Metabolomic Profile of Primary Turkey and Rat Hepatocytes and Two Cell Lines after Chloramphenicol Exposure


Abstract
Simple Summary
Abstract
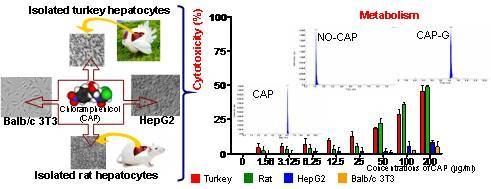
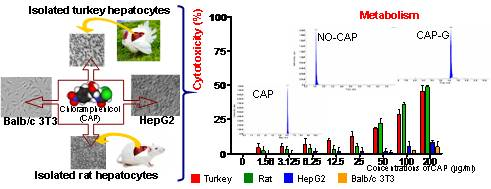
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Compound Preparation and Exposure
2.3. Cellular Models
2.3.1. The Primary Turkey Hepatocyte Cultures
Animals
Turkey Hepatocytes Isolation and Culture Conditions
2.3.2. Rat Hepatocyte Isolation and Primary Hepatocyte Culture Conditions
2.3.3. Cell Lines and Culture Conditions
2.4. Determination of CAP and Its Metabolized Products by HPLC-MS/MS
2.5. Cytotoxicity Assessment
2.5.1. LDH Assay
2.5.2. MTT Assay
2.5.3. NRU Assay
2.6. Statistical Analysis
3. Results
3.1. Primary Turkey and Rat Hepatocyte Cultures
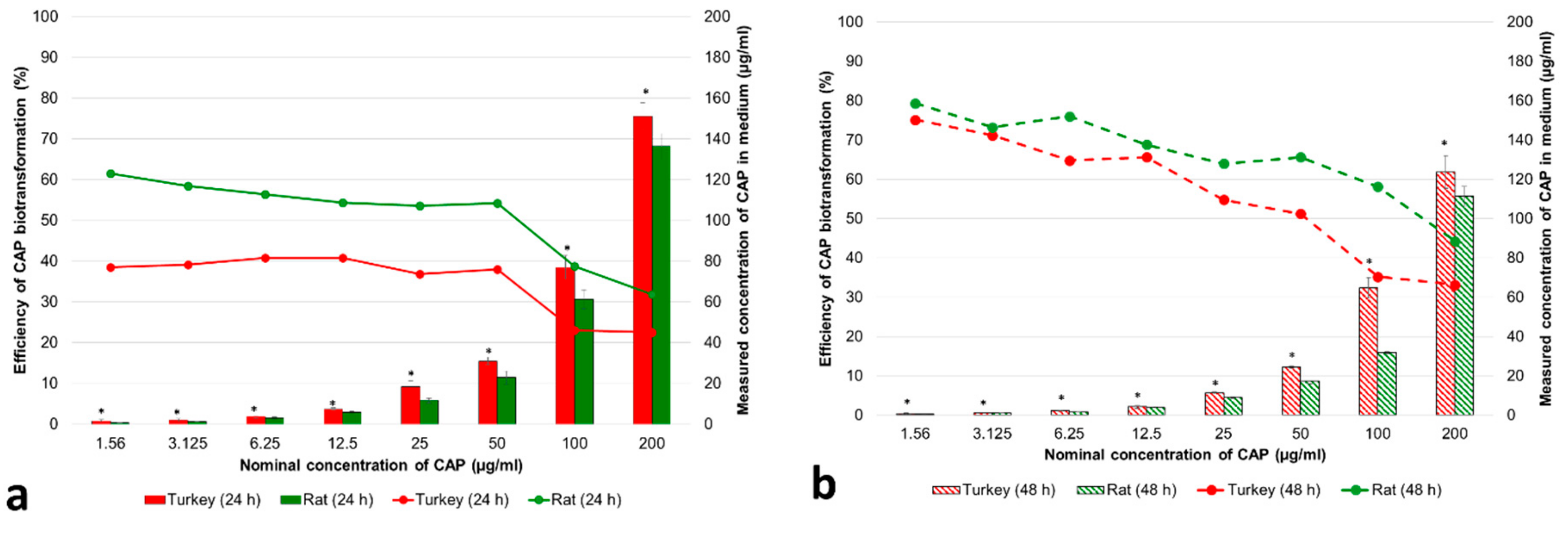
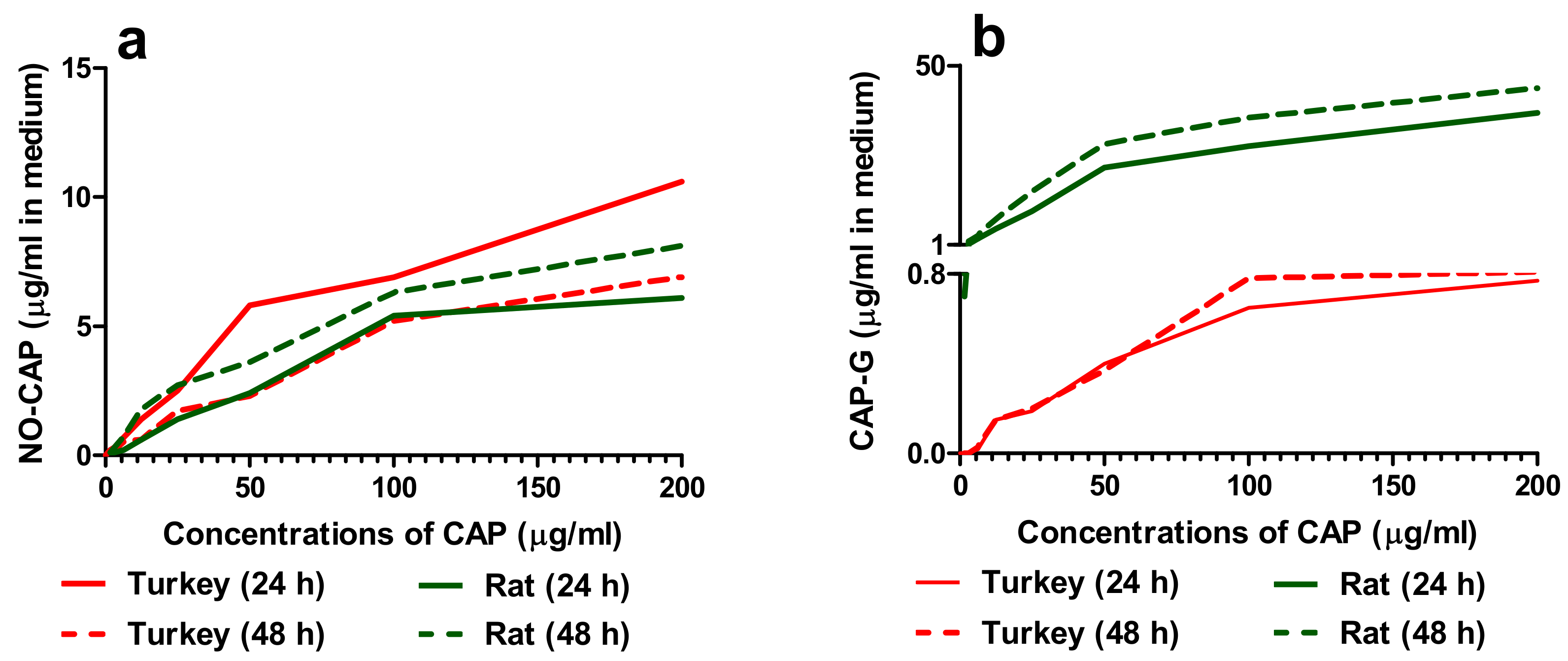
3.1.1. Concentrations of CAP and Its Metabolites in the Cell Medium
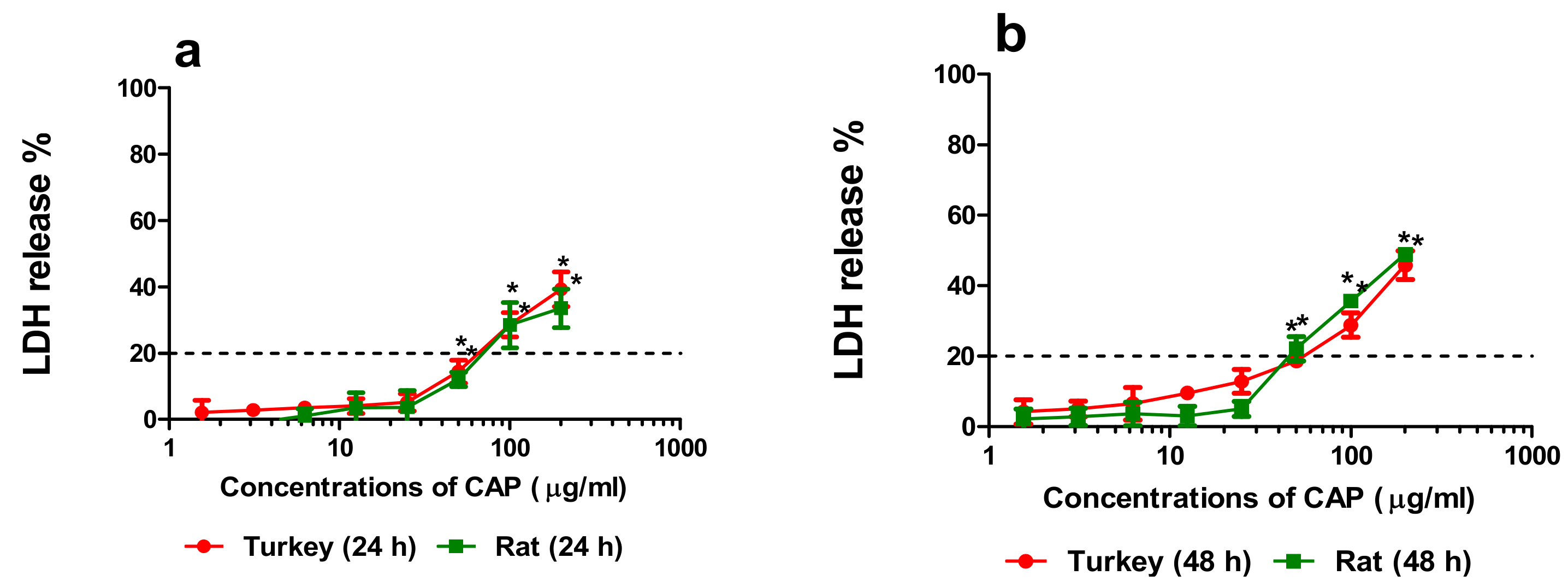
3.1.2. Cytotoxicity
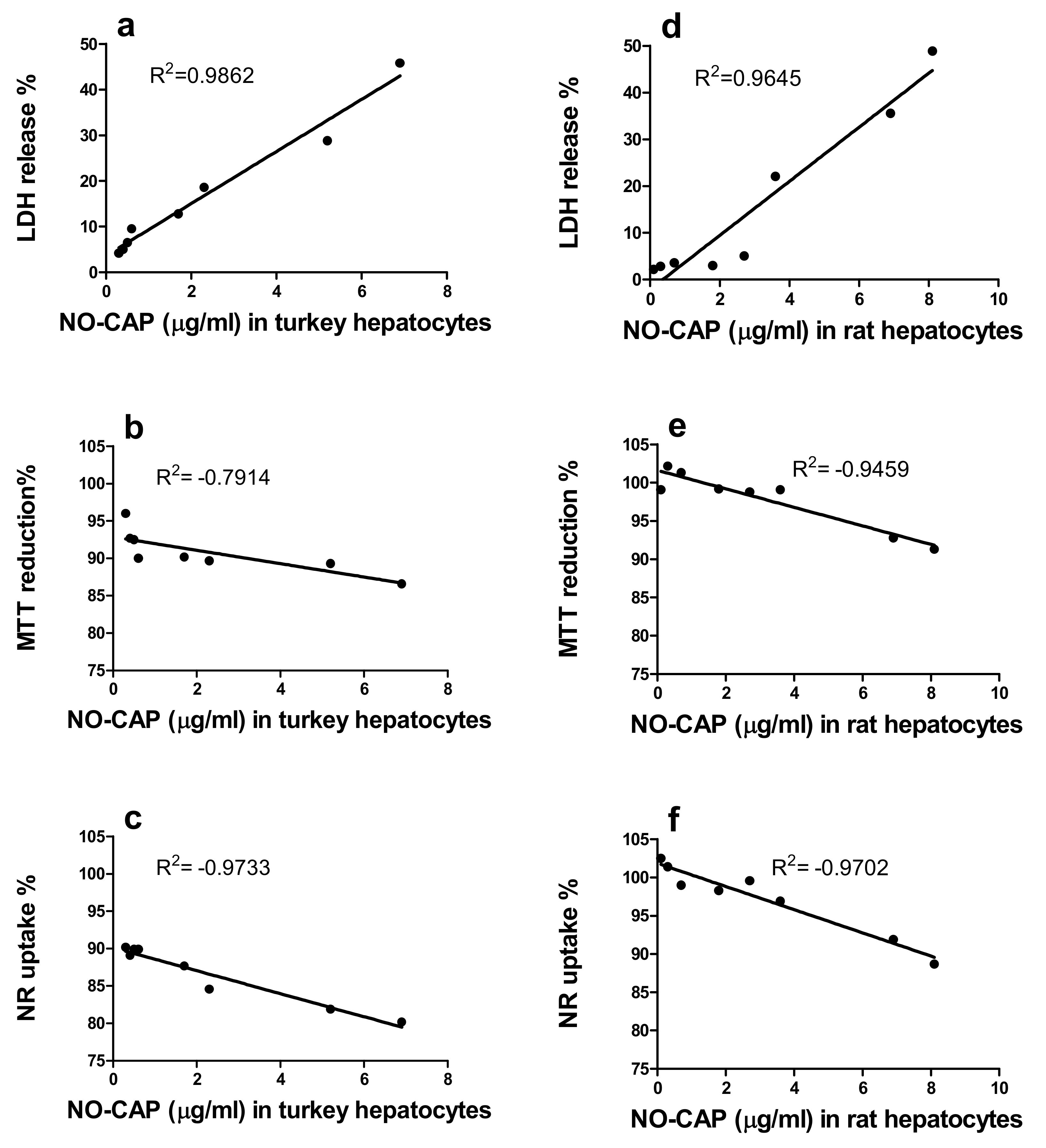
3.1.3. Correlation of the Quantity of NO-CAP and CAP-G in Medium with LDH Release, MTT Reduction, and NR Uptake in the Primary Hepatocytes
3.2. Human Hepatoma (HepG2) Cell and Balb/c 3T3 Fibroblast Cultures
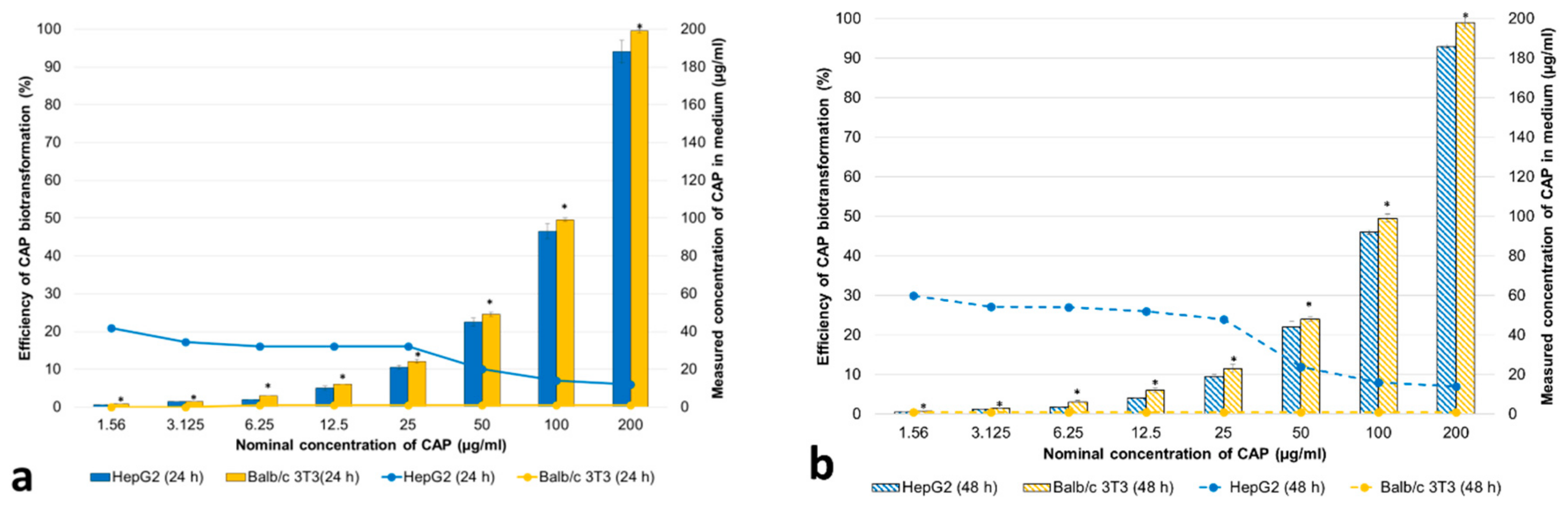
3.2.1. Concentrations of CAP and Its Metabolites in the Cell Medium
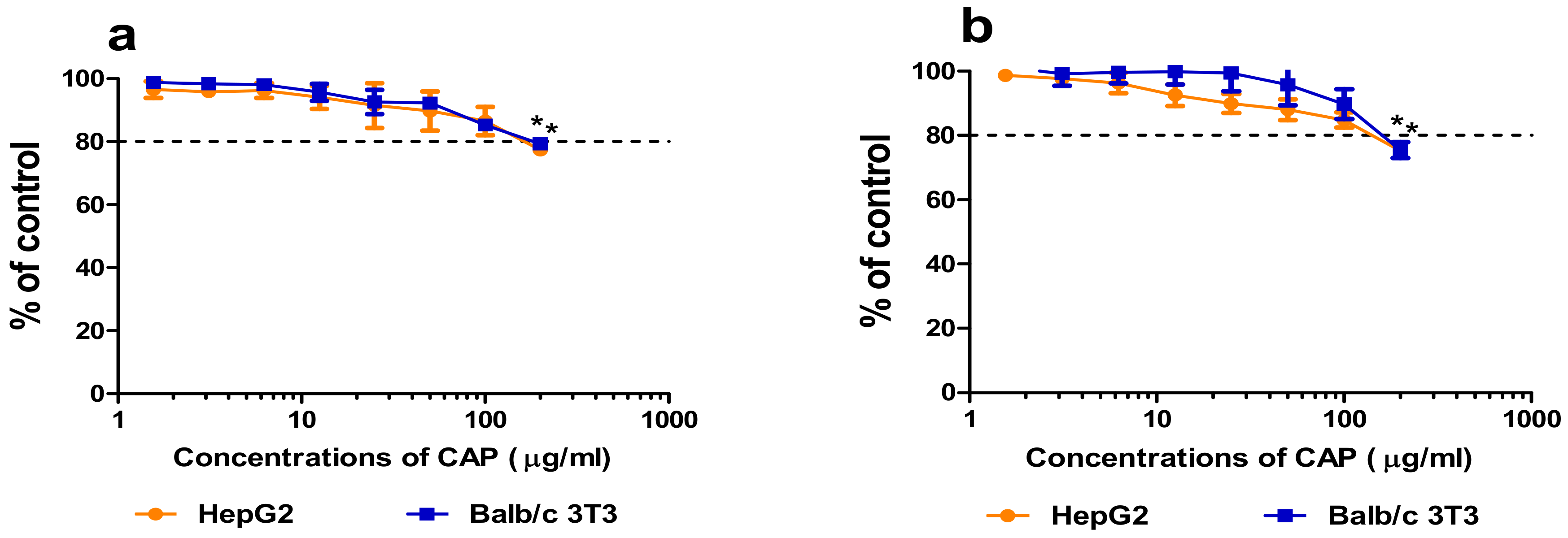
3.2.2. Cytotoxicity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- IARC. Monographs on the Evaluation of Carcinogenic Risk of Chemicals to Humans. Volume 50, Pharmaceutical Drugs; Chloramphenicol; IARC: Lyon, France, 1990; pp. 169–193. [Google Scholar]
- EFSA. Scientific opinion on chloramphenicol in food and feed. EFSA J. 2014, 12, 3907. [Google Scholar] [CrossRef]
