Mitogenome Diversity and Maternal Origins of Guangxi Cattle Breeds


Simple Summary
Abstract
1. Introduction
2. Materials and Methods
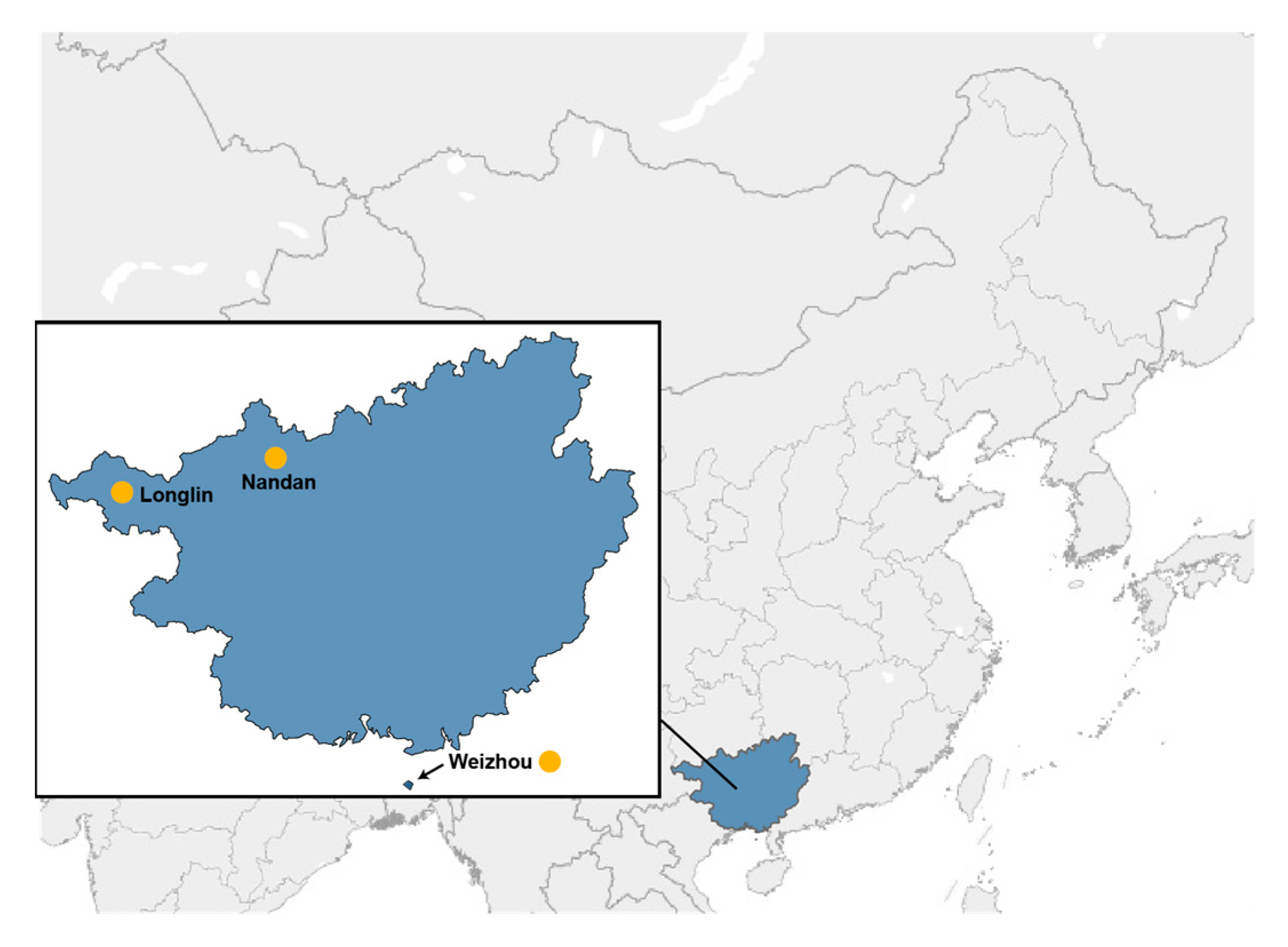
2.1. Animal Sampling and Ethics Statement
2.2. Illumina Sequencing and Reconstruction of Mitochondrial Genomes
2.3. Data Analysis
3. Results
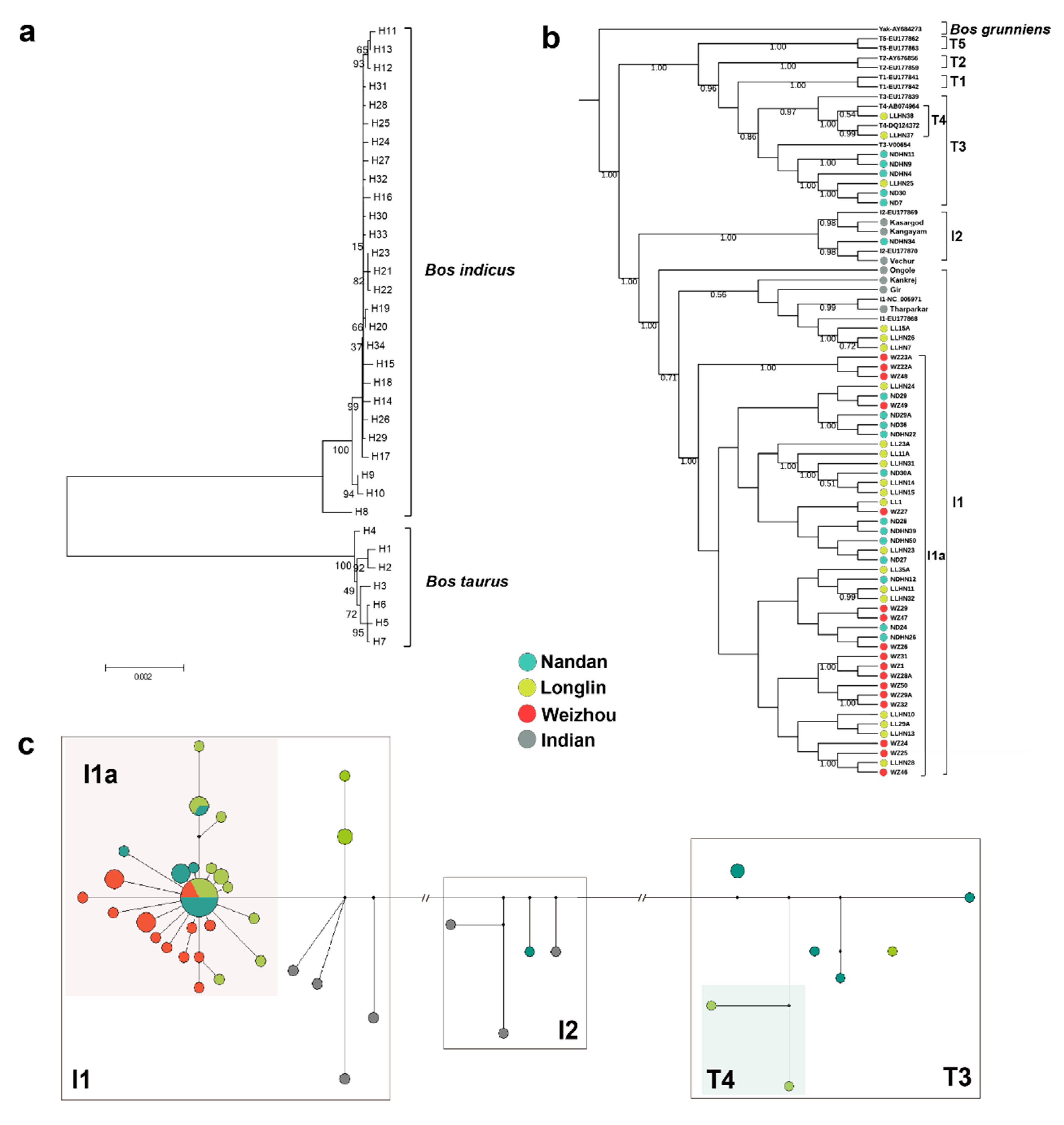
3.1. MtDNA Sequence Variation and Genetic Diversity
3.2. Population Phylogenetic Analysis
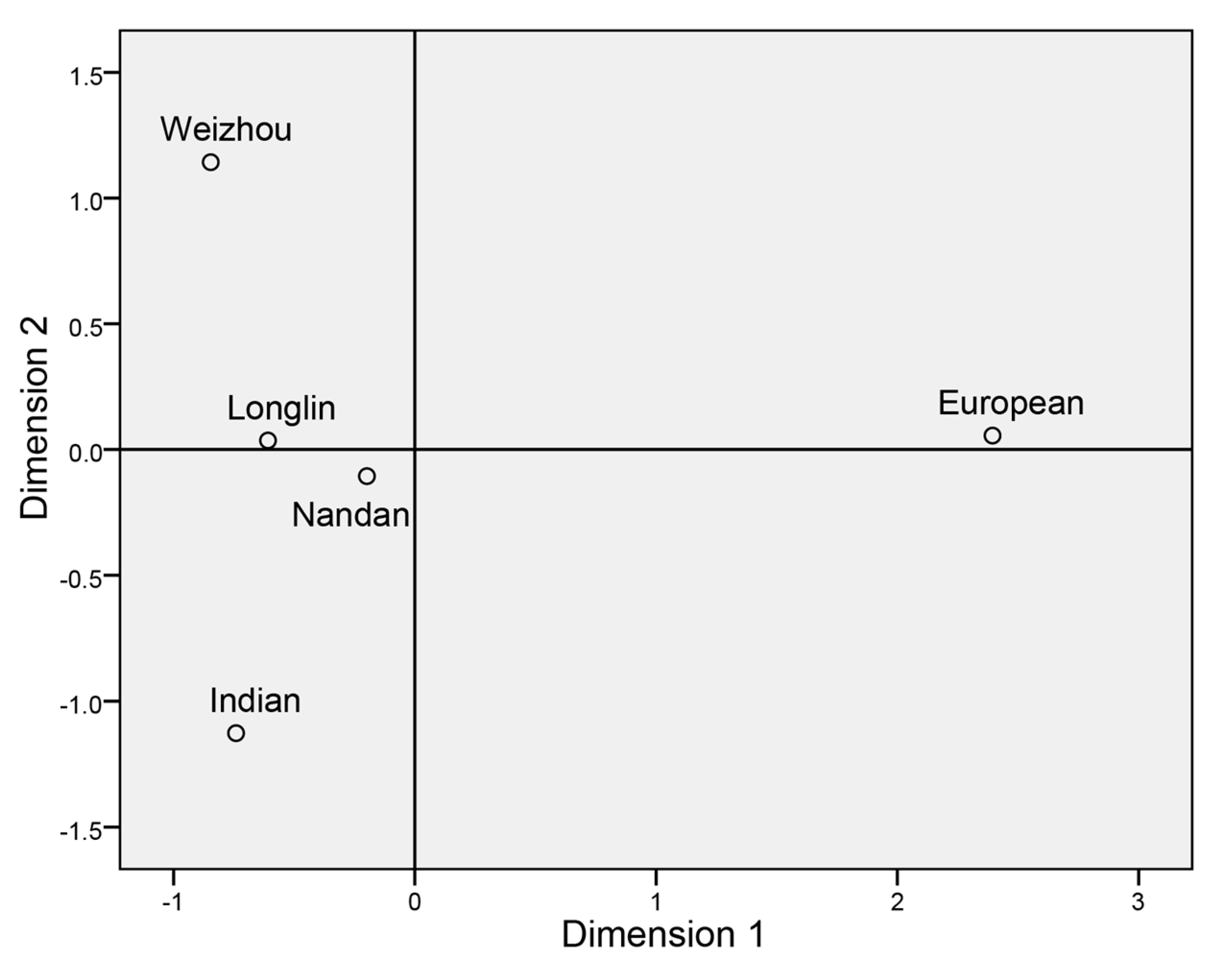
3.3. Population Genetic Structure
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yu, Y.; Nie, L.; He, Z.Q.; Wen, J.K.; Jian, C.S.; Zhang, Y.P. Mitochondrial DNA variation in cattle of south China: Origin and introgression. Anim. Genet. 1999, 30, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Lei, C.Z.; Chen, H.; Zhang, H.C.; Cai, X.; Liu, R.Y.; Luo, L.Y.; Wang, C.F.; Zhang, W.; Ge, Q.L.; Zhang, R.F.; et al. Origin and phylogeographical structure of Chinese cattle. Anim. Genet. 2006, 37, 579–582. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhang, X.M.; Campana, M.G.; Huang, J.P.; Chang, Z.H.; Qi, X.B.; Shi, H.; Su, B.; Zhang, R.F.; Lan, X.Y.; et al. Paternal origins of Chinese cattle. Anim. Genet. 2013, 44, 446–449. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Cai, Y.; Chen, Q.; Li, R.; Wang, K.; Huang, Y.; Hu, S.; Huang, S.; Zhang, H.; Zheng, Z.; et al. Whole-genome resequencing reveals world-wide ancestry and adaptive introgression events of domesticated cattle in East Asia. Nature 2018, 9, 2337. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Lin, B.Z.; Baig, M.; Mitra, B.; Lopes, R.J.; Santos, A.M.; Magee, D.A.; Azevedo, M.; Tarroso, P.; Sasazaki, S.; et al. Zebu cattle are an exclusive legacy of the South Asia neolithic. Mol. Biol. Evol. 2010, 27, 1–6. [Google Scholar] [CrossRef]
- Loftus, R.T.; Machugh, D.E.; Bradley, D.G.; Sharp, P.M.; Cunningham, P. Evidence for Two Independent Domestications of Cattle. PNAS 1994, 91, 2757–2761. [Google Scholar] [CrossRef]
- Lai, S.J.; Liu, Y.P.; Liu, Y.X.; Li, X.W.; Yao, Y.G. Genetic diversity and origin of Chinese cattle revealed by mtDNA D-loop sequence variation. Mol. Phylogenetics Evol. 2006, 38, 146–154. [Google Scholar] [CrossRef]
- Zhang, Y. Animal Genetic Resources in China-Bovines (in Chinese); China Agriculture Press: Beijing, China, 2011. [Google Scholar]
- Chen, Y. Characteristics of Chinese Yellow Cattle Ecospecies and Their Course of Utilization; China Agriculture Press: Beijing, China, 1990. [Google Scholar]
- Achilli, A.; Bonfiglio, S.; Olivieri, A.; Malusà, A.; Pala, M.; Kashani, B.H.; Perego, U.A.; Ajmone-Marsan, P.; Liotta, L.; Semino, O.; et al. The Multifaceted Origin of Taurine Cattle Reflected by the Mitochondrial Genome. PLoS ONE 2009, 4, e5753. [Google Scholar] [CrossRef]
