
High Throughput Analysis Reveals Changes in Gut Microbiota and Specific Fecal Metabolomic Signature in Hematopoietic Stem Cell Transplant Patients

 ,
,  , , ,
, , ,  ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Cohort
2.2. DNA Extraction, First-Step PCR Amplification, and Purification
2.3. Second-Step PCR Amplification (Indexing) and Purification
2.4. Bioinformatic and Statistical Analysis
2.5. GC-MS Sample Preparation and Metabolite Extraction
2.6. GC-MS Analysis and Metabolite Detection
2.7. Identification and Comparison of Volatile Compounds
2.8. Data Analysis
2.9. Selection of Biomarkers
3. Results
3.1. Hematopoietic Stem Cell Transplant Cohort
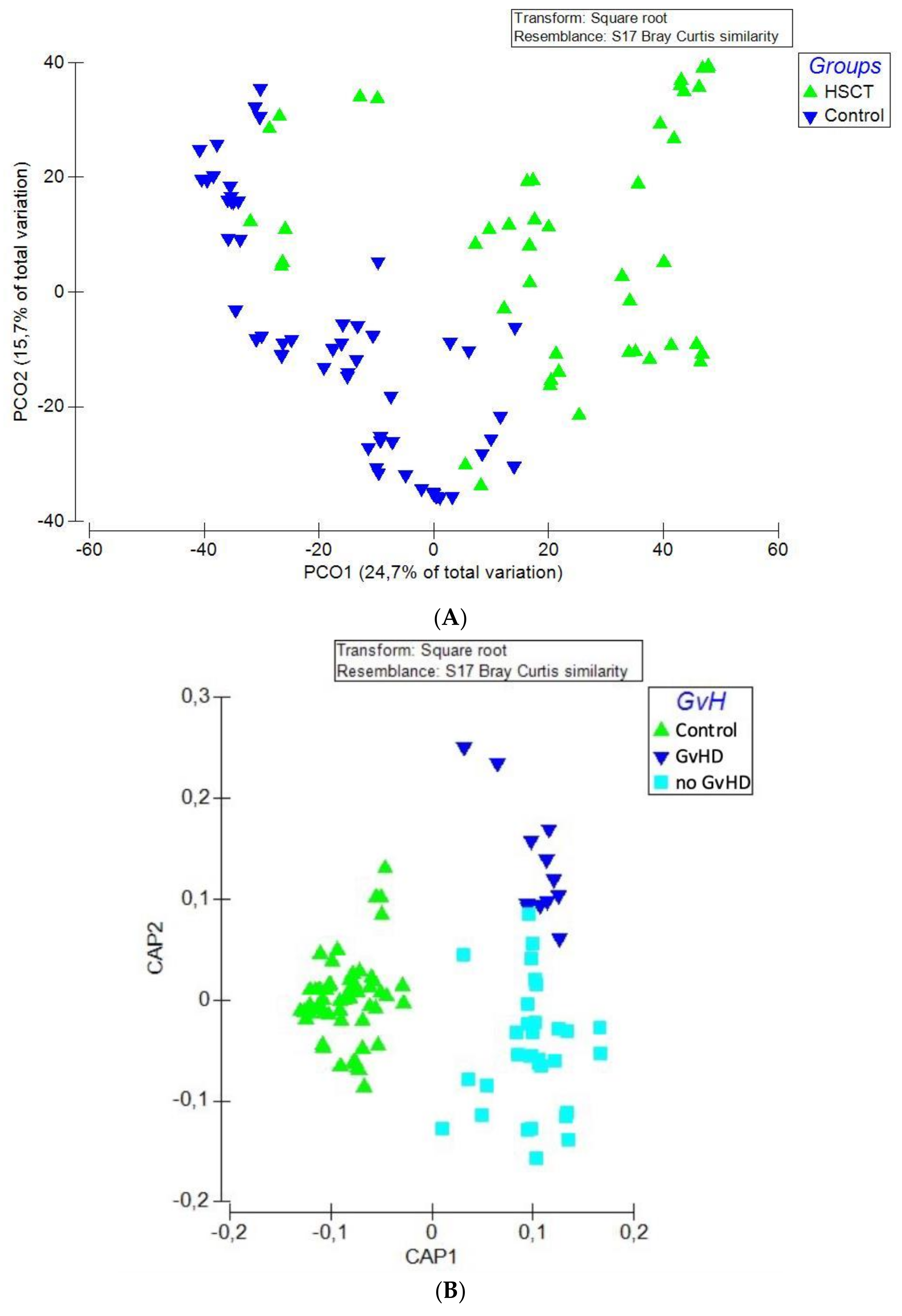
