GBS Data Identify Pigmentation-Specific Genes of Potential Role in Skin-Photosensitization in Two Tunisian Sheep Breeds

 , ,
, ,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Sheep Breeds
2.3. Genotyping-by-Sequencing, SNP Calling and Filtering
2.4. Population Stratification
2.5. Detection of Selection Signals
3. Results
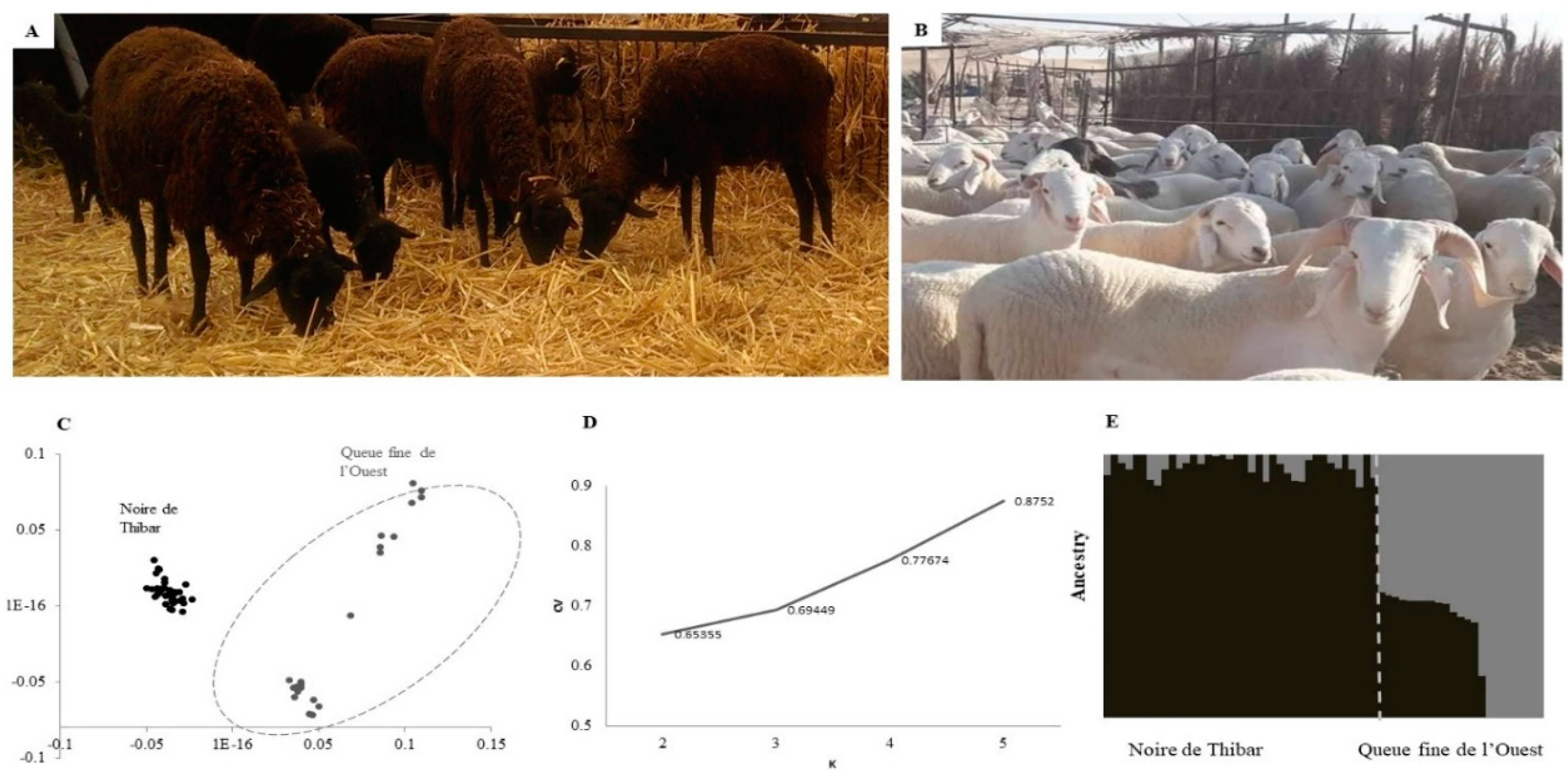
3.1. Population Differentiation
3.2. Detection of FST Outlier Loci
4. Discussion
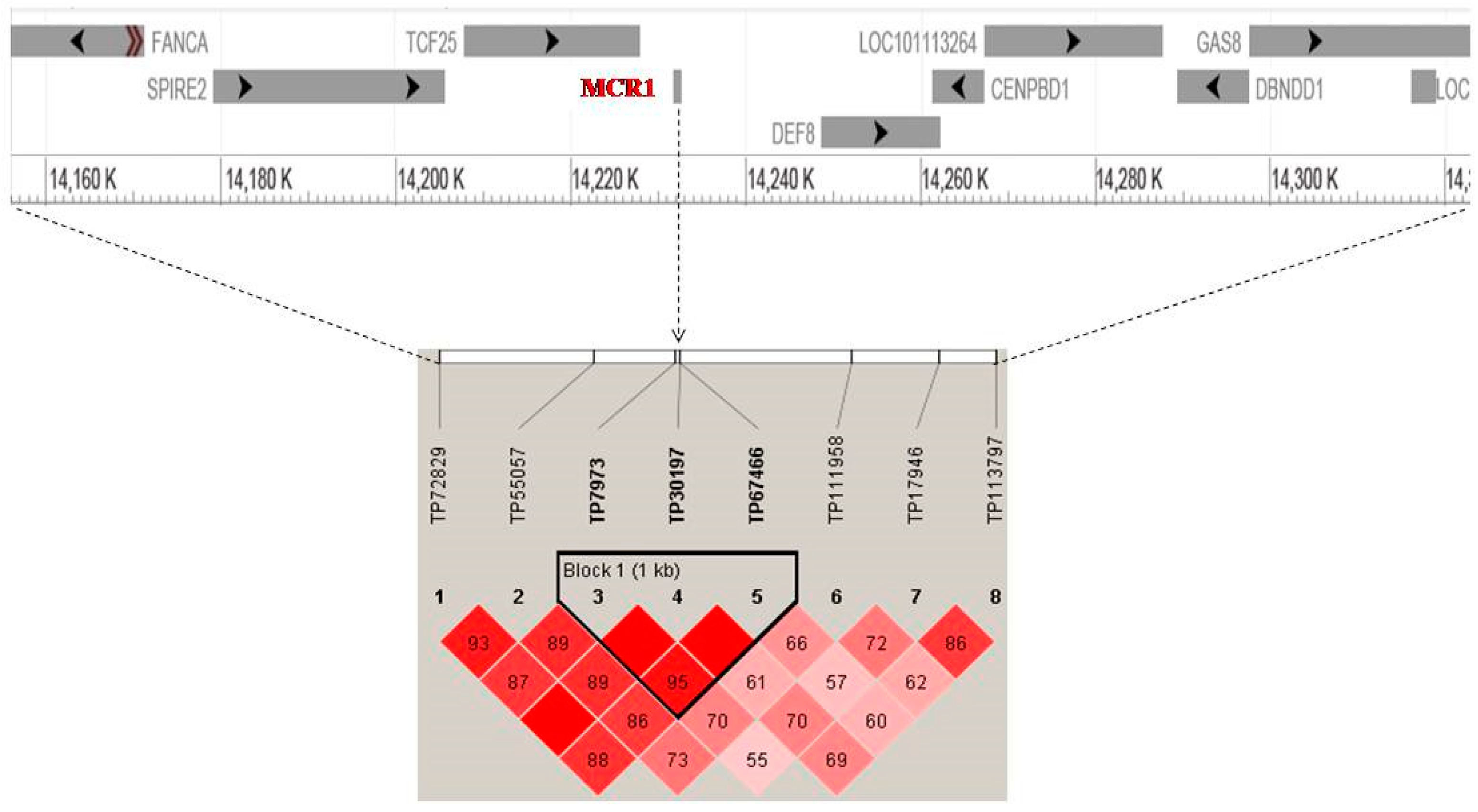
High Signals of Selection in Pigmentation Candidate Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kijas, J.W.; Lenstra, J.A.; Hayes, B.; Boitard, S.; Neto, L.R.P.; San Cristobal, M.; Servin, B.; McCulloch, R.; Whan, V.; Gietzen, K. Genome-wide analysis of the world’s sheep breeds reveals high levels of historic mixture and strong recent selection. PLoS Biol. 2012, 10, e1001258. [Google Scholar] [CrossRef] [PubMed]
- Rowe, L.D. Photosensitization problems in livestock. Vet. Clin. North Am. Food Anim. Pract. 1989, 5, 301–323. [Google Scholar] [CrossRef]
- Schempp, C.; Lüdtke, R.; Winghofer, B.; Simon, J. Effect of topical application of Hypericum perforatum extract (St. John’s wort) on skin sensitivity to solar simulated radiation. Photodermatol. Photoimmunol. Photomed. 2000, 16, 125–128. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.C.; Kessell, A.; Weston, L.A. Secondary plant products causing photosensitization in grazing herbivores: Their structure, activity and regulation. Int. J. Mol. Sci. 2014, 15, 1441–1465. [Google Scholar] [CrossRef] [PubMed]
- Schempp, C.; Müller, K.; Winghofer, B.; Schöpf, E.; Simon, J. St. John’s wort (Hypericum perforatum L.). A plant with relevance for dermatology. Der Hautarzt Z. Dermatol. Venerol. Verwandte Geb. 2002, 53, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Kallal, A. Lemoutonnoirde Thibar. Ph.D. Thesis, Ecole Nationale Vétérinaire de Toulouse, Toulouse, France, 1968; p. 62. Available online: https://books.google.tn/books?id=a8LcGwAACAAJ&dq=inauthor:%22Abdellatif+Kallal%22&hl=fr&sa=X&ved=0ahUKEwj6v726r7zmAhXRzaQKHVtjCDAQ6AEIJTAA (accessed on 17 December 2019).
- Chalh, A.; El Gazzah, M.; Djemali, M.; Chalbi, N. Genetic and phenotypic characterization of the Tunisian Noire De Thibar lambs on their growth traits. J. Biol. Sci. 2007, 7, 1347–1353. [Google Scholar]
- Rekik, M.; Aloulou, R.; Hamouda, B.M. Small ruminant breeds of Tunisia. Charact. Small Rumin. Breeds West Asia North Afr. 2005, 2, 91–140. [Google Scholar]