- Troy, C.S.; Machugh, D.E.; Bailey, J.F.; Magee, D.A.; Loftus, R.T.; Cunningham, P.; Chamberlain, A.T.; Sykes, B.C.; Bradley, D.G. Genetic evidence for Near-Eastern origins of European cattle. Nature 2001, 410, 1088–1091. [Google Scholar] [CrossRef]
- Mannen, H.; Kohno, M.; Nagata, Y.; Tsuji, S.; Bradley, D.G.; Yeo, J.S.; Nyamsamba, D.; Zagdsuren, Y.; Yokohama, M.; Nomura, K.; et al. Independent mitochondrial origin and historical genetic differentiation in North Eastern Asian cattle. Mol. Phylogenetics Evol. 2004, 32, 539–544. [Google Scholar] [CrossRef]
- Xi, X.; Qu, K.; Zhang, G.; Jia, Y.; Ma, Z.; Zhao, X.; Huang, Y.; Chen, H.; Huang, B.; Lei, C. Comprehensive analysis of the mitochondrial DNA diversity in Chinese cattle. Anim. Genet. 2019, 50, 70–73. [Google Scholar]
- Jia, S.; Chen, H.; Zhang, G.; Wang, Z.; Lei, C.; Yao, R.; Han, X. Genetic variation of mitochondrial D-loop region and evolution analysis in some Chinese cattle breeds. J. Genet. Genom. 2007, 34, 510–518. [Google Scholar] [CrossRef]
- Jia, S.G.; Zhou, Y.; Lei, C.; Yao, R.; Zhang, Z.; Fang, X.; Chen, H. A new insight into cattle’s maternal origin in six Asian countries. J. Genet. Genom. 2010, 37, 173–180. [Google Scholar] [CrossRef]
