
Current and Emerging Therapies to Combat Cystic Fibrosis Lung Infections


Abstract
:1. Introduction—The Cystic Fibrosis Lung Environment
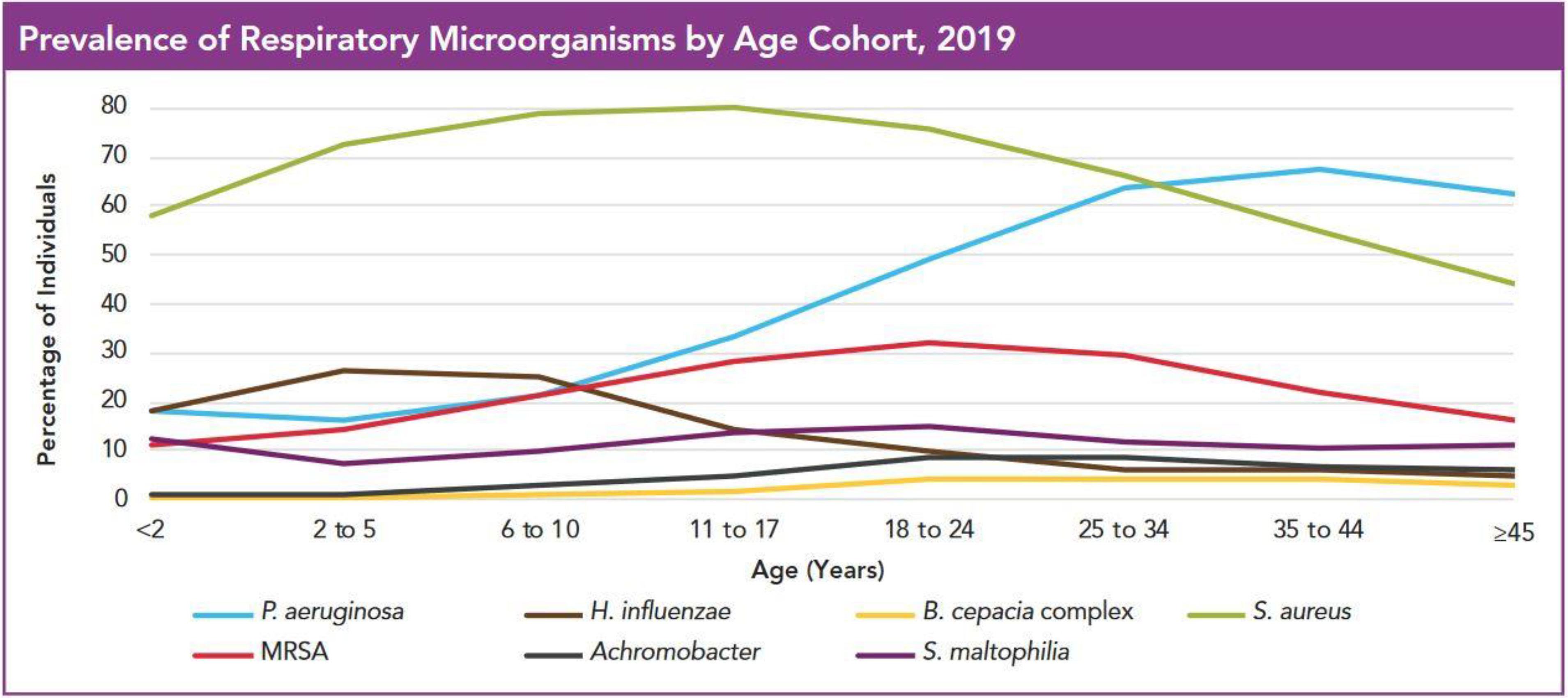
2. Infecting Species by Age Cohort
2.1. The Challenges of S. aureus Infection in Pre-Adolescents
2.2. Non-Typable Haemophilus Influenzae and H. parainfluenzae—A Significant Childhood Threat
2.3. P. aeruginosa—Slow to Start but Bound to Dominate
2.4. Acquisition Prevention and Early Eradication Strategies
2.5. Surveillance
3. Species and Prevalence in Adolescence and Young Adulthood
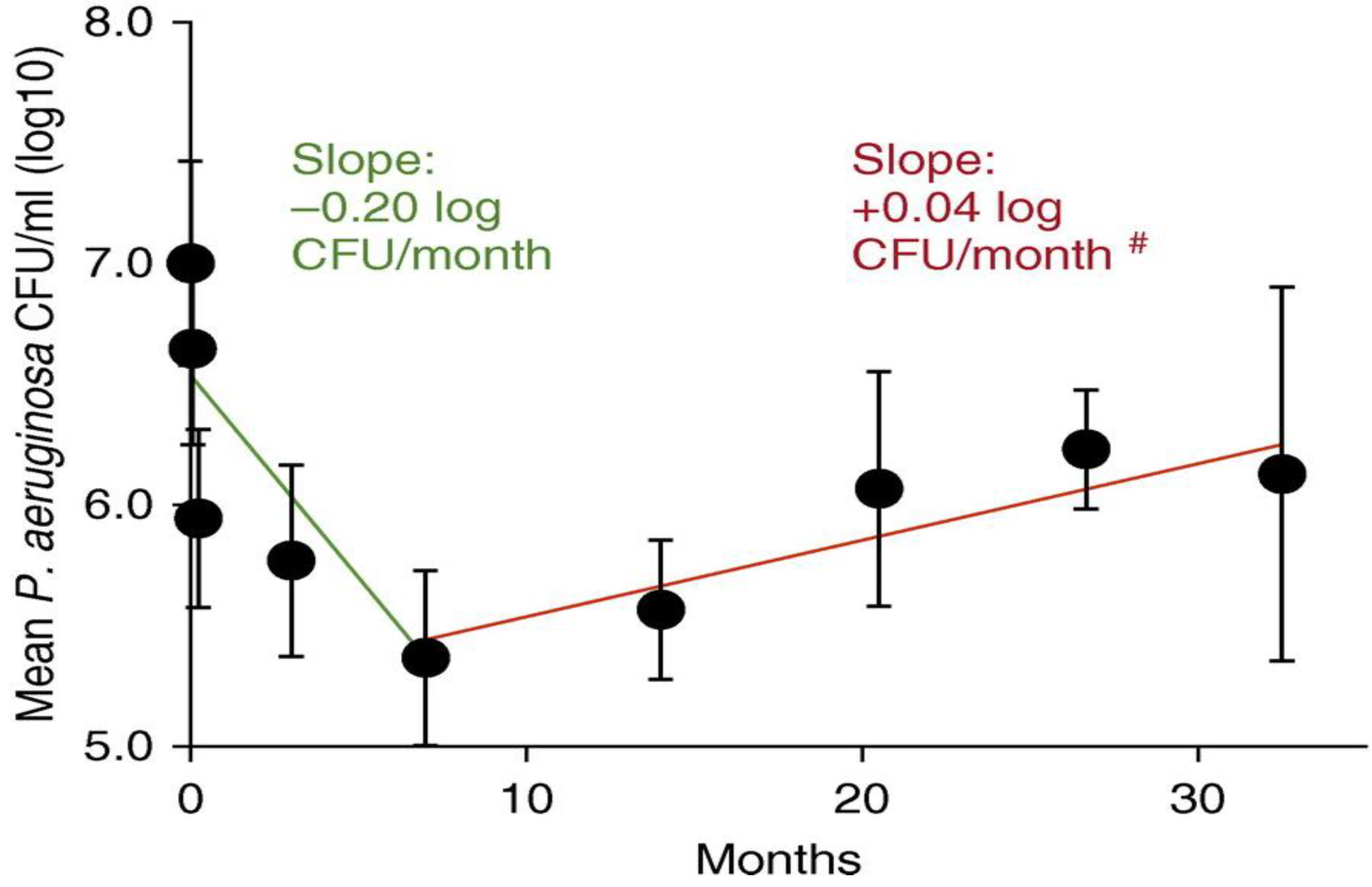
3.1. Changes in Pre-Existing Species
3.2. Achromobacter xylosoxidans and Stenotrophomonas maltophilia
3.3. Burkholderia Cepacia Complex (BCC) and Non-Tuberculous Mycobacterium (NTM)
4. Species and Prevalence in Adulthood
Differences in Treatment Strategies between Age Groups
5. Current Treatments for Bacterial Infection in CF
5.1. Non-Antibiotic Treatments—Physical Treatments to Promote Airway Clearance
5.1.1. Postural Drainage
5.1.2. Percussion Therapy
5.1.3. Forced Expiratory Methods (FET and PEP)
5.1.4. ACBT
5.1.5. Exercise Training
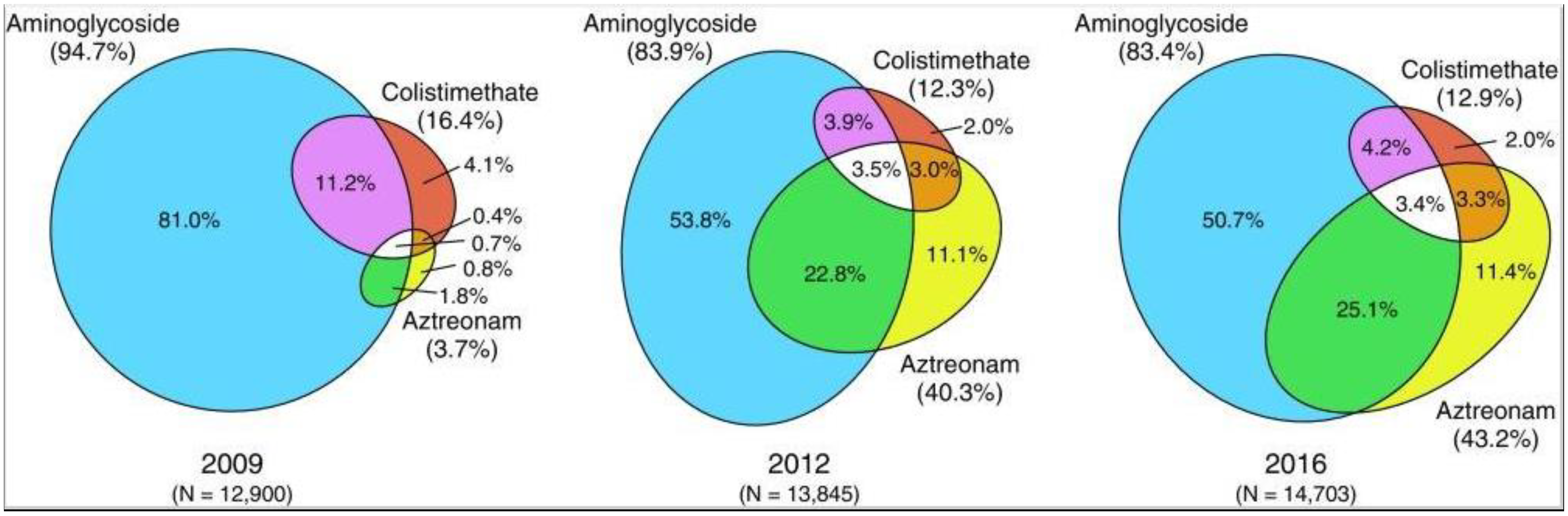
5.2. Antibiotics
5.3. Hypertonic Saline
5.4. Combined Treatments with Antibiotics
6. Emerging Treatments for Bacterial Infection in CF
6.1. Non-Steroidal Anti-Inflammatory Compounds
6.2. Quorum-Sensing Inhibitors
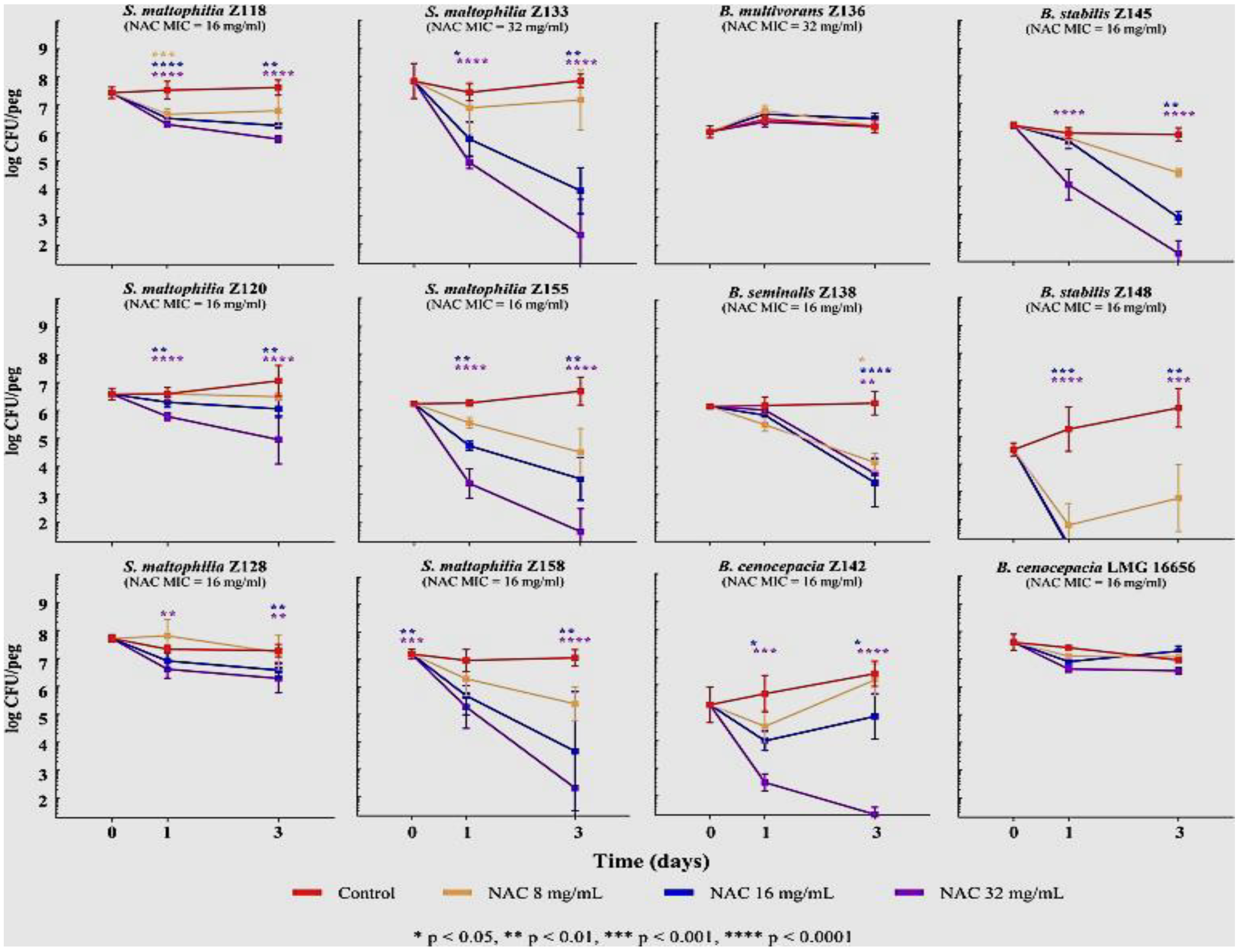
6.3. Antioxidants and Biofilm Disruption: Glutathione, N-acetylcysteine and Ascorbic Acid
6.4. Silver Nanoparticles
6.5. Bacteriophage Therapy
7. Attempts to Modify the CF Lung Environment
7.1. CFTR-Modifying Drugs and Bacterial Infection
7.2. Lung Transplant and Reinfection
8. Summary—Treatment of Bacterial CF Infections
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Davies, J.C.; Ebdon, A.M.; Orchard, C. Recent advances in the management of cystic fibrosis. Arch. Dis. Child. 2014, 99, 1033–1036. [Google Scholar] [CrossRef] [Green Version]
- Mall, M.A.; Hartl, D. CFTR: Cystic fibrosis and beyond. Eur. Respir. J. 2014, 44, 1042–1054. [Google Scholar] [CrossRef] [Green Version]