- Bedhiaf-Romdhani, S.; Djemali, M.; Zaklouta, M.; Iniguez, L. Monitoring crossbreeding trends in native Tunisian sheep breeds. Small Rumin. Res. 2008, 74, 274–278. [Google Scholar] [CrossRef]
- Djemali, M. Genetic improvement objectives of sheep and goats in Tunisia. Lessons learned. In Proceedings of the Options Méditerranéennes, Série ASéminaires Méditerranéens, Zaragoza, Spain, 18–20 November 1999; pp. 121–127. [Google Scholar]
- Koseniuk, A.; Ropka-Molik, K.; Rubiś, D.; Smołucha, G. Genetic background of coat colour in sheep. Arch. Anim. Breed. 2018, 61, 173–178. [Google Scholar] [CrossRef]
- Solano, F. Melanins: Skin pigments and much more—types, structural models, biological functions, and formation routes. New J. Sci. 2014, 2014, 28. [Google Scholar] [CrossRef]
- Hernando, B.; Sanz-Page, E.; Pitarch, G.; Mahiques, L.; Valcuende-Cavero, F.; Martinez-Cadenas, C. Genetic variants associated with skin photosensitivity in a southern European population from Spain. Photodermatol. Photoimmunol. Photomed. 2018, 34, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Khalesi, M.; Whiteman, D.C.; Tran, B.; Kimlin, M.G.; Olsen, C.M.; Neale, R.E. A meta-analysis of pigmentary characteristics, sun sensitivity, freckling and melanocytic nevi and risk of basal cell carcinoma of the skin. Cancer Epidemiol. 2013, 37, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Gurgul, A.; Miksza-Cybulska, A.; Szmatoła, T.; Jasielczuk, I.; Piestrzyńska-Kajtoch, A.; Fornal, A.; Semik-Gurgul, E.; Bugno-Poniewierska, M. Genotyping-by-sequencing performance in selected livestock species. Genomics 2019, 111, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Ma, H.; Xu, L.; Zhu, B.; Liu, Y.; Bordbar, F.; Chen, Y.; Zhang, L.; Gao, X.; Gao, H. Genome-wide scan identifies selection signatures in chinese wagyu cattle using a high-density SNP array. Animals 2019, 9, 296. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wu, P.; Yang, Q.; Chen, D.; Zhou, J.; Jiang, A.; Ma, J.; Tang, Q.; Xiao, W.; Jiang, Y. Detection of selection signatures in Chinese Landrace and Yorkshire pigs based on genotyping-by-sequencing data. Front. Genet. 2018, 9, 119. [Google Scholar] [CrossRef]
- Wu, P.; Yang, Q.; Wang, K.; Zhou, J.; Ma, J.; Tang, Q.; Jin, L.; Xiao, W.; Jiang, A.; Jiang, Y. Single step genome-wide association studies based on genotyping by sequence data reveals novel loci for the litter traits of domestic pigs. Genomics 2018, 110, 171–179. [Google Scholar] [CrossRef]