- Xia, X.; Yao, Y.; Li, C.; Zhang, F.; Qu, K.; Chen, H.; Huang, B.; Lei, C. Genetic diversity of Chinese cattle revealed by Y-SNP and Y-STR markers. Anim. Genet. 2019, 50, 64–69. [Google Scholar] [CrossRef]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual; Huang, P.T., Translator; China Science Press: Beijing, China, 2002. [Google Scholar]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinform. Online 2007, 1, 47. [Google Scholar] [CrossRef]
- Tamura, K.; Peterson, D.; Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
- Bandelt, H.J.; Forster, P.; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37. [Google Scholar] [CrossRef]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A Software Platform for Bayesian Evolutionary Analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef]
- Pramod, R.K.; Velayutham, D.; Sajesh, P.K.; Beena, P.S.; Zachariah, A.; Zachariah, A.; Chandramohan, B.; Sujith, S.S.; Santhosh, S.; Iype, S.; et al. The complete mitochondrial genome of Indian cattle (Bos indicus). Mitochondrial DNA Part B 2018, 3, 207–208. [Google Scholar] [CrossRef]
- Xia, X.; Qu, K.; Li, F.; Jia, P.; Chen, Q.; Chen, N.; Zhang, J.; Chen, H.; Huang, B.; Lei, C. Abundant Genetic Diversity of Yunling Cattle Based on Mitochondrial. Animals 2019, 9, 641. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Breed | N | S | H | Haplogroup | Hd ± SD | π ± SD | k | |||