- Hoegger, M.J.; Fischer, A.J.; McMenimen, J.D.; Ostedgaard, L.S.; Tucker, A.J.; Awadalla, M.A.; Moninger, T.O.; Michalski, A.S.; Hoffman, E.A.; Zabner, J.; et al. Impaired mucus detachment disrupts mucociliary transport in a piglet model of cystic fibrosis. Science 2014, 345, 818–822. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.Y.; Muallem, D.; Kiselyov, K.; Lee, M.G.; Thomas, P.J.; Muallem, S. Aberrant CFTR-dependent HCO3- transport in mutations associated with cystic fibrosis. Nature 2001, 410, 94–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berkebile, A.R.; McCray, P.B., Jr. Effects of airway surface liquid pH on host defense in cystic fibrosis. Int. J. Biochem. Cell Biol. 2014, 52, 124–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.X.; Ostedgaard, L.S.; Hoegger, M.J.; Moninger, T.O.; Karp, P.H.; McMenimen, J.D.; Choudhury, B.; Varki, A.; Stoltz, D.A.; Welsh, M.J. Acidic pH increases airway surface liquid viscosity in cystic fibrosis. J. Clin. Investig. 2016, 126, 879–891. [Google Scholar] [CrossRef] [Green Version]
- Dean, S.N.; Bishop, B.M.; van Hoek, M.L. Natural and synthetic cathelicidin peptides with anti-microbial and anti-biofilm activity against Staphylococcus aureus. BMC Microbiol. 2011, 11, 114. [Google Scholar] [CrossRef] [Green Version]
- Overhage, J.; Campisano, A.; Bains, M.; Torfs, E.C.; Rehm, B.H.; Hancock, R.E. Human host defense peptide LL-37 prevents bacterial biofilm formation. Infect. Immun. 2008, 76, 4176–4182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Worlitzsch, D.; Tarran, R.; Ulrich, M.; Schwab, U.; Cekici, A.; Meyer, K.C.; Birrer, P.; Bellon, G.; Berger, J.; Weiss, T.; et al. Effects of reduced mucus oxygen concentration in airway Pseudomonas infections of cystic fibrosis patients. J. Clin. Investig. 2002, 109, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Polke, M.; Seiler, F.; Lepper, P.M.; Kamyschnikow, A.; Langer, F.; Monz, D.; Herr, C.; Bals, R.; Beisswenger, C. Hypoxia and the hypoxia-regulated transcription factor HIF-1alpha suppress the host defence of airway epithelial cells. Innate Immun. 2017, 23, 373–380. [Google Scholar] [CrossRef]
- Quinn, R.A.; Adem, S.; Mills, R.H.; Comstock, W.; DeRight Goldasich, L.; Humphrey, G.; Aksenov, A.A.; Melnik, A.V.; da Silva, R.; Ackermann, G.; et al. Neutrophilic proteolysis in the cystic fibrosis lung correlates with a pathogenic microbiome. Microbiome 2019, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Cystic Fibrosis Foundation Patient Registry 2019 Annual Data Report; ©2020 Cystic Fibrosis Foundation: Bethesda, MD, USA, 2020.