- Pértille, F.; Zanella, R.; Felício, A.; Ledur, M.; PEIXOTO, J.d.O.; Coutinho, L.L. Identification of polymorphisms associated with production traits on chicken (Gallus gallus) chromosome 4. Embrapa Suínos E Aves-Artig. Em Periódico Indexado (Alice) 2015, 14, 10717–10728. [Google Scholar] [CrossRef]
- Bahbahani, H.; Musa, H.H.; Wragg, D.; Shuiep, E.S.; Almathen, F.; Hanotte, O. Genome diversity and signatures of selection for production and performance traits in dromedary camels. Front. Genet. 2019, 10, 893. [Google Scholar] [CrossRef]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1989. [Google Scholar]
- Dodds, K.G.; McEwan, J.C.; Brauning, R.; Anderson, R.M.; van Stijn, T.C.; Kristjánsson, T.; Clarke, S.M. Construction of relatedness matrices using genotyping-by-sequencing data. BMC Genom. 2015, 16, 1047. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef]
- Lu, F.; Lipka, A.E.; Glaubitz, J.; Elshire, R.; Cherney, J.H.; Casler, M.D.; Buckler, E.S.; Costich, D.E. Switchgrass genomic diversity, ploidy, and evolution: Novel insights from a network-based SNP discovery protocol. PLoS Genet. 2013, 9, e1003215. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.; Daly, M.J. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Lange, K. Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinform. 2011, 12, 246. [Google Scholar] [CrossRef] [PubMed]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef]
- Jiang, Y.; Xie, M.; Chen, W.; Talbot, R.; Maddox, J.F.; Faraut, T.; Wu, C.; Muzny, D.M.; Li, Y.; Zhang, W. The sheep genome illuminates biology of the rumen and lipid metabolism. Science 2014, 344, 1168–1173. [Google Scholar] [CrossRef]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 2004, 21, 263–265. [Google Scholar] [CrossRef]
- Sassi-Zaidy, Y.B.; Maretto, F.; Charfi-Cheikrouha, F.; Cassandro, M. Genetic diversity, structure, and breed relationships in Tunisian sheep. Small Rumin. Res. 2014, 119, 52–56. [Google Scholar] [CrossRef]
- Chang, L.-Y.; Toghiani, S.; Ling, A.; Aggrey, S.E.; Rekaya, R. High density marker panels, SNPs prioritizing and accuracy of genomic selection. BMC Genet. 2018, 19, 4. [Google Scholar]
- Smith, J.M.; Haigh, J. The hitch-hiking effect of a favourable gene. Genet. Res. 1974, 23, 23–35. [Google Scholar] [CrossRef]