- Śniegocki, T.; Sell, B.; Giergiel, M.; Posyniak, A. QuEChERS and HPLC-MS/MS Combination for the Determination of Chloramphenicol in Twenty Two Different Matrices. Molecules 2019, 24, 384. [Google Scholar] [CrossRef] [PubMed]
- Śniegocki, T.; Gbylik-Sikorska, M.; Posyniak, A. Transfer of chloramphenicol from milk to commercial dairy products – experimental proof. Food Control 2015, 57, 411–418. [Google Scholar] [CrossRef]
- Rønning, H.T.; Einarsen, K.; Asp, T.N. Determination of chloramphenicol residue in meat, seafood, egg, honey, milk, plasma and urine with liquid chromatography-tandem mass spectrometry, and the validation of the method based on 2002/657/EC. J. Chromatogr. A 2006, 1118, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Aresta, A.; Bianchi, D.; Calvano, C.D.; Zambonin, C.G. Solid phase microextraction-liquid chromatography (SPME-LC) determination of chloramphenicol in urine and environmental water samples. J. Pharm. Biomed. Anal. 2010, 53, 440–444. [Google Scholar] [CrossRef]
- Choi, K.; Kim, Y.; Jung, J.; Kim, M.H.; Kim, Ch.S.; Kim, N.H.; Park, J. Occurrences and ecological risks of roxithromycin, trimethoprim, and chloramphenicol in the Han River, Korea. Environ. Toxicol. Chem. 2008, 27, 711–719. [Google Scholar] [CrossRef]
- Hanekamp, J.C.; Bast, A. Antibiotics exposure and health risks: Chloramphenicol. Environ. Toxicol. Pharmacol. 2015, 39, 213–220. [Google Scholar] [CrossRef]
- Berendsen, B.; Stolker, L.; de Jong, J.; Nielen, M.; Tserendorj, E.; Sodnomdarjaa, R. Evidence of natural occurrence of the banned antibiotic chloramphenicol in herbs and grass. Anal. BioAnal. Chem. 2010, 397, 1955–1963. [Google Scholar] [CrossRef]
- Holt, D.E.; Bajoria, R. The role of nitro-reduction and nitric oxide in the toxicity of chloramphenicol. Hum. Exp. Toxicol. 1999, 18, 111–118. [Google Scholar] [CrossRef]