- Pillarisetti, N.; Williamson, E.; Linnane, B.; Skoric, B.; Robertson, C.F.; Robinson, P.; Massie, J.; Hall, G.L.; Sly, P.; Stick, S.; et al. Infection, inflammation, and lung function decline in infants with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2011, 184, 75–81. [Google Scholar] [CrossRef]
- Esposito, S.; Terranova, L.; Zampiero, A.; Ierardi, V.; Rios, W.P.; Pelucchi, C.; Principi, N. Oropharyngeal and nasal Staphylococcus aureus carriage by healthy children. BMC Infect. Dis. 2014, 14, 723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mika, M.; Korten, I.; Qi, W.; Regamey, N.; Frey, U.; Casaulta, C.; Latzin, P.; Hilty, M. The nasal microbiota in infants with cystic fibrosis in the first year of life: A prospective cohort study. Lancet. Respir. Med. 2016, 4, 627–635. [Google Scholar] [CrossRef] [Green Version]
- Harik, N.S.; Com, G.; Tang, X.; Melguizo Castro, M.; Stemper, M.E.; Carroll, J.L. Clinical characteristics and epidemiology of methicillin-resistant Staphylococcus aureus (MRSA) in children with cystic fibrosis from a center with a high MRSA prevalence. Am. J. Infect. Control. 2016, 44, 409–415. [Google Scholar] [CrossRef]
- Muhlebach, M.S.; Heltshe, S.L.; Popowitch, E.B.; Miller, M.B.; Thompson, V.; Kloster, M.; Ferkol, T.; Hoover, W.C.; Schechter, M.S.; Saiman, L.; et al. Multicenter Observational Study on Factors and Outcomes Associated with Various Methicillin-Resistant Staphylococcus aureus Types in Children with Cystic Fibrosis. Ann. Am. Thorac. Soc. 2015, 12, 864–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starner, T.D.; Zhang, N.; Kim, G.; Apicella, M.A.; McCray, P.B., Jr. Haemophilus influenzae forms biofilms on airway epithelia: Implications in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2006, 174, 213–220. [Google Scholar] [CrossRef] [Green Version]
- King, P. Haemophilus influenzae and the lung (Haemophilus and the lung). Clin. Transl. Med. 2012, 1, 10. [Google Scholar] [CrossRef] [Green Version]
- King, P.T.; Sharma, R.; O’Sullivan, K.; Selemidis, S.; Lim, S.; Radhakrishna, N.; Lo, C.; Prasad, J.; Callaghan, J.; McLaughlin, P.; et al. Nontypeable Haemophilus influenzae induces sustained lung oxidative stress and protease expression. PLoS ONE 2015, 10, e0120371. [Google Scholar] [CrossRef] [Green Version]
- Craig, J.E.; Cliffe, A.; Garnett, K.; High, N.J. Survival of nontypeable Haemophilus influenzae in macrophages. FEMS Microbiol. Lett. 2001, 203, 55–61. [Google Scholar] [CrossRef] [Green Version]
- Cardines, R.; Giufre, M.; Pompilio, A.; Fiscarelli, E.; Ricciotti, G.; Di Bonaventura, G.; Cerquetti, M. Haemophilus influenzae in children with cystic fibrosis: Antimicrobial susceptibility, molecular epidemiology, distribution of adhesins and biofilm formation. Int. J. Med. Microbiol. 2012, 302, 45–52. [Google Scholar] [CrossRef]
- Ebbing, R.; Robertson, C.F.; Robinson, P.J. Haemophilus influenzae and Haemophilus parainfluenza in Cystic Fibrosis: 15 Years Experience. J. Med. Microbiol. Diagn. 2015, S5, 4. [Google Scholar] [CrossRef]
- Emerson, J.; Rosenfeld, M.; McNamara, S.; Ramsey, B.; Gibson, R.L. Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis. Pediatr. Pulmonol. 2002, 34, 91–100. [Google Scholar] [CrossRef]
- Ratjen, F.; Munck, A.; Kho, P.; Angyalosi, G.; Group, E.S. Treatment of early Pseudomonas aeruginosa infection in patients with cystic fibrosis: The ELITE trial. Thorax 2010, 65, 286–291. [Google Scholar] [CrossRef] [Green Version]
- Blanchard, A.C.; Horton, E.; Stanojevic, S.; Taylor, L.; Waters, V.; Ratjen, F. Effectiveness of a stepwise Pseudomonas aeruginosa eradication protocol in children with cystic fibrosis. J. Cyst. Fibros. 2017, 16, 395–400. [Google Scholar] [CrossRef]
- Ratjen, F.; Moeller, A.; McKinney, M.L.; Asherova, I.; Alon, N.; Maykut, R.; Angyalosi, G. Eradication of early P. aeruginosa infection in children <7 years of age with cystic fibrosis: The early study. J. Cyst. Fibros. 2019, 18, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Mogayzel, P.J., Jr.; Naureckas, E.T.; Robinson, K.A.; Brady, C.; Guill, M.; Lahiri, T.; Lubsch, L.; Matsui, J.; Oermann, C.M.; Ratjen, F.; et al. Cystic Fibrosis Foundation pulmonary guideline. pharmacologic approaches to prevention and eradication of initial Pseudomonas aeruginosa infection. Ann. Am. Thorac. Soc. 2014, 11, 1640–1650. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Kosorok, M.R.; Farrell, P.M.; Laxova, A.; West, S.E.; Green, C.G.; Collins, J.; Rock, M.J.; Splaingard, M.L. Longitudinal development of mucoid Pseudomonas aeruginosa infection and lung disease progression in children with cystic fibrosis. JAMA 2005, 293, 581–588. [Google Scholar] [CrossRef] [Green Version]
- Sanders, D.B. Phenotypes that matter: Pseudomonas aeruginosa and progression of cystic fibrosis lung disease. Am. J. Respir. Crit. Care Med. 2014, 190, 245–246. [Google Scholar] [CrossRef] [PubMed]
- Sanders, D.B.; Li, Z.; Laxova, A.; Rock, M.J.; Levy, H.; Collins, J.; Ferec, C.; Farrell, P.M. Risk factors for the progression of cystic fibrosis lung disease throughout childhood. Ann. Am. Thorac. Soc. 2014, 11, 63–72. [Google Scholar] [CrossRef] [Green Version]
- Heltshe, S.L.; Khan, U.; Beckett, V.; Baines, A.; Emerson, J.; Sanders, D.B.; Gibson, R.L.; Morgan, W.; Rosenfeld, M. Longitudinal development of initial, chronic and mucoid Pseudomonas aeruginosa infection in young children with cystic fibrosis. J. Cyst. Fibros. 2018, 17, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Talwalkar, J.S.; Murray, T.S. The Approach to Pseudomonas aeruginosa in Cystic Fibrosis. Clin. Chest. Med. 2016, 37, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Treggiari, M.M.; Retsch-Bogart, G.; Mayer-Hamblett, N.; Khan, U.; Kulich, M.; Kronmal, R.; Williams, J.; Hiatt, P.; Gibson, R.L.; Spencer, T.; et al. Comparative efficacy and safety of 4 randomized regimens to treat early Pseudomonas aeruginosa infection in children with cystic fibrosis. Arch. Pediatr. Adolesc. Med. 2011, 165, 847–856. [Google Scholar] [CrossRef] [Green Version]
- Breuer, O.; Schultz, A.; Turkovic, L.; de Klerk, N.; Keil, A.D.; Brennan, S.; Harrison, J.; Robertson, C.; Robinson, P.J.; Sly, P.D.; et al. Changing Prevalence of Lower Airway Infections in Young Children with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 200, 590–599. [Google Scholar] [CrossRef] [PubMed]
- Schelstraete, P.; Haerynck, F.; Van daele, S.; Deseyne, S.; De Baets, F. Eradication therapy for Pseudomonas aeruginosa colonization episodes in cystic fibrosis patients not chronically colonized by P. aeruginosa. J. Cyst. Fibros. 2013, 12, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaver, C.M.; Hauser, A.R. Relative contributions of Pseudomonas aeruginosa ExoU, ExoS, and ExoT to virulence in the lung. Infect. Immun. 2004, 72, 6969–6977. [Google Scholar] [CrossRef] [Green Version]
- Gawel, J.; Pogorzelski, A.; Dzialek-Smetek, E.; Sochan, B.; Ligarska, R.; Lacka, M.; Mazurek, H. Distribution of antibodies to selected antigens of Pseudomonas aeruginosa in children and young adults with cystic fibrosis. Pneumonol. Alergol. Pol. 2014, 82, 336–341. [Google Scholar] [CrossRef]
- Manos, J.; Hu, H.; Rose, B.R.; Wainwright, C.E.; Zablotska, I.B.; Cheney, J.; Turnbull, L.; Whitchurch, C.B.; Grimwood, K.; Harmer, C.; et al. Virulence factor expression patterns in Pseudomonas aeruginosa strains from infants with cystic fibrosis. Eur. J. Clin. Microbiol. Infect. Dis. 2013, 32, 1583–1592. [Google Scholar] [CrossRef] [PubMed]
- Vidya, P.; Smith, L.; Beaudoin, T.; Yau, Y.C.; Clark, S.; Coburn, B.; Guttman, D.S.; Hwang, D.M.; Waters, V. Chronic infection phenotypes of Pseudomonas aeruginosa are associated with failure of eradication in children with cystic fibrosis. Eur. J. Clin. Microbiol. Infect. Dis. 2016, 35, 67–74. [Google Scholar] [CrossRef]
- Marvig, R.L.; Sommer, L.M.; Molin, S.; Johansen, H.K. Convergent evolution and adaptation of Pseudomonas aeruginosa within patients with cystic fibrosis. Nat. Genet. 2015, 47, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Conway, S. Segregation is good for patients with cystic fibrosis. J. R. Soc. Med. 2008, 101 (Suppl. 1), S31–S35. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, A.L.; Jamsen, K.; Carlin, J.B.; Grimwood, K.; Carzino, R.; Robinson, P.J.; Massie, J.; Armstrong, D.S. Effects of segregation on an epidemic Pseudomonas aeruginosa strain in a cystic fibrosis clinic. Am. J. Respir. Crit. Care Med. 2005, 171, 1020–1025. [Google Scholar] [CrossRef]
- Kevat, A.; Carzino, R.; Massie, J.; Harrison, J.; Griffiths, A.L. Elimination of Australian epidemic strain (AES1) Pseudomonas aeruginosa in a pediatric cystic fibrosis center. Pediatr. Pulmonol. 2018, 53, 1498–1503. [Google Scholar] [CrossRef]
- van Mansfeld, R.; de Vrankrijker, A.; Brimicombe, R.; Heijerman, H.; Teding van Berkhout, F.; Spitoni, C.; Grave, S.; van der Ent, C.; Wolfs, T.; Willems, R.; et al. The Effect of Strict Segregation on Pseudomonas aeruginosa in Cystic Fibrosis Patients. PLoS ONE 2016, 11, e0157189. [Google Scholar] [CrossRef] [Green Version]
- Geddes, D. Segregation is not good for patients with cystic fibrosis. J. R. Soc. Med. 2008, 101 (Suppl. 1), S36–S38. [Google Scholar] [CrossRef] [Green Version]
- Mayer-Hamblett, N.; Kloster, M.; Rosenfeld, M.; Gibson, R.L.; Retsch-Bogart, G.Z.; Emerson, J.; Thompson, V.; Ramsey, B.W. Impact of Sustained Eradication of New Pseudomonas aeruginosa Infection on Long-term Outcomes in Cystic Fibrosis. Clin. Infect. Dis. 2015, 61, 707–715. [Google Scholar] [CrossRef]
- Hu, H.; Manos, J. Pulsed-field gel electrophoresis of Pseudomonas aeruginosa. Methods Mol. Biol. 2015, 1301, 157–170. [Google Scholar] [CrossRef]
- Hery-Arnaud, G.; Nowak, E.; Caillon, J.; David, V.; Dirou, A.; Revert, K.; Munck, M.R.; Frachon, I.; Haloun, A.; Horeau-Langlard, D.; et al. Evaluation of quantitative PCR for early diagnosis of Pseudomonas aeruginosa infection in cystic fibrosis: A prospective cohort study. Clin. Microbiol. Infect. 2017, 23, 203–207. [Google Scholar] [CrossRef] [Green Version]
- Chmiel, J.F.; Aksamit, T.R.; Chotirmall, S.H.; Dasenbrook, E.C.; Elborn, J.S.; LiPuma, J.J.; Ranganathan, S.C.; Waters, V.J.; Ratjen, F.A. Antibiotic management of lung infections in cystic fibrosis. I. The microbiome, methicillin-resistant Staphylococcus aureus, gram-negative bacteria, and multiple infections. Ann. Am. Thorac. Soc. 2014, 11, 1120–1129. [Google Scholar] [CrossRef] [Green Version]
- Australian Cystic Fibrosis Data Registry. Available online: https://www.cysticfibrosis.org.au/getmedia/24e94d66-29fa-4e3f-8e65-21ee24ed2e5a/ACFDR-2017-Annual-Report_highres_singles.pdf.aspx (accessed on 1 March 2020).