- Grossman, S.R.; Shylakhter, I.; Karlsson, E.K.; Byrne, E.H.; Morales, S.; Frieden, G.; Hostetter, E.; Angelino, E.; Garber, M.; Zuk, O. A composite of multiple signals distinguishes causal variants in regions of positive selection. Science 2010, 327, 883–886. [Google Scholar] [CrossRef]
- Gupta, R.C. Veterinary Toxicology: Basic and Clinical Principles; Academic Press: Cambridge, MA, USA, 2012. [Google Scholar]
- Fu, P.P.; Xia, Q.; Zhao, Y.; Wang, S.; Yu, H.; Chiang, H.-M. Phototoxicity of herbal plants and herbal products. J. Environ. Sci. HealthPart C 2013, 31, 213–255. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Quinn, J.C.; Weston, L.A.; Loukopoulos, P. The aetiology, prevalence and morbidity of outbreaks of photosensitisation in livestock: A review. PLoS ONE 2019, 14, e0211625. [Google Scholar] [CrossRef]
- Li, J.; Yang, H.; Li, J.; Li, H.; Ning, T.; Pan, X.; Shi, P.; Zhang, Y. Artificial selection of the melanocortin receptor 1 gene in Chinese domestic pigs during domestication. Heredity 2010, 105, 274. [Google Scholar] [CrossRef] [PubMed]
- Gouveia, J.J.d.S.; Silva, M.V.G.B.d.; Paiva, S.R.; Oliveira, S.M.P.d. Identification of selection signatures in livestock species. Genet. Mol. Biol. 2014, 37, 330–342. [Google Scholar] [CrossRef] [PubMed]
- Kijas, J.; Serrano, M.; McCulloch, R.; Li, Y.; Salces Ortiz, J.; Calvo, J.; Pérez-Guzmán, M.; Consortium, I.S.G. Genomewide association for a dominant pigmentation gene in sheep. J. Anim. Breed. Genet. 2013, 130, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Fontanesi, L.; Dall’Olio, S.; Beretti, F.; Portolano, B.; Russo, V. Coat colours in the Massese sheep breed are associated with mutations in the agouti signalling protein (ASIP) and melanocortin 1 receptor (MC1R) genes. Animal 2011, 5, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Kijas, J.; Moller, M.; Plastow, G.; Andersson, L. A frameshift mutation in MC1R and a high frequency of somatic reversions cause black spotting in pigs. Genetics 2001, 158, 779–785. [Google Scholar] [PubMed]
- Kennedy, C.; ter Huurne, J.; Berkhout, M.; Gruis, N.; Bastiaens, M.; Bergman, W.; Willemze, R.; Bavinck, J.N.B. Melanocortin 1 receptor (MC1R) gene variants are associated with an increased risk for cutaneous melanoma which is largely independent of skin type and hair color. J. Investig. Dermatol. 2001, 117, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Latreille, J.; Ezzedine, K.; Elfakir, A.; Ambroisine, L.; Gardinier, S.; Galan, P.; Hercberg, S.; Gruber, F.; Rees, J.; Tschachler, E. MC1R gene polymorphism affects skin color and phenotypic features related to sun sensitivity in a population of French adult women. Photochem. Photobiol. 2009, 85, 1451–1458. [Google Scholar] [CrossRef] [PubMed]