- Büdingen, F.V. Relevance of liver failure for anti-infective agents: From pharmacokinetic alterations to dosage adjustments. Ther. Adv. Infect. Dis. 2014, 2, 17–42. [Google Scholar] [CrossRef]
- Isildar, M.; Abou-Khalil, W.H.; Jimenez, J.J.; Abou-Khalil, S.; Yunis, A.A. Aerobic nitroreduction of dehydrochloramphenicol by bone marrow. Toxicol. Appl. Pharmacol. 1988, 94, 305–310. [Google Scholar] [CrossRef]
- Switała, M.; Hrynyk, R.; Smutkiewicz, A.; Jaworski, K.; Pawlowski, P.; Okoniewski, P.; Grabowski, T.; Debowy, J. Pharmacokinetics of florfenicol, thiamphenicol, and chloramphenicol in turkeys. J. Vet. Pharmacol. Ther. 2007, 30, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Van den Hof, W.F.P.M.; Van Summeren, A.; Lommen, A.; Coonen, M.L.; Brauers, K.; van Herwijnen, M.; Wodzig, W.K.; Kleinjans, J.C. Integrative cross-omics analysis in primary mouse hepatocytes unravels mechanisms of cyclosporin A-induced hepatotoxicity. Toxicology 2014, 324, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Vermeersch, K.A.; Wang, L.; Mezencev, R.; McDonald, J.F.; Styczynski, M.P. OVCAR-3 spheroid-derived cells display distinct metabolic profiles. PLoS ONE 2015, 10, e0118262. [Google Scholar] [CrossRef]
- Bouhifd, M.; Hartung, T.; Hogberg, H.T.; Kleensang, A.; Zhao, L. Review: Toxicometabolomics. J. Appl. Toxicol. 2013, 33, 1365–1383. [Google Scholar] [CrossRef]
- Gerets, H.H.J.; Hanon, E.; Cornet, M.; Dhalluin, S.; Depelchin, O.; Canning, M.; Atienzar, F.A. Selection of cytotoxicity markers for the screening of new chemical entities in a pharmaceutical context: A preliminary study using a multiplexing approach. Toxicol. Vitr. 2009, 23, 319–332. [Google Scholar] [CrossRef]