- Gemelll, J.W.; Rempe, K.A. Haemophilus influenzae. In Principles and Practice of Pediatric Infectious Diseases; Long, S.S., Fischer, M., Prober, C.G., Eds.; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar] [CrossRef]
- Lo, D.K.H.; Muhlebach, M.S.; Smyth, A.R. Interventions for the eradication of meticillin-resistant Staphylococcus aureus (MRSA) in people with cystic fibrosis. Cochrane Database Syst. Rev. 2018, 14, 1858. [Google Scholar] [CrossRef]
- Cox, M.J.; Allgaier, M.; Taylor, B.; Baek, M.S.; Huang, Y.J.; Daly, R.A.; Karaoz, U.; Andersen, G.L.; Brown, R.; Fujimura, K.E.; et al. Airway microbiota and pathogen abundance in age-stratified cystic fibrosis patients. PLoS ONE 2010, 5, e11044. [Google Scholar] [CrossRef]
- Fodor, A.A.; Klem, E.R.; Gilpin, D.F.; Elborn, J.S.; Boucher, R.C.; Tunney, M.M.; Wolfgang, M.C. The adult cystic fibrosis airway microbiota is stable over time and infection type, and highly resilient to antibiotic treatment of exacerbations. PLoS ONE 2012, 7, e45001. [Google Scholar] [CrossRef]
- Frayman, K.B.; Armstrong, D.S.; Carzino, R.; Ferkol, T.W.; Grimwood, K.; Storch, G.A.; Teo, S.M.; Wylie, K.M.; Ranganathan, S.C. The lower airway microbiota in early cystic fibrosis lung disease: A longitudinal analysis. Thorax 2017, 72, 1104–1112. [Google Scholar] [CrossRef]
- Cameron, L.C.; Bonis, B.; Phan, C.Q.; Kent, L.A.; Lee, A.K.; Hunter, R.C. A putative enoyl-CoA hydratase contributes to biofilm formation and the antibiotic tolerance of Achromobacter xylosoxidans. NPJ Biofilms Microbiomes 2019, 5, 20. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, S.M.; Penstoft, L.N.; Norskov-Lauritsen, N. Motility, Biofilm Formation and Antimicrobial Efflux of Sessile and Planktonic Cells of Achromobacter xylosoxidans. Pathogens 2019, 8, 14. [Google Scholar] [CrossRef] [Green Version]
- Isler, B.; Kidd, T.J.; Stewart, A.G.; Harris, P.; Paterson, D.L. Achromobacter Infections and Treatment Options. Antimicrob. Agents Chemother. 2020, 64, e01025-20. [Google Scholar] [CrossRef] [PubMed]
- Steinkamp, G.; Wiedemann, B.; Rietschel, E.; Krahl, A.; Gielen, J.; Barmeier, H.; Ratjen, F.; Emerging Bacteria Study, G. Prospective evaluation of emerging bacteria in cystic fibrosis. J. Cyst. Fibros. 2005, 4, 41–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waters, V.; Atenafu, E.G.; Lu, A.; Yau, Y.; Tullis, E.; Ratjen, F. Chronic Stenotrophomonas maltophilia infection and mortality or lung transplantation in cystic fibrosis patients. J. Cyst. Fibros. 2013, 12, 482–486. [Google Scholar] [CrossRef] [Green Version]
- Sfeir, M.M. Burkholderia cepacia complex infections: More complex than the bacterium name suggest. J. Infect. 2018, 77, 166–170. [Google Scholar] [CrossRef]
- De Soyza, A.; Meachery, G.; Hester, K.L.; Nicholson, A.; Parry, G.; Tocewicz, K.; Pillay, T.; Clark, S.; Lordan, J.L.; Schueler, S.; et al. Lung transplantation for patients with cystic fibrosis and Burkholderia cepacia complex infection: A single-center experience. J. Heart Lung Transplant. 2010, 29, 1395–1404. [Google Scholar] [CrossRef]
- Salsgiver, E.L.; Fink, A.K.; Knapp, E.A.; LiPuma, J.J.; Olivier, K.N.; Marshall, B.C.; Saiman, L. Changing Epidemiology of the Respiratory Bacteriology of Patients with Cystic Fibrosis. Chest 2016, 149, 390–400. [Google Scholar] [CrossRef] [Green Version]
- Floto, R.A.; Olivier, K.N.; Saiman, L.; Daley, C.L.; Herrmann, J.L.; Nick, J.A.; Noone, P.G.; Bilton, D.; Corris, P.; Gibson, R.L.; et al. US Cystic Fibrosis Foundation and European Cystic Fibrosis Society consensus recommendations for the management of non-tuberculous mycobacteria in individuals with cystic fibrosis: Executive summary. Thorax 2016, 71, 88–90. [Google Scholar] [CrossRef] [Green Version]
- Henderson, A.G.; Ehre, C.; Button, B.; Abdullah, L.H.; Cai, L.H.; Leigh, M.W.; DeMaria, G.C.; Matsui, H.; Donaldson, S.H.; Davis, C.W.; et al. Cystic fibrosis airway secretions exhibit mucin hyperconcentration and increased osmotic pressure. J. Clin. Investig. 2014, 124, 3047–3060. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Barat, L.; Ciofu, O.; Kragh, K.N.; Pressler, T.; Johansen, U.; Motos, A.; Torres, A.; Hoiby, N. Phenotypic shift in Pseudomonas aeruginosa populations from cystic fibrosis lungs after 2-week antipseudomonal treatment. J. Cyst. Fibros. 2017, 16, 222–229. [Google Scholar] [CrossRef] [Green Version]
- Lutz, L.; Leao, R.S.; Ferreira, A.G.; Pereira, D.C.; Raupp, C.; Pitt, T.; Marques, E.A.; Barth, A.L. Hypermutable Pseudomonas aeruginosa in Cystic fibrosis patients from two Brazilian cities. J. Clin. Microbiol. 2013, 51, 927–930. [Google Scholar] [CrossRef] [Green Version]
- Mena, A.; Smith, E.E.; Burns, J.L.; Speert, D.P.; Moskowitz, S.M.; Perez, J.L.; Oliver, A. Genetic adaptation of Pseudomonas aeruginosa to the airways of cystic fibrosis patients is catalyzed by hypermutation. J. Bacteriol. 2008, 190, 7910–7917. [Google Scholar] [CrossRef] [Green Version]
- Rees, V.E.; Deveson Lucas, D.S.; Lopez-Causape, C.; Huang, Y.; Kotsimbos, T.; Bulitta, J.B.; Rees, M.C.; Barugahare, A.; Peleg, A.Y.; Nation, R.L.; et al. Characterization of Hypermutator Pseudomonas aeruginosa Isolates from Patients with Cystic Fibrosis in Australia. Antimicrob. Agents Chemother. 2019, 63, 18. [Google Scholar] [CrossRef] [Green Version]
- Waine, D.J.; Honeybourne, D.; Smith, E.G.; Whitehouse, J.L.; Dowson, C.G. Association between hypermutator phenotype, clinical variables, mucoid phenotype, and antimicrobial resistance in Pseudomonas aeruginosa. J. Clin. Microbiol. 2008, 46, 3491–3493. [Google Scholar] [CrossRef] [Green Version]
- Cabot, G.; Zamorano, L.; Moya, B.; Juan, C.; Navas, A.; Blazquez, J.; Oliver, A. Evolution of Pseudomonas aeruginosa Antimicrobial Resistance and Fitness under Low and High Mutation Rates. Antimicrob. Agents Chemother. 2016, 60, 1767–1778. [Google Scholar] [CrossRef] [Green Version]
- Breidenstein, E.B.; de la Fuente-Nunez, C.; Hancock, R.E. Pseudomonas aeruginosa: All roads lead to resistance. Trends Microbiol. 2011, 19, 419–426. [Google Scholar] [CrossRef]
- Davies, E.V.; James, C.E.; Brockhurst, M.A.; Winstanley, C. Evolutionary diversification of Pseudomonas aeruginosa in an artificial sputum model. BMC Microbiol. 2017, 17, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klockgether, J.; Cramer, N.; Fischer, S.; Wiehlmann, L.; Tummler, B. Long-Term Microevolution of Pseudomonas aeruginosa Differs between Mildly and Severely Affected Cystic Fibrosis Lungs. Am. J. Respir. Cell Mol. Biol. 2018, 59, 246–256. [Google Scholar] [CrossRef] [PubMed]
- Hatziagorou, E.; Orenti, A.; Drevinek, P.; Kashirskaya, N.; Mei-Zahav, M.; De Boeck, K. Changing epidemiology of the respiratory bacteriology of patients with cystic fibrosis-data from the European cystic fibrosis society patient registry. J. Cyst. Fibros. 2019, 10, 1016. [Google Scholar] [CrossRef]
- Chmiel, J.F.; Davis, P.B. State of the art: Why do the lungs of patients with cystic fibrosis become infected and why can’t they clear the infection? Respir. Res. 2003, 4, 8. [Google Scholar] [CrossRef] [Green Version]
- Defraine, V.; Fauvart, M.; Michiels, J. Fighting bacterial persistence: Current and emerging anti-persister strategies and therapeutics. Drug Resist. Updat. 2018, 38, 12–26. [Google Scholar] [CrossRef]
- Stepanyan, K.; Wenseleers, T.; Duenez-Guzman, E.A.; Muratori, F.; Van den Bergh, B.; Verstraeten, N.; De Meester, L.; Verstrepen, K.J.; Fauvart, M.; Michiels, J. Fitness trade-offs explain low levels of persister cells in the opportunistic pathogen Pseudomonas aeruginosa. Mol. Ecol. 2015, 24, 1572–1583. [Google Scholar] [CrossRef] [Green Version]
- Wagener, J.S.; Kupfer, O. Dornase alfa (Pulmozyme). Curr. Opin. Pulm. Med. 2012, 18, 609–614. [Google Scholar] [CrossRef]
- Mogayzel, P.J., Jr.; Naureckas, E.T.; Robinson, K.A.; Mueller, G.; Hadjiliadis, D.; Hoag, J.B.; Lubsch, L.; Hazle, L.; Sabadosa, K.; Marshall, B.; et al. Cystic fibrosis pulmonary guidelines. Chronic medications for maintenance of lung health. Am. J. Respir. Crit. Care Med. 2013, 187, 680–689. [Google Scholar] [CrossRef] [PubMed]
- Maselli, D.J.; Keyt, H.; Restrepo, M.I. Inhaled Antibiotic Therapy in Chronic Respiratory Diseases. Int. J. Mol. Sci. 2017, 18, 1062. [Google Scholar] [CrossRef] [Green Version]
- Warnock, L.; Gates, A. Chest physiotherapy compared to no chest physiotherapy for cystic fibrosis. Cochrane Database Syst. Rev. 2015, 14, 1401. [Google Scholar] [CrossRef]
- Morrow, B.M. Airway clearance therapy in acute paediatric respiratory illness: A state-of-the-art review. S Afr. J. Physiother. 2019, 75, 1295. [Google Scholar] [CrossRef] [Green Version]
- Kirilloff, L.H.; Owens, G.R.; Rogers, R.M.; Mazzocco, M.C. Does chest physical therapy work? Chest 1985, 88, 436–444. [Google Scholar] [CrossRef]
- Sutton, P.P. Chest physiotherapy: Time for reappraisal. Br. J. Dis. Chest 1988, 82, 127–137. [Google Scholar] [CrossRef]
- Webber, B.A.; Hofmeyr, J.L.; Morgan, M.D.; Hodson, M.E. Effects of postural drainage, incorporating the forced expiration technique, on pulmonary function in cystic fibrosis. Br. J. Dis. Chest 1986, 80, 353–359. [Google Scholar] [CrossRef]
- Hospital, U.H.A.F.C. Cystic Fibrosis (CF) Treatment: Manual Chest Physiotherapy. 2018. Available online: https://www.ecfs.eu/sites/default/files/general-content-files/working-groups/IPG%20CF_Blue%20Booklet_5th%20edition%202018.pdf (accessed on 1 March 2020).