|---|---|---|---|---|---|---|---|---|---|---|
| I1 | I2 | T3 | T4 | |||||||
| Longlin | 21 | 272 | 15 | 9 | 1 | 5 | 0.957 ± 0.030 | 0.00409 ± 0.00163 | 66.7524 | |
| Nandan | 18 | 267 | 10 | 12 | 1 | 2 | 0.876 ± 0.063 | 0.00646 ± 0.00143 | 105.5229 | |
| Weizhou | 17 | 22 | 12 | 12 | 0.949 ± 0.037 | 0.00024 ± 0.00003 | 3.8529 | |||
| Total | 56 | 308 | 34 | 33 | 1 | 6 | 2 | 0.947 ± 0.021 | 0.00394 ± 0.00098 | 64.2792 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xia, X.; Huang, G.; Wang, Z.; Sun, J.; Wu, Z.; Chen, N.; Lei, C.; Hanif, Q. Mitogenome Diversity and Maternal Origins of Guangxi Cattle Breeds. Animals 2020, 10, 19. https://doi.org/10.3390/ani10010019
Xia X, Huang G, Wang Z, Sun J, Wu Z, Chen N, Lei C, Hanif Q. Mitogenome Diversity and Maternal Origins of Guangxi Cattle Breeds. Animals. 2020; 10(1):19. https://doi.org/10.3390/ani10010019
Chicago/Turabian StyleXia, Xiaoting, Guangyun Huang, Zihao Wang, Junli Sun, Zhuyue Wu, Ningbo Chen, Chuzhao Lei, and Quratulain Hanif. 2020. "Mitogenome Diversity and Maternal Origins of Guangxi Cattle Breeds" Animals 10, no. 1: 19. https://doi.org/10.3390/ani10010019
APA StyleXia, X., Huang, G., Wang, Z., Sun, J., Wu, Z., Chen, N., Lei, C., & Hanif, Q. (2020). Mitogenome Diversity and Maternal Origins of Guangxi Cattle Breeds. Animals, 10(1), 19. https://doi.org/10.3390/ani10010019