- Ivanova, L.; Uhlig, S. A bioassay for the simultaneous measurement of metabolic activity, membrane integrity, and lysosomal activity in cell cultures. Anal. BioChem. 2008, 379, 16–19. [Google Scholar] [CrossRef]
- Pohjala, L.; Tammela, P.; Samanta, S.K.; Yli-Kauhaluoma, J.; Vuorela, P. Assessing the data quality in predictive toxicology using a panel of cell lines and cytotoxicity assays. Anal. BioChem. 2007, 362, 221–228. [Google Scholar] [CrossRef]
- Fotakis, G.; Timbrell, J.A. In vitro cytotoxicity assays: Comparison of LDH, neutral red, MTT and protein assay in hepatoma cell lines following exposure to cadmium chloride. Toxicol. Lett. 2006, 160, 171–177. [Google Scholar] [CrossRef]
- Weyermann, J.; Lohmann, D.; Zimmer, A. A practical note on the use of cytotoxicity assays. Int. J. Pharm. 2005, 288, 369–376. [Google Scholar] [CrossRef]
- Hewitt, N.J.; Lechón, M.J.; Houston, J.B.; Hallifax, D.; Brown, H.S.; Maurel, P.; Kenna, J.G.; Gustavsson, L.; Lohmann, C.; Skonberg, C.; et al. Primary hepatocytes: Current understanding of the regulation of metabolic enzymes and transporter proteins, and pharmaceutical practice for the use of hepatocytes in metabolism, enzyme induction, transporter, clearance, and hepatotoxicity studies. Drug Metab. Rev. 2007, 39, 159–234. [Google Scholar] [CrossRef] [PubMed]
- Shulman, M.; Nahmias, Y. Long-term culture and coculture of primary rat and human hepatocytes. Methods Mol. Biol. 2013, 945, 287–302. [Google Scholar] [CrossRef] [PubMed]
- Radko, L.; Minta, M.; Stypuła-Trębas, S. Differential toxicities of albendazole and its two main metabolites to Balb/c 3T3, HepG2, and FaO lines and rat hepatocytes. J. Vet. Res. 2016, 60, 495–500. [Google Scholar] [CrossRef][Green Version]