- Flume, P.A.; Mogayzel, P.J., Jr.; Robinson, K.A.; Goss, C.H.; Rosenblatt, R.L.; Kuhn, R.J.; Marshall, B.C.; Bujan, J.; Downs, A.; Finder, J.; et al. Cystic fibrosis pulmonary guidelines: Treatment of pulmonary exacerbations. Am. J. Respir. Crit. Care Med. 2009, 180, 802–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reix, P.; Aubert, F.; Werck-Gallois, M.C.; Toutain, A.; Mazzocchi, C.; Moreux, N.; Bellon, G.; Rabilloud, M.; Kassai, B. Exercise with incorporated expiratory manoeuvres was as effective as breathing techniques for airway clearance in children with cystic fibrosis: A randomised crossover trial. J. Physiother. 2012, 58, 241–247. [Google Scholar] [CrossRef] [Green Version]
- McIlwaine, M.B.B.; Nevitt, S.J. Positive expiratory pressure physiotherapy for airway clearance in people with cystic fibrosis. Cochrane Database Syst. Rev. 2019, 14, 3147. [Google Scholar] [CrossRef]
- Mckoy, N.A.; Wilson, L.M.; Saldanha, I.J.; Odelola, O.A.; Robinson, K.A. Active cycle of breathing technique for cystic fibrosis. Cochrane Database Syst. Rev. 2016, 14, 1858. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, P.A.; Dodds, M.; Phillips, B.; Poole, J.; Webb, A.K. Regular exercise and reduction of breathlessness in patients with cystic fibrosis. Br. J. Dis. Chest 1987, 81, 62–69. [Google Scholar] [CrossRef]
- Orenstein, D.M.; Higgins, L.W. Update on the role of exercise in cystic fibrosis. Curr. Opin. Pulm. Med. 2005, 11, 519–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagener, J.S.; Rasouliyan, L.; VanDevanter, D.R.; Pasta, D.J.; Regelmann, W.E.; Morgan, W.J.; Konstan, M.W. for the Investigators and Coordinators of the Epidemiologic Study of Cystic Fibrosis. Oral, inhaled, and intravenous antibiotic choice for treating pulmonary exacerbations in cystic fibrosis. Pediatr. Pulmonol. 2013, 48, 666–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, G.; Singh, M.; Dwan, K. Inhaled antibiotics for long-term therapy in cystic fibrosis. Cochrane Database Syst. Rev. 2011, 14, 1021. [Google Scholar] [CrossRef]
- Nichols, D.P.; Durmowicz, A.G.; Field, A.; Flume, P.A.; VanDevanter, D.R.; Mayer-Hamblett, N. Developing Inhaled Antibiotics in Cystic Fibrosis: Current Challenges and Opportunities. Ann. Am. Thorac. Soc. 2019, 16, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Aparicio, M.; Saleta Canosa, J.L.; Valino Lopez, P.; Martin Egana, M.T.; Vidal Garcia, I.; Montero Martinez, C. Eradication of Pseudomonas aeruginosa with inhaled colistin in adults with non-cystic fibrosis bronchiectasis. Chron. Respir. Dis. 2019, 16, 1479973119872513. [Google Scholar] [CrossRef] [Green Version]
- Plant, B.J.; Downey, D.G.; Eustace, J.A.; Gunaratnam, C.; Haworth, C.S.; Jones, A.M.; McKone, E.F.; Peckham, D.G.; Ketchell, R.I.; Bilton, D. A treatment evaluator tool to monitor the real-world effectiveness of inhaled aztreonam lysine in cystic fibrosis. J. Cyst. Fibros. 2017, 16, 695–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vardakas, K.Z.; Voulgaris, G.L.; Samonis, G.; Falagas, M.E. Inhaled colistin monotherapy for respiratory tract infections in adults without cystic fibrosis: A systematic review and meta-analysis. Int. J. Antimicrob. Agents 2018, 51, 1–9. [Google Scholar] [CrossRef] [PubMed]
- VanDyke, R.D.; McPhail, G.L.; Huang, B.; Fenchel, M.C.; Amin, R.S.; Carle, A.C.; Chini, B.A.; Seid, M. Inhaled tobramycin effectively reduces FEV1 decline in cystic fibrosis. An instrumental variables analysis. Ann. Am. Thorac. Soc. 2013, 10, 205–212. [Google Scholar] [CrossRef] [Green Version]
- Van de Kerkhove, C.; Goeminne, P.C.; Kicinski, M.; Nawrot, T.S.; Lorent, N.; Van Bleyenbergh, P.; De Boeck, K.; Dupont, L.J. Continuous alternating inhaled antibiotic therapy in CF: A single center retrospective analysis. J. Cyst. Fibros. 2016, 15, 802–808. [Google Scholar] [CrossRef] [Green Version]
- Harrison, M.J.; McCarthy, M.; Fleming, C.; Hickey, C.; Shortt, C.; Eustace, J.A.; Murphy, D.M.; Plant, B.J. Inhaled versus nebulised tobramycin: A real world comparison in adult cystic fibrosis (CF). J. Cyst. Fibros. 2014, 13, 692–698. [Google Scholar] [CrossRef] [Green Version]
- Demoly, P.; Hagedoorn, P.; de Boer, A.H.; Frijlink, H.W. The clinical relevance of dry powder inhaler performance for drug delivery. Respir. Med. 2014, 108, 1195–1203. [Google Scholar] [CrossRef] [Green Version]
- Elkins, M.R.; Robinson, M.; Rose, B.R.; Harbour, C.; Moriarty, C.P.; Marks, G.B.; Belousova, E.G.; Xuan, W.; Bye, P.T.; National Hypertonic Saline in Cystic Fibrosis Study, G. A controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis. N. Engl. J. Med. 2006, 354, 229–240. [Google Scholar] [CrossRef] [Green Version]
- Wark, P.; McDonald, V.M. Nebulised hypertonic saline for cystic fibrosis. Cochrane Database Syst. Rev. 2018, 9, CD001506. [Google Scholar] [CrossRef]
- Stahl, M.; Wielputz, M.O.; Ricklefs, I.; Dopfer, C.; Barth, S.; Schlegtendal, A.; Graeber, S.Y.; Sommerburg, O.; Diekmann, G.; Husing, J.; et al. Preventive Inhalation of Hypertonic Saline in Infants with Cystic Fibrosis (PRESIS). A Randomized, Double-Blind, Controlled Study. Am. J. Respir. Crit. Care Med. 2019, 199, 1238–1248. [Google Scholar] [CrossRef]
- Klemmer, A.; Kramer, I.; Kamin, W. Physicochemical compatibility of nebulizable drug admixtures containing budesonide and colistimethate or hypertonic saline. Int. J. Pharm. Compd. 2013, 17, 254–261. [Google Scholar]
- Kramer, I.; Schwabe, A.; Lichtinghagen, R.; Kamin, W. Physicochemical compatibility of mixtures of dornase alfa and tobramycin containing nebulizer solutions. Pediatr. Pulmonol. 2009, 44, 134–141. [Google Scholar] [CrossRef]
- Deacon, J.; Abdelghany, S.M.; Quinn, D.J.; Schmid, D.; Megaw, J.; Donnelly, R.F.; Jones, D.S.; Kissenpfennig, A.; Elborn, J.S.; Gilmore, B.F.; et al. Antimicrobial efficacy of tobramycin polymeric nanoparticles for Pseudomonas aeruginosa infections in cystic fibrosis: Formulation, characterisation and functionalisation with dornase alfa (DNase). J. Control. Release 2015, 198, 55–61. [Google Scholar] [CrossRef] [Green Version]
- Chmiel, J.F.; Konstan, M.W.; Elborn, J.S. Antibiotic and anti-inflammatory therapies for cystic fibrosis. Cold Spring Harb. Perspect. Med. 2013, 3, a009779. [Google Scholar] [CrossRef] [Green Version]
- Konstan, M.W.; Byard, P.J.; Hoppel, C.L.; Davis, P.B. Effect of high-dose ibuprofen in patients with cystic fibrosis. N. Engl. J. Med. 1995, 332, 848–854. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.N.; Marshall-Batty, K.R.; Smolen, J.A.; Tagaev, J.A.; Chen, Q.; Rodesney, C.A.; Le, H.H.; Gordon, V.D.; Greenberg, D.E.; Cannon, C.L. Antimicrobial Activity of Ibuprofen against Cystic Fibrosis-Associated Gram-Negative Pathogens. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lands, L.C.; Stanojevic, S. Oral non-steroidal anti-inflammatory drug therapy for lung disease in cystic fibrosis. Cochrane Database Syst. Rev. 2019, 14, 1858. [Google Scholar] [CrossRef] [PubMed]
- Defoirdt, T. Quorum-Sensing Systems as Targets for Antivirulence Therapy. Trends Microbiol. 2018, 26, 313–328. [Google Scholar] [CrossRef]
- Jakobsen, T.H.; van Gennip, M.; Phipps, R.K.; Shanmugham, M.S.; Christensen, L.D.; Alhede, M.; Skindersoe, M.E.; Rasmussen, T.B.; Friedrich, K.; Uthe, F.; et al. Ajoene, a sulfur-rich molecule from garlic, inhibits genes controlled by quorum sensing. Antimicrob. Agents Chemother. 2012, 56, 2314–2325. [Google Scholar] [CrossRef] [Green Version]