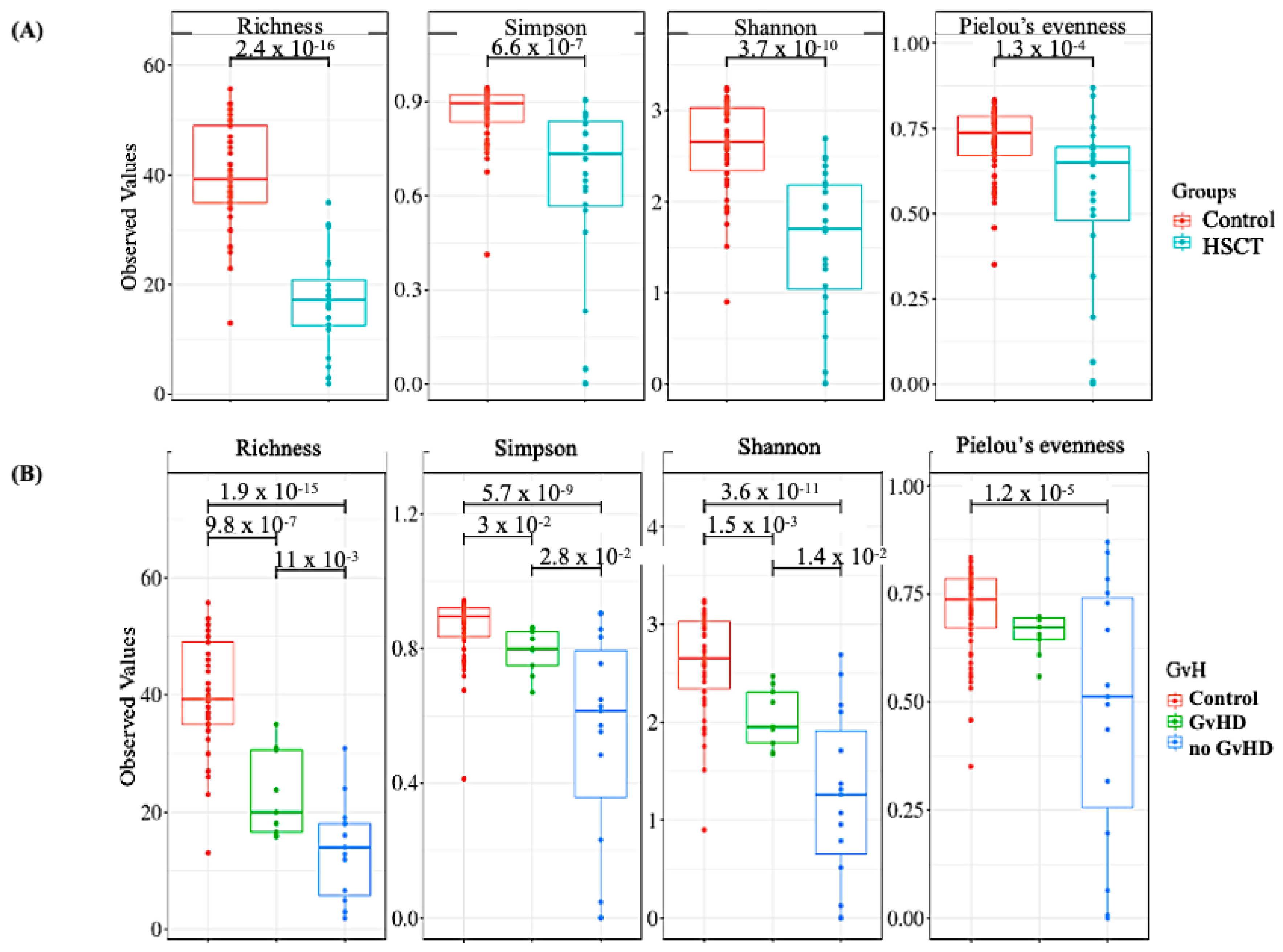
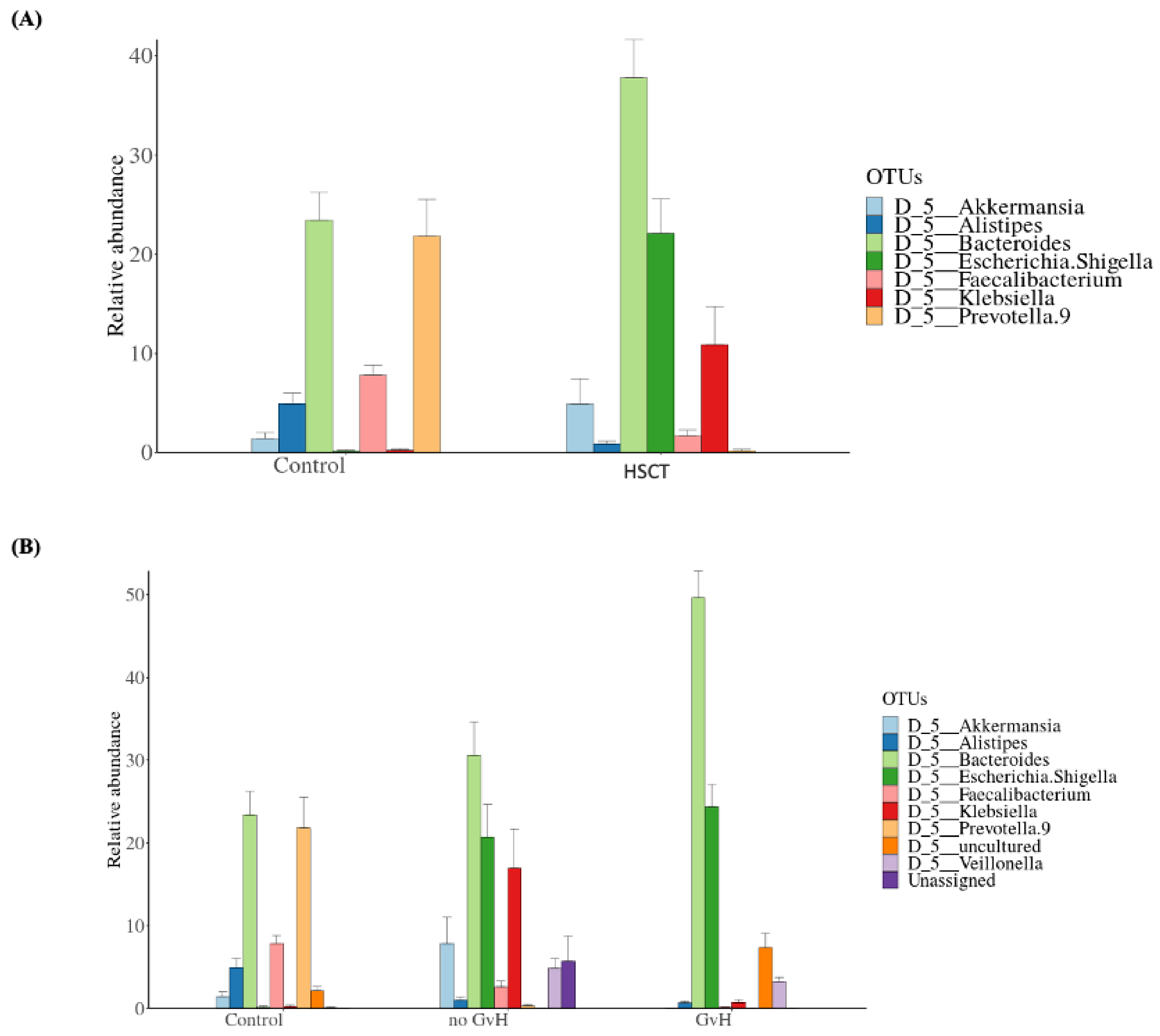
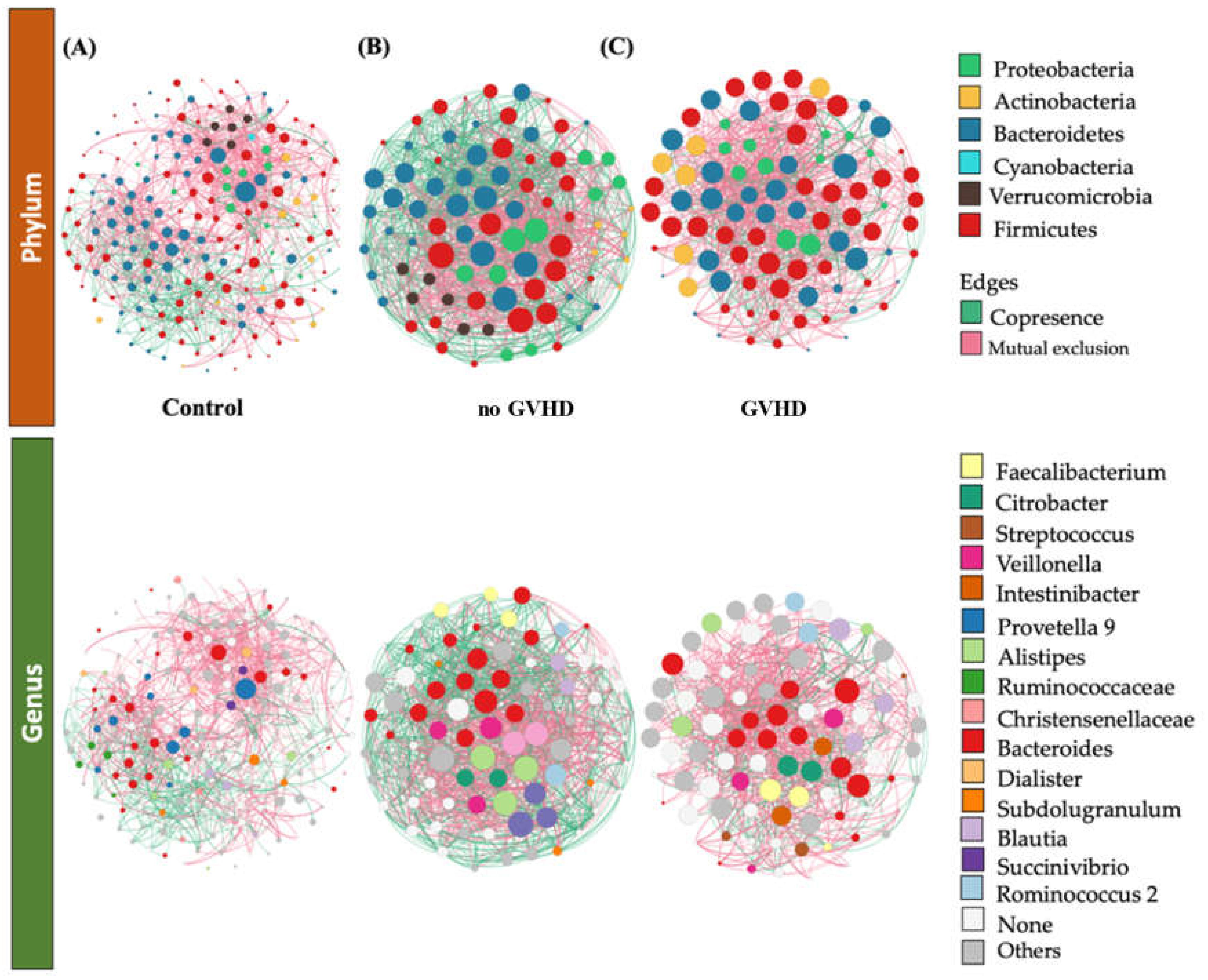
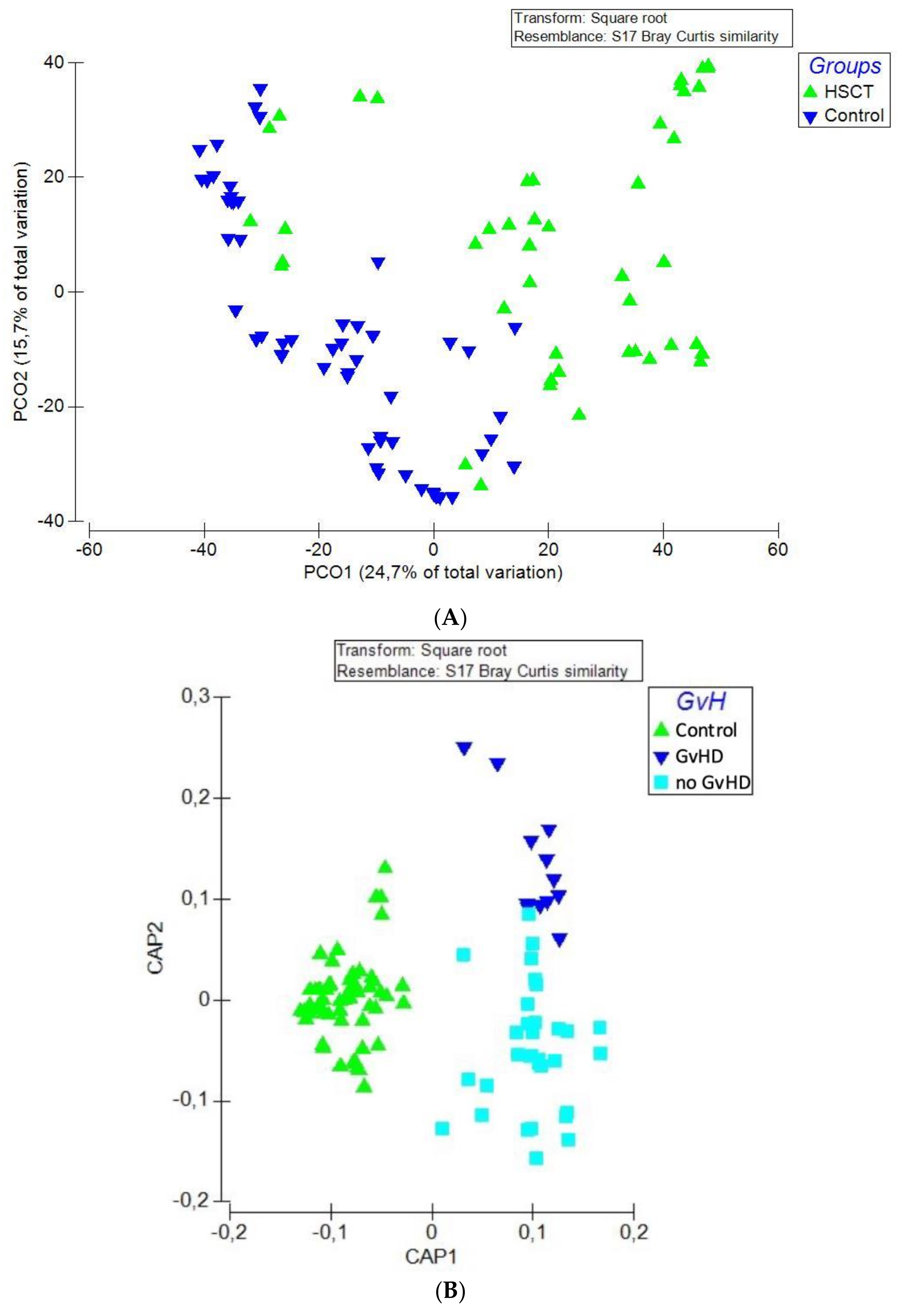