- Radko, L.; Minta, M.; Jedziniak, P.; Stypuła-Trębas, S. Comparison of albendazole cytotoxicity in terms of metabolite formation in four model systems. J. Vet. Res. 2017, 61, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Donato, M.T.; Lahoz, A.; Castell, J.V.; Gómez-Lechón, M.J. Cell lines: A tool for in vitro drug metabolism studies. Curr. Drug Metab. 2008, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Scheers, E.M.; Ekwall, B.; Dierickx, P.J. In vitro long-term cytotoxicity testing of 27 MEIC chemicals on HepG2 cells and comparison with acute human toxicity data. Toxicol. Vitr. 2001, 15, 153–161. [Google Scholar] [CrossRef]
- Schoonen, W.G.E.J.; de Roos, J.A.D.M.; Westerink, W.M.A.; Débiton, E. Cytotoxic effects of 110 reference compounds on HepG2 cells and for 60 compounds on HeLa, ECC-1, and CHO cells. II Mechanistic assays on NAD(P)H, ATP and DNA contents. Toxicol. Vitr. 2005, 19, 491–503. [Google Scholar] [CrossRef]
- Schoonen, W.G.E.J.; Westerink, W.M.A.; de Roos, J.A.D.M.; Débiton, E. Cytotoxic effects of 100 reference compounds on HepG2 and HeLa cells and of 60 compounds on ECC-1 and CHO cells. I Mechanistic assays on ROS, glutathione depletion, and calcium uptake. Toxicol. Vitr. 2005, 19, 505–516. [Google Scholar] [CrossRef]
- Ramirez, T.; Strigun, A.; Verlohner, A.; Huener, H.A.; Peter, E.; Herold, M.; Bordag, N.; Mellert, W.; Walk, T.; Spitzer, M.; et al. Prediction of liver toxicity and mode of action using metabolomics in vitro in HepG2 cells. Arch. Toxicol. 2018, 92, 893–906. [Google Scholar] [CrossRef]
- OECD. Guidance Document on Using Cytotoxicity Tests to Estimate Starting Doses for Acute Oral Systemic Toxicity Tests; no. 129. Report No.: ENV/JM/MONO(2010)20; OECD: Paris, France, 2010. [Google Scholar]