- Jakobsen, T.H.; Warming, A.N.; Vejborg, R.M.; Moscoso, J.A.; Stegger, M.; Lorenzen, F.; Rybtke, M.; Andersen, J.B.; Petersen, R.; Andersen, P.S.; et al. A broad range quorum sensing inhibitor working through sRNA inhibition. Sci. Rep. 2017, 7, 9857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hentzer, M.; Riedel, K.; Rasmussen, T.B.; Heydorn, A.; Andersen, J.B.; Parsek, M.R.; Rice, S.A.; Eberl, L.; Molin, S.; Hoiby, N.; et al. Inhibition of quorum sensing in Pseudomonas aeruginosa biofilm bacteria by a halogenated furanone compound. Microbiology 2002, 148, 87–102. [Google Scholar] [CrossRef] [Green Version]
- Shetye, G.S.; Singh, N.; Gao, X.; Bandyopadhyay, D.; Yan, A.; Luk, Y.-Y. Structures and biofilm inhibition activities of brominated furanones for Escherichia coli and Pseudomonas aeruginosa. Med. Chem. Comm. 2013, 4, 1079–1084. [Google Scholar] [CrossRef]
- Paczkowski, J.E.; Mukherjee, S.; McCready, A.R.; Cong, J.P.; Aquino, C.J.; Kim, H.; Henke, B.R.; Smith, C.D.; Bassler, B.L. Flavonoids Suppress Pseudomonas aeruginosa Virulence through Allosteric Inhibition of Quorum-sensing Receptors. J. Biol. Chem. 2017, 292, 4064–4076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Park, J.H.; Cho, H.S.; Joo, S.W.; Cho, M.H.; Lee, J. Anti-biofilm activities of quercetin and tannic acid against Staphylococcus aureus. Biofouling 2013, 29, 491–499. [Google Scholar] [CrossRef] [PubMed]
- da Costa Junior, S.D.; de Oliveira Santos, J.V.; de Almeida Campos, L.A.; Pereira, M.A.; Santos Magalhães, N.S.; Ferro Cavalcanti, I.M. Antibacterial and antibiofilm activities of quercetin against clinical isolates of Staphyloccocus aureus and Staphylococcus saprophyticus with resistance profile. Int. J. Environ. Agric. Biotechnol. 2018, 3, 1948–1958. [Google Scholar]
- Jakobsen, T.H.; Bragason, S.K.; Phipps, R.K.; Christensen, L.D.; van Gennip, M.; Alhede, M.; Skindersoe, M.; Larsen, T.O.; Hoiby, N.; Bjarnsholt, T.; et al. Food as a source for quorum sensing inhibitors: Iberin from horseradish revealed as a quorum sensing inhibitor of Pseudomonas aeruginosa. Appl. Environ. Microbiol. 2012, 78, 2410–2421. [Google Scholar] [CrossRef] [Green Version]
- Brackman, G.; Breyne, K.; De Rycke, R.; Vermote, A.; Van Nieuwerburgh, F.; Meyer, E.; Van Calenbergh, S.; Coenye, T. The Quorum Sensing Inhibitor Hamamelitannin Increases Antibiotic Susceptibility of Staphylococcus aureus Biofilms by Affecting Peptidoglycan Biosynthesis and eDNA Release. Sci. Rep. 2016, 6, 20321. [Google Scholar] [CrossRef]
- Udine, C.; Brackman, G.; Bazzini, S.; Buroni, S.; Van Acker, H.; Pasca, M.R.; Riccardi, G.; Coenye, T. Phenotypic and genotypic characterisation of Burkholderia cenocepacia J2315 mutants affected in homoserine lactone and diffusible signal factor-based quorum sensing systems suggests interplay between both types of systems. PLoS ONE 2013, 8, e55112. [Google Scholar] [CrossRef] [Green Version]
- Suppiger, A.; Schmid, N.; Aguilar, C.; Pessi, G.; Eberl, L. Two quorum sensing systems control biofilm formation and virulence in members of the Burkholderia cepacia complex. Virulence 2013, 4, 400–409. [Google Scholar] [CrossRef] [Green Version]
- Buroni, S.; Scoffone, V.C.; Fumagalli, M.; Makarov, V.; Cagnone, M.; Trespidi, G.; De Rossi, E.; Forneris, F.; Riccardi, G.; Chiarelli, L.R. Investigating the Mechanism of Action of Diketopiperazines Inhibitors of the Burkholderia cenocepacia Quorum Sensing Synthase CepI: A Site-Directed Mutagenesis Study. Front. Pharmacol. 2018, 9, 836. [Google Scholar] [CrossRef]
- Scoffone, V.C.; Chiarelli, L.R.; Makarov, V.; Brackman, G.; Israyilova, A.; Azzalin, A.; Forneris, F.; Riabova, O.; Savina, S.; Coenye, T.; et al. Discovery of new diketopiperazines inhibiting Burkholderia cenocepacia quorum sensing in vitro and in vivo. Sci. Rep. 2016, 6, 32487. [Google Scholar] [CrossRef] [Green Version]
- Cheluvappa, R.; Shimmon, R.; Dawson, M.; Hilmer, S.N.; Le Couteur, D.G. Reactions of Pseudomonas aeruginosa pyocyanin with reduced glutathione. Acta Biochim. Pol. 2008, 55, 571–580. [Google Scholar] [CrossRef] [Green Version]
- Roum, J.H.; Buhl, R.; McElvaney, N.G.; Borok, Z.; Crystal, R.G. Systemic deficiency of glutathione in cystic fibrosis. J. Appl. Physiol. 1993, 75, 2419–2424. [Google Scholar] [CrossRef]
- Klare, W.; Das, T.; Ibugo, A.; Buckle, E.; Manefield, M.; Manos, J. Glutathione-Disrupted Biofilms of Clinical Pseudomonas aeruginosa Strains Exhibit an Enhanced Antibiotic Effect and a Novel Biofilm Transcriptome. Antimicrob. Agents Chemother. 2016, 60, 4539–4551. [Google Scholar] [CrossRef] [Green Version]
- Das, T.; Simone, M.; Ibugo, A.I.; Witting, P.K.; Manefield, M.; Manos, J. Glutathione Enhances Antibiotic Efficiency and Effectiveness of DNase I in Disrupting Pseudomonas aeruginosa Biofilms While Also Inhibiting Pyocyanin Activity, Thus Facilitating Restoration of Cell Enzymatic Activity, Confluence and Viability. Front. Microbiol. 2017, 8, 2429. [Google Scholar] [CrossRef] [Green Version]
- Das, T.; Paino, D.; Manoharan, A.; Farrell, J.; Whiteley, G.; Kriel, F.H.; Glasbey, T.; Manos, J. Conditions Under Which Glutathione Disrupts the Biofilms and Improves Antibiotic Efficacy of Both ESKAPE and Non-ESKAPE Species. Front. Microbiol. 2019, 10, 2000. [Google Scholar] [CrossRef] [Green Version]
- Ratjen, F.; Grasemann, H. New therapies in cystic fibrosis. Curr. Pharm. Des. 2012, 18, 614–627. [Google Scholar] [CrossRef]
- Tirouvanziam, R.; Conrad, C.K.; Bottiglieri, T.; Herzenberg, L.A.; Moss, R.B.; Herzenberg, L.A. High-dose oral N-acetylcysteine, a glutathione prodrug, modulates inflammation in cystic fibrosis. Proc. Natl. Acad. Sci. USA 2006, 103, 4628–4633. [Google Scholar] [CrossRef] [Green Version]
- Dauletbaev, N.; Fischer, P.; Aulbach, B.; Gross, J.; Kusche, W.; Thyroff-Friesinger, U.; Wagner, T.O.; Bargon, J. A phase II study on safety and efficacy of high-dose N-acetylcysteine in patients with cystic fibrosis. Eur. J. Med. Res. 2009, 14, 352–358. [Google Scholar] [CrossRef] [Green Version]
- Rubin, B.K. Secretion properties, clearance, and therapy in airway disease. Transl. Respir. Med. 2014, 2, 6. [Google Scholar] [CrossRef] [Green Version]
- Therapeutic Goods Administration. Omegapharm Acetylcysteine Solution for Administration—Product Information; Therapeutic Goods Administration: Woden, ACT, Australia, 2011. [Google Scholar]
- Palmer, L.A.; Doctor, A.; Chhabra, P.; Sheram, M.L.; Laubach, V.E.; Karlinsey, M.Z.; Forbes, M.S.; Macdonald, T.; Gaston, B. S-nitrosothiols signal hypoxia-mimetic vascular pathology. J. Clin. Investig. 2007, 117, 2592–2601. [Google Scholar] [CrossRef]
- Conrad, C.; Lymp, J.; Thompson, V.; Dunn, C.; Davies, Z.; Chatfield, B.; Nichols, D.; Clancy, J.; Vender, R.; Egan, M.E.; et al. Long-term treatment with oral N-acetylcysteine: Affects lung function but not sputum inflammation in cystic fibrosis subjects. A phase II randomized placebo-controlled trial. J. Cyst. Fibros. 2015, 14, 219–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blasi, F.; Page, C.; Rossolini, G.M.; Pallecchi, L.; Matera, M.G.; Rogliani, P.; Cazzola, M. The effect of N-acetylcysteine on biofilms: Implications for the treatment of respiratory tract infections. Respir. Med. 2016, 117, 190–197. [Google Scholar] [CrossRef] [Green Version]
- Perez-Giraldo, C.; Rodriguez-Benito, A.; Moran, F.J.; Hurtado, C.; Blanco, M.T.; Gomez-Garcia, A.C. Influence of N-acetylcysteine on the formation of biofilm by Staphylococcus epidermidis. J. Antimicrob. Chemother. 1997, 39, 643–646. [Google Scholar] [CrossRef]
- Zhao, T.; Liu, Y. N-acetylcysteine inhibit biofilms produced by Pseudomonas aeruginosa. BMC Microbiol. 2010, 10, 140. [Google Scholar] [CrossRef] [Green Version]
