.I.; Druml, T.; von Niederhäusern, R.; Leeb, T.; Neuditschko, M. Genome-Wide Association Studies Based on Equine Joint Angle Measurements Reveal New QTL Affecting the Conformation of Horses. Genes 2019, 10, 370. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, L.A. Horse Conformation Analysis; Cooperative Extension, Washington State University: Pullman, WA, USA, 2001. [Google Scholar]
- Clayton, H.M.; Hobbs, S.J. An exploration of strategies used by dressage horses to control moments around the center of mass when performing passage. PeerJ 2017, 5, e3866. [Google Scholar] [CrossRef] [PubMed]
- Mallo, M.; Alonso, C.R. The regulation of Hox gene expression during animal development. Development 2013, 140, 3951–3963. [Google Scholar] [CrossRef] [PubMed]
- Pineault, K.M.; Wellik, D.M. Hox genes and limb musculoskeletal development. Curr. Osteoporos. Rep. 2014, 12, 420–427. [Google Scholar] [CrossRef]
- Xu, B.; Wellik, D.M. Axial Hox9 activity establishes the posterior field in the developing forelimb. Proc. Natl. Acad. Sci. USA 2011, 108, 4888–4891. [Google Scholar] [CrossRef]
- Barreto, R.d.S.N.; Rodrigues, M.N.; Carvalho, R.C.; De Oliveira, E.; Silva, F.M.; Rigoglio, N.N.; Jacob, J.C.F.; Gastal, E.L.; Miglino, M.A. Organogenesis of the Musculoskeletal System in Horse Embryos and Early Fetuses. Anat. Rec. 2016, 299, 722–729. [Google Scholar] [CrossRef]
- Bobbert, M.F.; Santamaría, S. Contribution of the forelimbs and hindlimbs of the horse to mechanical energy changes in jumping. J. Exp. Biol. 2005, 208, 249–260. [Google Scholar] [CrossRef]
- Schröder, W.; Klostermann, A.; Distl, O. Candidate genes for physical performance in the horse. Vet. J. 2011, 190, 39–48. [Google Scholar] [CrossRef]
- Stock, K.F.; Distl, O. Genetic correlations between conformation traits and radiographic findings in the limbs of German Warmblood riding horses. Genet. Sel. Evol. 2006, 38, 657. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biological Process | Genes | FDR |
|---|---|---|
| appendage development (GO:0048736) | HOXD9, HOXD10, HOXD12, HOXD13, and LNPK | 0.008 |
| appendage morphogenesis (GO:0035107) | HOXD9, HOXD10, HOXD12, HOXD13, and LNPK | 0.006 |
| embryonic appendage morphogenesis (GO:0035113) | HOXD9, HOXD10, HOXD12, HOXD13, and LNPK | 0.006 |
| embryonic limb morphogenesis (GO:0030326) | HOXD9, HOXD10, HOXD12, HOXD13, and LNPK | 0.011 |
| embryonic morphogenesis (GO:0048598) | HOXD9, HOXD10, HOXD12, HOXD13, MAFB, FBN1, and LNPK | 0.013 |
| forelimb morphogenesis (GO:0035136) | HOXD9, HOXD10, and LNPK | 0.032 |
| limb development (GO:0060173) | HOXD9, HOXD10, HOXD12, HOXD13, and LNPK | 0.01 |
| limb morphogenesis (GO:0035108) | HOXD9, HOXD10, HOXD12, HOXD13, and LNPK | 0.008 |
| pattern specification process (GO:0007389) | HOXD8, HOXD9, HOXD10, HOXD12, HOXD13, and MAFB | 0.015 |
| skeletal system development (GO:0001501) | HOXD8, HOXD9, HOXD10, HOXD12, HOXD13, and FBN1 | 0.03 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salek Ardestani, S.; Aminafshar, M.; Zandi Baghche Maryam, M.B.; Banabazi, M.H.; Sargolzaei, M.; Miar, Y. Whole-Genome Signatures of Selection in Sport Horses Revealed Selection Footprints Related to Musculoskeletal System Development Processes. Animals 2020, 10, 53. https://doi.org/10.3390/ani10010053
Salek Ardestani S, Aminafshar M, Zandi Baghche Maryam MB, Banabazi MH, Sargolzaei M, Miar Y. Whole-Genome Signatures of Selection in Sport Horses Revealed Selection Footprints Related to Musculoskeletal System Development Processes. Animals. 2020; 10(1):53. https://doi.org/10.3390/ani10010053
Chicago/Turabian StyleSalek Ardestani, Siavash, Mehdi Aminafshar, Mohammad Bagher Zandi Baghche Maryam, Mohammad Hossein Banabazi, Mehdi Sargolzaei, and Younes Miar. 2020. "Whole-Genome Signatures of Selection in Sport Horses Revealed Selection Footprints Related to Musculoskeletal System Development Processes" Animals 10, no. 1: 53. https://doi.org/10.3390/ani10010053
APA StyleSalek Ardestani, S., Aminafshar, M., Zandi Baghche Maryam, M. B., Banabazi, M. H., Sargolzaei, M., & Miar, Y. (2020). Whole-Genome Signatures of Selection in Sport Horses Revealed Selection Footprints Related to Musculoskeletal System Development Processes. Animals, 10(1), 53. https://doi.org/10.3390/ani10010053