- Fraslin, J.M.; Touquette, L.; Douaire, M.; Menezo, Y.; Guillemot, J.C.; Mallard, J. Isolation and long-term maintenance of differentiated adult chicken hepatocytes in primary culture. Vitr. Cell Dev. Biol. 1992, 28, 615–620. [Google Scholar] [CrossRef]
- Sante, E. Guidance Document on Analytical Quality Control and Method Validation Procedures for Pesticides Residues Analysis in Food and Feed. 2015 SANTE/11945/2015. Available online: http://www.eurl-pesticides. eu/library/docs/allcrl/AqcGuidance_SANTE_2015_11945.pdf (accessed on 19 October 2019).
- Korzeniewski, C.; Calleawert, D.M. An enzyme-release assay for natural cytotoxicity. J. Immunol. Methods 1983, 64, 313–320. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assay. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Borenfreund, E.; Puerner, J.A. Toxicity determined in vitro by morphological alterations and neutral red absorption. Toxicol. Lett. 1985, 24, 119–124. [Google Scholar] [CrossRef]
- Duewelhenk, N.; Krut, O.; Eysel, P. Influence on mitochondria and cytotoxicity of different antibiotics administered in high concentrations on primary human osteoblasts and cell lines. Antimicrob. Agents Chemo.Ther. 2007, 51, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Robbana-Barnat, S.; Decloite, F.; Frayssinet, C.; Seigneurin, J.M.; Toucas, L.; Lafarge-Frayssinet, C. Use of human lymphoblastoid cells to detect the toxic effect of chloramphenicol and metabolites possibly involved in aplastic anemia in man. Drug Chem. Toxicol. 1997, 20, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Lafarge-Frayssinet, L.; Robbana-Barnat, S.; Fryssinet, C.; Toucas, L.; Decloitre, F. Cytotoxicity and DNA damaging potency of chloramphenicol and six metabolites: A new evaluation in human lymphocytes and Raji cells. Mutat. Res. 1994, 320, 207–215. [Google Scholar] [CrossRef]