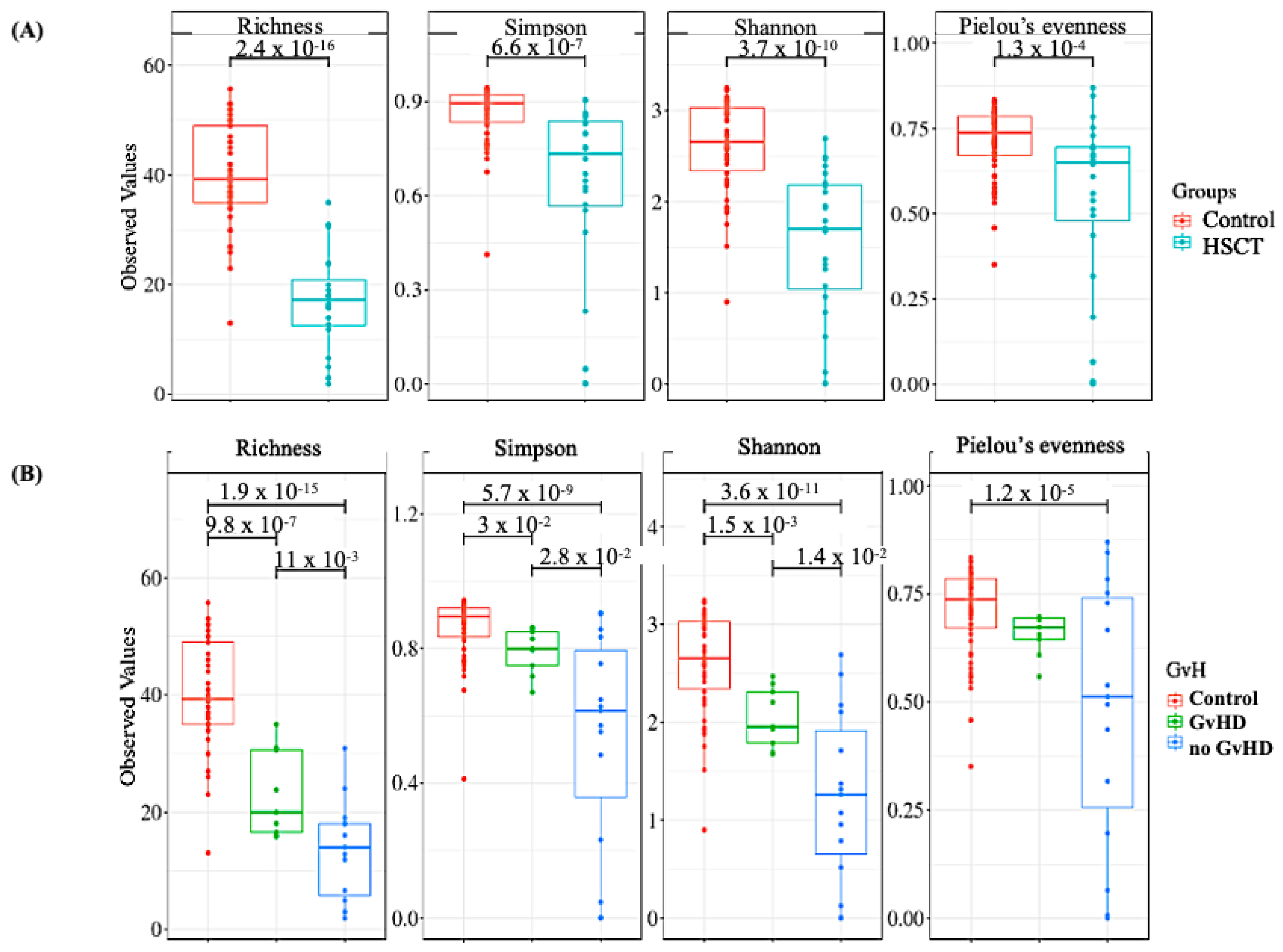
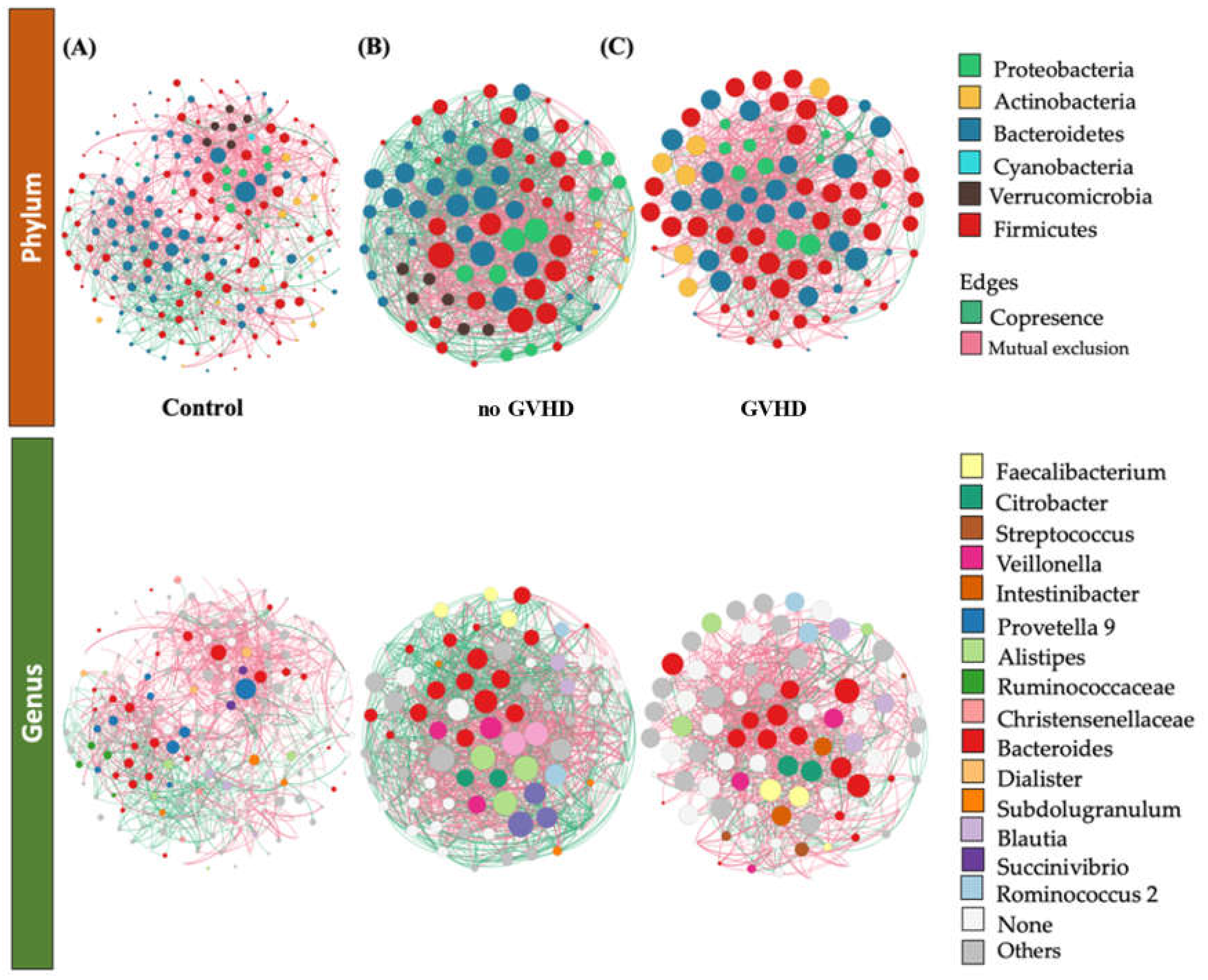
3.2. Structure of the Gut Bacterial Community