- Hanna, L.L.H.; Sanders, J.O.; Riley, D.G.; Abbey, C.A.; Gill, C.A. Identification of a major locus interacting with MC1R and modifying black coat color in an F2 Nellore-Angus population. Genet. Sel. Evol. 2014, 46, 4. [Google Scholar] [CrossRef] [PubMed]
- Nazari-Ghadikolaei, A.; Mehrabani-Yeganeh, H.; Miarei-Aashtiani, S.R.; Staiger, E.A.; Rashidi, A.; Huson, H.J. Genome-wide association studies identify candidate genes for coat color and mohair traits in the iranian markhoz goat. Front. Genet. 2018, 9, 105. [Google Scholar] [CrossRef] [PubMed]
- Sulem, P.; Gudbjartsson, D.F.; Stacey, S.N.; Helgason, A.; Rafnar, T.; Magnusson, K.P.; Manolescu, A.; Karason, A.; Palsson, A.; Thorleifsson, G. Genetic determinants of hair, eye and skin pigmentation in Europeans. Nat. Genet. 2007, 39, 1443. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Kraft, P.; Nan, H.; Guo, Q.; Chen, C.; Qureshi, A.; Hankinson, S.E.; Hu, F.B.; Duffy, D.L.; Zhao, Z.Z. A genome-wide association study identifies novel alleles associated with hair color and skin pigmentation. PLoS Genet. 2008, 4, e1000074. [Google Scholar] [CrossRef]
- Praetorius, C.; Grill, C.; Stacey, S.N.; Metcalf, A.M.; Gorkin, D.U.; Robinson, K.C.; Van Otterloo, E.; Kim, R.S.; Bergsteinsdottir, K.; Ogmundsdottir, M.H. A polymorphism in IRF4 affects human pigmentation through a tyrosinase-dependent MITF/TFAP2A pathway. Cell 2013, 155, 1022–1033. [Google Scholar] [CrossRef]
- Guo, J.; Tao, H.; Li, P.; Li, L.; Zhong, T.; Wang, L.; Ma, J.; Chen, X.; Song, T.; Zhang, H. Whole-genome sequencing reveals selection signatures associated with important traits in six goat breeds. Sci. Rep. 2018, 8, 10405. [Google Scholar] [CrossRef]
- Seroussi, E.; Rosov, A.; Shirak, A.; Lam, A.; Gootwine, E. Unveiling genomic regions that underlie differences between Afec-Assaf sheep and its parental Awassi breed. Genet. Sel. Evol. 2017, 49, 19. [Google Scholar] [CrossRef]
- Fariello, M.-I.; Servin, B.; Tosser-Klopp, G.; Rupp, R.; Moreno, C.; San Cristobal, M.; Boitard, S.; Consortium, I.S.G. Selection signatures in worldwide sheep populations. PLoS ONE 2014, 9, e103813. [Google Scholar] [CrossRef]