- Martelli, A.; Mattioli, F.; Pastorino, G.; Robbiano, L.; Allavena, A.; Brambilla, G. Genotoxicity testing of chloramphenicol in rodent and human cells. Mutat. Res. 1991, 260, 65–72. [Google Scholar] [CrossRef]
- Chen, M.; LeDuc, B.; Kerr, S.; Howe, D.; Williams, D.A. Identification of human UGT2B7 as the major isoform involved in the O-glucuronidation of chloramphenicol. Drug Metab. Dispos. 2010, 38, 368–375. [Google Scholar] [CrossRef]
- Anadon, A.; Bringas, P.; Martinez-Larranaga, M.R.; Diaz, M.J. Bioavailability pharmacokinetics and residue of chloramphenicol in the chicken. J. Vet. Pharmacol. Ther. 1994, 17, 52–58. [Google Scholar] [CrossRef]
- Denbow, D.M. Gastrointestinal anatomy and physiology. In Sturkie’s Avian Physiology, 6th ed.; Scanes, G.C., Ed.; Academic Press: London, UK, 2000; pp. 299–325. [Google Scholar] [CrossRef]
- Ascherly, M.; Eyer, P.; Kampffmeyer, H. Formation and disposition of nitrosochloramphenicol in rat liver. BioChem. Pharmacol. 1985, 35, 3755–3763. [Google Scholar] [CrossRef]
- Wu, C.; Clift, P.; Fry, C.H.; Henry, J.A. Membrane action of chloramphenicol measured by protozoan motility inhibition. Arch. Toxicol. 1996, 70, 850–853. [Google Scholar] [CrossRef] [PubMed]
- Yunis, A.A. Chloramphenicol: Relation of structure to activity and toxicity. Annu. Rev. Pharmacol. Toxicol. 1988, 28, 83–100. [Google Scholar] [CrossRef] [PubMed]
- Castell, J.V.; Jover, R.; Martinez-Jiménez, C.P.; Gómez-Lechón, M.J. Hepatocyte cell lines: Their use, scope, and limitations in drug metabolism studies. Expert Opin. Drug Metab. Toxicol. 2006, 2, 183–212. [Google Scholar] [CrossRef] [PubMed]
- Westerink, W.M.; Schoonen, W.G. Phase II enzyme levels in HepG2 cells and cryopreserved primary human hepatocytes and their induction in HepG2 cells. Toxicol. Vitr. 2007, 21, 1592–1602. [Google Scholar] [CrossRef]