- Pollini, S.; Di Pilato, V.; Landini, G.; Di Maggio, T.; Cannatelli, A.; Sottotetti, S.; Cariani, L.; Aliberti, S.; Blasi, F.; Sergio, F.; et al. In vitro activity of N-acetylcysteine against Stenotrophomonas maltophilia and Burkholderia cepacia complex grown in planktonic phase and biofilm. PLoS ONE 2018, 13, e0203941. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Li, P.; Chen, X.; Zou, Y.; Li, H.; Yuan, G.; Hu, H. Activity of Sodium Lauryl Sulfate, Rhamnolipids, and N-Acetylcysteine Against Biofilms of Five Common Pathogens. Microb. Drug Resist. 2020, 26, 290–299. [Google Scholar] [CrossRef]
- Harrison, J.J.; Stremick, C.A.; Turner, R.J.; Allan, N.D.; Olson, M.E.; Ceri, H. Microtiter susceptibility testing of microbes growing on peg lids: A miniaturized biofilm model for high-throughput screening. Nat. Protoc. 2010, 5, 1236–1254. [Google Scholar] [CrossRef]
- Lababidi, N.; Ofosu Kissi, E.; Elgaher, W.A.M.; Sigal, V.; Haupenthal, J.; Schwarz, B.C.; Hirsch, A.K.H.; Rades, T.; Schneider, M. Spray-drying of inhalable, multifunctional formulations for the treatment of biofilms formed in cystic fibrosis. J. Control. Release 2019, 314, 62–71. [Google Scholar] [CrossRef]
- Aslam, S.; Darouiche, R.O. Role of antibiofilm-antimicrobial agents in controlling device-related infections. Int. J. Artif. Organs. 2011, 34, 752–758. [Google Scholar] [CrossRef] [Green Version]
- Manoharan, A.; Das, T.; Whiteley, G.S.; Glasbey, T.; Kriel, F.H.; Manos, J. The effect of N-acetylcysteine in a combined antibiofilm treatment against antibiotic-resistant Staphylococcus aureus. J. Antimicrob. Chemother. 2020, 75, 1787–1798. [Google Scholar] [CrossRef]
- Kallio, J.; Jaakkola, M.; Maki, M.; Kilpelainen, P.; Virtanen, V. Vitamin C inhibits Staphylococcus aureus growth and enhances the inhibitory effect of quercetin on growth of Escherichia coli in vitro. Planta Med. 2012, 78, 1824–1830. [Google Scholar] [CrossRef] [Green Version]
- Diaz De Rienzo, M.A.; Stevenson, P.S.; Marchant, R.; Banat, I.M. Pseudomonas aeruginosa biofilm disruption using microbial surfactants. J. Appl. Microbiol. 2016, 120, 868–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandit, S.; Ravikumar, V.; Abdel-Haleem, A.M.; Derouiche, A.; Mokkapati, V.; Sihlbom, C.; Mineta, K.; Gojobori, T.; Gao, X.; Westerlund, F.; et al. Low Concentrations of Vitamin C Reduce the Synthesis of Extracellular Polymers and Destabilize Bacterial Biofilms. Front. Microbiol. 2017, 8, 2599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalton, J.P.; Uy, B.; Phummarin, N.; Copp, B.R.; Denny, W.A.; Swift, S.; Wiles, S. Effect of common and experimental anti-tuberculosis treatments on Mycobacterium tuberculosis growing as biofilms. PeerJ 2016, 4, e2717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razvi, S.; Saiman, L. Nontuberculous mycobacteria in cystic fibrosis. Pediatr. Infect. Dis. J. 2007, 26, 263–264. [Google Scholar] [CrossRef]
- Syal, K.; Bhardwaj, N.; Chatterji, D. Vitamin C targets (p)ppGpp synthesis leading to stalling of long-term survival and biofilm formation in Mycobacterium smegmatis. FEMS Microbiol. Lett. 2017, 364. [Google Scholar] [CrossRef] [Green Version]
- Fraser, J.F.; Bodman, J.; Sturgess, R.; Faoagali, J.; Kimble, R.M. An in vitro study of the anti-microbial efficacy of a 1% silver sulphadiazine and 0.2% chlorhexidine digluconate cream, 1% silver sulphadiazine cream and a silver coated dressing. Burns 2004, 30, 35–41. [Google Scholar] [CrossRef]
- Pruitt, B.A., Jr.; McManus, A.T.; Kim, S.H.; Goodwin, C.W. Burn wound infections: Current status. World J. Surg. 1998, 22, 135–145. [Google Scholar] [CrossRef]
- Radzig, M.A.; Nadtochenko, V.A.; Koksharova, O.A.; Kiwi, J.; Lipasova, V.A.; Khmel, I.A. Antibacterial effects of silver nanoparticles on gram-negative bacteria: Influence on the growth and biofilms formation, mechanisms of action. Colloids Surf. B Biointerfaces 2013, 102, 300–306. [Google Scholar] [CrossRef]
- Mohanty, S.; Mishra, S.; Jena, P.; Jacob, B.; Sarkar, B.; Sonawane, A. An investigation on the antibacterial, cytotoxic, and antibiofilm efficacy of starch-stabilized silver nanoparticles. Nanomedicine 2012, 8, 916–924. [Google Scholar] [CrossRef]
- Habash, M.B.; Park, A.J.; Vis, E.C.; Harris, R.J.; Khursigara, C.M. Synergy of silver nanoparticles and aztreonam against Pseudomonas aeruginosa PAO1 biofilms. Antimicrob. Agents Chemother. 2014, 58, 5818–5830. [Google Scholar] [CrossRef] [Green Version]
- Habash, M.B.; Goodyear, M.C.; Park, A.J.; Surette, M.D.; Vis, E.C.; Harris, R.J.; Khursigara, C.M. Potentiation of Tobramycin by Silver Nanoparticles against Pseudomonas aeruginosa Biofilms. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [Green Version]
- Jeannet, N.; Fierz, M.; Schneider, S.; Kunzi, L.; Baumlin, N.; Salathe, M.; Burtscher, H.; Geiser, M. Acute toxicity of silver and carbon nanoaerosols to normal and cystic fibrosis human bronchial epithelial cells. Nanotoxicology 2016, 10, 279–291. [Google Scholar] [CrossRef]
- Pompilio, A.; Geminiani, C.; Bosco, D.; Rana, R.; Aceto, A.; Bucciarelli, T.; Scotti, L.; Di Bonaventura, G. Electrochemically Synthesized Silver Nanoparticles Are Active Against Planktonic and Biofilm Cells of Pseudomonas aeruginosa and Other Cystic Fibrosis-Associated Bacterial Pathogens. Front. Microbiol. 2018, 9, 1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Smolen, J.A.; Zhang, S.; Li, R.; Shah, P.N.; Cho, S.; Wang, H.; Raymond, J.E.; Cannon, C.L.; Wooley, K.L. Degradable polyphosphoester-based silver-loaded nanoparticles as therapeutics for bacterial lung infections. Nanoscale 2015, 7, 2265–2270. [Google Scholar] [CrossRef] [PubMed]
- Hraiech, S.; Bregeon, F.; Rolain, J.M. Bacteriophage-based therapy in cystic fibrosis-associated Pseudomonas aeruginosa infections: Rationale and current status. Drug Des. Devel. Ther. 2015, 9, 3653–3663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanlon, G.W. Bacteriophages: An appraisal of their role in the treatment of bacterial infections. Int. J. Antimicrob. Agents 2007, 30, 118–128. [Google Scholar] [CrossRef]
- Semler, D.D.; Goudie, A.D.; Finlay, W.H.; Dennis, J.J. Aerosol phage therapy efficacy in Burkholderia cepacia complex respiratory infections. Antimicrob. Agents Chemother. 2014, 58, 4005–4013. [Google Scholar] [CrossRef] [Green Version]
- Debarbieux, L.; Leduc, D.; Maura, D.; Morello, E.; Criscuolo, A.; Grossi, O.; Balloy, V.; Touqui, L. Bacteriophages can treat and prevent Pseudomonas aeruginosa lung infections. J. Infect. Dis. 2010, 201, 1096–1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golshahi, L.; Lynch, K.H.; Dennis, J.J.; Finlay, W.H. In vitro lung delivery of bacteriophages KS4-M and PhiKZ using dry powder inhalers for treatment of Burkholderia cepacia complex and Pseudomonas aeruginosa infections in cystic fibrosis. J. Appl. Microbiol. 2011, 110, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Hoe, S.; Semler, D.D.; Goudie, A.D.; Lynch, K.H.; Matinkhoo, S.; Finlay, W.H.; Dennis, J.J.; Vehring, R. Respirable bacteriophages for the treatment of bacterial lung infections. J. Aerosol. Med. Pulm. Drug Deliv. 2013, 26, 317–335. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.Y.K.; Das, T.; Manos, J.; Kutter, E.; Morales, S.; Chan, H.K. Bacteriophage PEV20 and Ciprofloxacin Combination Treatment Enhances Removal of Pseudomonas aeruginosa Biofilm Isolated from Cystic Fibrosis and Wound Patients. AAPS J. 2019, 21, 49. [Google Scholar] [CrossRef] [PubMed]
- Gainey, A.B.; Burch, A.K.; Brownstein, M.J.; Brown, D.E.; Fackler, J.; Horne, B.; Biswas, B.; Bivens, B.N.; Malagon, F.; Daniels, R. Combining bacteriophages with cefiderocol and meropenem/vaborbactam to treat a pan-drug resistant Achromobacter species infection in a pediatric cystic fibrosis patient. Pediatr. Pulmonol. 2020, 55, 2990–2994. [Google Scholar] [CrossRef]