- Gunnarsson, U.; Kerje, S.; Bed’hom, B.; Sahlqvist, A.S.; Ekwall, O.; Tixier-Boichard, M.; Kämpe, O.; Andersson, L. The Dark brown plumage color in chickens is caused by an 8.3-kb deletion upstream of SOX10. Pigment Cell Melanoma Res. 2011, 24, 268–274. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Region | OAR | Significant SNP | Genomic Region | ||||
|---|---|---|---|---|---|---|---|
| FST 1 | Position (pb) 2 | Markers 3 | Length (Kb) | Genes 4 | Candidate Genes | ||
| 1 | 14 | 0.97 | 14231897 | 8 | 576 | 24 | MC1R |
| 2 | 6 | 0.78 | 70008012 | 5 | 487 | 5 | PDGFRA, KIT |
| 3 | 20 | 0.72 | 50553857 | 10 | 611 | 8 | IRF4, PICK1 |
| 4 | 3 | 0.63 | 213795317 | 3 | 416 | 17 | SOX10, PICK1 |
| 5 | 20 | 0.54 | 38835509 | 1 | 400 | 8 | - |
| 6 | 11 | 0.52 | 32377178 | 1 | 400 | 7 | - |
| 7 | 10 | 0.51 | 35837558 | 1 | 400 | 14 | - |
| 8 | 13 | 0.51 | 56477753 | 1 | 400 | 6 | EDN3 |
| 9 | 4 | 0.50 | 97859387 | 1 | 400 | 8 | - |
| 10 | 11 | 0.49 | 53654935 | 1 | 400 | 8 | - |
| 11 | 2 | 0.48 | 247265800 | 1 | 400 | 4 | - |
| 12 | 20 | 0.46 | 44022745 | 1 | 400 | 5 | - |
| 13 | 17 | 0.46 | 44834726 | 1 | 400 | 7 | - |
| 14 | 10 | 0.46 | 36042096 | 1 | 400 | 8 | - |
| 15 | 22 | 0.46 | 50353516 | 1 | 400 | 10 | - |
| 16 | 25 | 0.45 | 1327751 | 1 | 400 | 1 | - |
| 17 | 16 | 0.45 | 28645498 | 1 | 400 | 5 | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baazaoui, I.; McEwan, J.; Anderson, R.; Brauning, R.; McCulloch, A.; Van Stijn, T.; Bedhiaf-Romdhani, S. GBS Data Identify Pigmentation-Specific Genes of Potential Role in Skin-Photosensitization in Two Tunisian Sheep Breeds. Animals 2020, 10, 5. https://doi.org/10.3390/ani10010005
Baazaoui I, McEwan J, Anderson R, Brauning R, McCulloch A, Van Stijn T, Bedhiaf-Romdhani S. GBS Data Identify Pigmentation-Specific Genes of Potential Role in Skin-Photosensitization in Two Tunisian Sheep Breeds. Animals. 2020; 10(1):5. https://doi.org/10.3390/ani10010005
Chicago/Turabian StyleBaazaoui, Imen, John McEwan, Rayna Anderson, Rudiger Brauning, Alan McCulloch, Tracey Van Stijn, and Sonia Bedhiaf-Romdhani. 2020. "GBS Data Identify Pigmentation-Specific Genes of Potential Role in Skin-Photosensitization in Two Tunisian Sheep Breeds" Animals 10, no. 1: 5. https://doi.org/10.3390/ani10010005
APA StyleBaazaoui, I., McEwan, J., Anderson, R., Brauning, R., McCulloch, A., Van Stijn, T., & Bedhiaf-Romdhani, S. (2020). GBS Data Identify Pigmentation-Specific Genes of Potential Role in Skin-Photosensitization in Two Tunisian Sheep Breeds. Animals, 10(1), 5. https://doi.org/10.3390/ani10010005