- Karbowski, M.; Kurono, C.; Wozniak, M.; Ostrowski, M.; Teranishi, M.; Soji, T.; Wakabayashi, T. Cycloheximide and 4-OH-TEMPO suppress chloramphenicol-induced apoptosis in RL-34 cells via the suppression of the formation of megamitochondia. Biochim. Biophys. Acta Mol. Cell Res. 1999, 1449, 25–40. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LDH | MTT | NRU | ||||
|---|---|---|---|---|---|---|
| 24 h | 48 h | 24 h | 48 h | 24 h | 48 h | |
| PTH | 67.0 ± 3.4 a | 54.1 ± 2.4 a | ne | ne | ne | ne |
| PRH | 81.3 ± 3.6 b | 47.5 ± 1.9 b | ne | ne | ne | ne |
| LDH | MTT | NRU | ||||
|---|---|---|---|---|---|---|
| 24 h | 48 h | 24 h | 48 h | 24 h | 48 h | |
| HepG2 | ne | ne | ne | 169 ± 13.0 a | ne | 152 ± 4.3 a |
| Balb/c3T3 | ne | ne | ne | 182 ± 13.5 a | ne | 168 ± 13.0 a |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Radko, L.; Śniegocki, T.; Sell, B.; Posyniak, A. Metabolomic Profile of Primary Turkey and Rat Hepatocytes and Two Cell Lines after Chloramphenicol Exposure. Animals 2020, 10, 30. https://doi.org/10.3390/ani10010030
Radko L, Śniegocki T, Sell B, Posyniak A. Metabolomic Profile of Primary Turkey and Rat Hepatocytes and Two Cell Lines after Chloramphenicol Exposure. Animals. 2020; 10(1):30. https://doi.org/10.3390/ani10010030
Chicago/Turabian StyleRadko, Lidia, Tomasz Śniegocki, Bartosz Sell, and Andrzej Posyniak. 2020. "Metabolomic Profile of Primary Turkey and Rat Hepatocytes and Two Cell Lines after Chloramphenicol Exposure" Animals 10, no. 1: 30. https://doi.org/10.3390/ani10010030
APA StyleRadko, L., Śniegocki, T., Sell, B., & Posyniak, A. (2020). Metabolomic Profile of Primary Turkey and Rat Hepatocytes and Two Cell Lines after Chloramphenicol Exposure. Animals, 10(1), 30. https://doi.org/10.3390/ani10010030