- Lin, Y.; Quan, D.; Yoon Kyung Chang, R.; Chow, M.Y.T.; Wang, Y.; Li, M.; Morales, S.; Britton, W.J.; Kutter, E.; Li, J.; et al. Synergistic activity of phage PEV20-ciprofloxacin combination powder formulation-A proof-of-principle study in a P. aeruginosa lung infection model. Eur. J. Pharm. Biopharm. 2020. [Google Scholar] [CrossRef]
- Law, N.; Logan, C.; Yung, G.; Furr, C.L.; Lehman, S.M.; Morales, S.; Rosas, F.; Gaidamaka, A.; Bilinsky, I.; Grint, P.; et al. Successful adjunctive use of bacteriophage therapy for treatment of multidrug-resistant Pseudomonas aeruginosa infection in a cystic fibrosis patient. Infection 2019, 47, 665–668. [Google Scholar] [CrossRef] [PubMed]
- Pettit, R.S.; Fellner, C. CFTR Modulators for the Treatment of Cystic Fibrosis. Pharm. Ther. 2014, 39, 500–511. [Google Scholar]
- McKone, E.F.; Borowitz, D.; Drevinek, P.; Griese, M.; Konstan, M.W.; Wainwright, C.; Ratjen, F.; Sermet-Gaudelus, I.; Plant, B.; Munck, A.; et al. Long-term safety and efficacy of ivacaftor in patients with cystic fibrosis who have the Gly551Asp-CFTR mutation: A phase 3, open-label extension study (PERSIST). Lancet Respir. Med. 2014, 2, 902–910. [Google Scholar] [CrossRef]
- Heltshe, S.L.; Mayer-Hamblett, N.; Burns, J.L.; Khan, U.; Baines, A.; Ramsey, B.W.; Rowe, S.M. GOAL (the G551D Observation-AL) Investigators of the Cystic Fibrosis Foundation Therapeutics Development Network. Pseudomonas aeruginosa in cystic fibrosis patients with G551D-CFTR treated with ivacaftor. Clin. Infect. Dis. 2015, 60, 703–712. [Google Scholar] [CrossRef]
- Frost, F.J.; Nazareth, D.S.; Charman, S.C.; Winstanley, C.; Walshaw, M.J. Ivacaftor Is Associated with Reduced Lung Infection by Key Cystic Fibrosis Pathogens. A Cohort Study Using National Registry Data. Ann. Am. Thorac Soc. 2019, 16, 1375–1382. [Google Scholar] [CrossRef]
- Bernarde, C.; Keravec, M.; Mounier, J.; Gouriou, S.; Rault, G.; Ferec, C.; Barbier, G.; Hery-Arnaud, G. Impact of the CFTR-potentiator ivacaftor on airway microbiota in cystic fibrosis patients carrying a G551D mutation. PLoS ONE 2015, 10, e0124124. [Google Scholar] [CrossRef] [Green Version]
- Harris, J.K.; Wagner, B.D.; Zemanick, E.T.; Robertson, C.E.; Stevens, M.J.; Heltshe, S.L.; Rowe, S.M.; Sagel, S.D. Changes in Airway Microbiome and Inflammation with Ivacaftor Treatment in Patients with Cystic Fibrosis and the G551D Mutation. Ann. Am. Thorac. Soc. 2020, 17, 212–220. [Google Scholar] [CrossRef]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; De Boeck, K.; Flume, P.A.; et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. [Google Scholar] [CrossRef] [Green Version]
- McColley, S.A.; Konstan, M.W.; Ramsey, B.W.; Stuart Elborn, J.; Boyle, M.P.; Wainwright, C.E.; Waltz, D.; Vera-Llonch, M.; Marigowda, G.; Jiang, J.G.; et al. Lumacaftor/Ivacaftor reduces pulmonary exacerbations in patients irrespective of initial changes in FEV1. J. Cyst. Fibros. 2019, 18, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Tesell, M.A.; Alper, C.J.; Bacon, R.; Greenwood, B.C.; Lenz, K.; Jeffrey, P.L.; Stevens, K. Effect of Lumacaftor/Ivacaftor on Pulmonary Exacerbation Rates in Members with Cystic Fibrosis in a Medicaid Population. J. Manag. Care Spec. Pharm. 2019, 25, 1021–1025. [Google Scholar] [CrossRef]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef]
- Hisert, K.B.; Heltshe, S.L.; Pope, C.; Jorth, P.; Wu, X.; Edwards, R.M.; Radey, M.; Accurso, F.J.; Wolter, D.J.; Cooke, G.; et al. Restoring Cystic Fibrosis Transmembrane Conductance Regulator Function Reduces Airway Bacteria and Inflammation in People with Cystic Fibrosis and Chronic Lung Infections. Am. J. Respir. Crit. Care Med. 2017, 195, 1617–1628. [Google Scholar] [CrossRef]
- Yusen, R.D.; Edwards, L.B.; Kucheryavaya, A.Y.; Benden, C.; Dipchand, A.I.; Goldfarb, S.B.; Levvey, B.J.; Lund, L.H.; Meiser, B.; Rossano, J.W.; et al. The Registry of the International Society for Heart and Lung Transplantation: Thirty-second Official Adult Lung and Heart-Lung Transplantation Report--2015; Focus Theme: Early Graft Failure. J. Heart Lung Transplant. 2015, 34, 1264–1277. [Google Scholar] [CrossRef] [PubMed]
- Weill, D.; Benden, C.; Corris, P.A.; Dark, J.H.; Davis, R.D.; Keshavjee, S.; Lederer, D.J.; Mulligan, M.J.; Patterson, G.A.; Singer, L.G.; et al. A consensus document for the selection of lung transplant candidates: 2014—An update from the Pulmonary Transplantation Council of the International Society for Heart and Lung Transplantation. J. Heart Lung Transplant. 2015, 34, 1–15. [Google Scholar] [CrossRef]
- Lobo, L.J.; Chang, L.C.; Esther, C.R., Jr.; Gilligan, P.H.; Tulu, Z.; Noone, P.G. Lung transplant outcomes in cystic fibrosis patients with pre-operative Mycobacterium abscessus respiratory infections. Clin. Transplant. 2013, 27, 523–529. [Google Scholar] [CrossRef]
- Gottlieb, J.; Mattner, F.; Weissbrodt, H.; Dierich, M.; Fuehner, T.; Strueber, M.; Simon, A.; Welte, T. Impact of graft colonization with gram-negative bacteria after lung transplantation on the development of bronchiolitis obliterans syndrome in recipients with cystic fibrosis. Respir. Med. 2009, 103, 743–749. [Google Scholar] [CrossRef] [Green Version]
- Orfanos, S.; Gomez, C.; Baron, S.; Akkisetty, R.; Dufeu, N.; Coltey, B.; Thomas, P.A.; Rolain, J.M.; Reynaud-Gaubert, M. Impact of gram negative bacteria airway recolonization on the occurrence of chronic lung allograft dysfunction after lung transplantation in a population of cystic fibrosis patients. BMC Microbiol. 2018, 18, 88. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Part 3: Time since Isolation of Initial Strain (Adapted from Manos et al. 2013 Eur J Clin MicrobiolInfect Dis 32:1583–1592 [39]. | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Virulence Factor | Units | 0 Months | 1–11 Months | p Value a | 12–23 Months | p Value a | 24–35 Months | p Value a | +36 Months | p Value a |
| Pyocyanin | % | 18.03 | 20.00 | 0.822 | 19.05 | 0.918 | 0.00 | 0.061 | 33.33 | 0.202 |
| Pyoverdine | % | 40.98 | 26.67 | 0.180 | 25.40 | 0.171 | 18.18 | 0.166 | 26.67 | 0.309 |
| Swarming | % | 13.7 | 11.1 | 0.725 | 4.8 | 0.292 | 18.20 | 0.695 | 8.9 | 0.552 |
| Elastase | mm2 | 89.88 | 79.36 | 0.203 | 89.20 | 0.942 | 100.84 | 0.367 | 49.75 | 0.000 *** |
| Rhamnolipid | mm2 | 62.07 | 59.60 | 0.690 | 60.23 | 0.794 | 66.77 | 0.607 | 66.93 | 0.545 |
| PLC b | mm2 | 97.72 | 88.68 | 0.287 | 95.62 | 0.827 | 96.20 | 0.903 | 65.56 | 0.003 ** |
| Haemolysin | mm2 | 96.88 | 76.96 | 0.147 | 83.34 | 0.385 | 90.92 | 0.767 | 59.88 | 0.037 * |
| Total protease | mm2 | 91.50 | 84.34 | 0.249 | 88.99 | 0.722 | 94.35 | 0.755 | 73.82 | 0.028 * |
| Biofilm mass | % c | 95.07 | 70.61 | 0.033 * | 92.71 | 0.850 | 100.82 | 0.721 | 85.02 | 0.480 |
| Swimming | mm2 | 78.01 | 61.71 | 0.034 * | 68.68 | 0.284 | 75.34 | 0.812 | 58.90 | 0.054 † |
| Twitching | mm2 | 70.96 | 62.13 | 0.354 | 45.34 | 0.018 * | 55.89 | 0.282 | 42.67 | 0.054 † |
| Colony size | mm | 2.86 | 2.83 | 0.874 | 2.43 | 0.027 * | 2.64 | 0.375 | 1.93 | 0.000 *** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manos, J. Current and Emerging Therapies to Combat Cystic Fibrosis Lung Infections. Microorganisms 2021, 9, 1874. https://doi.org/10.3390/microorganisms9091874
Manos J. Current and Emerging Therapies to Combat Cystic Fibrosis Lung Infections. Microorganisms. 2021; 9(9):1874. https://doi.org/10.3390/microorganisms9091874
Chicago/Turabian StyleManos, Jim. 2021. "Current and Emerging Therapies to Combat Cystic Fibrosis Lung Infections" Microorganisms 9, no. 9: 1874. https://doi.org/10.3390/microorganisms9091874
APA StyleManos, J. (2021). Current and Emerging Therapies to Combat Cystic Fibrosis Lung Infections. Microorganisms, 9(9), 1874. https://doi.org/10.3390/microorganisms9091874