of the Hematopoietic Stem Cell Transplant Recipients
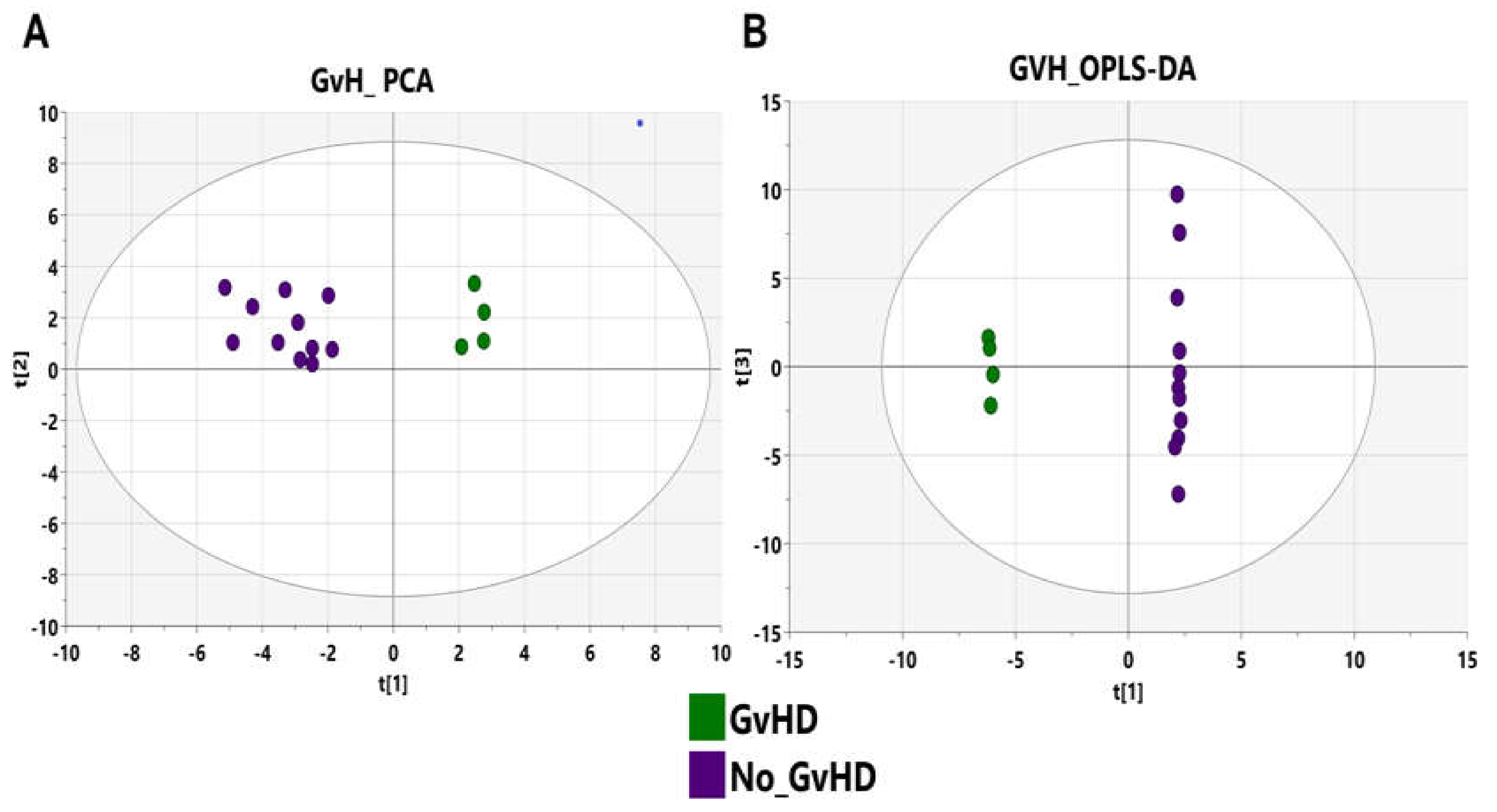
3.3. Bacteriome of Graft-Versus-Host Disease GvHD
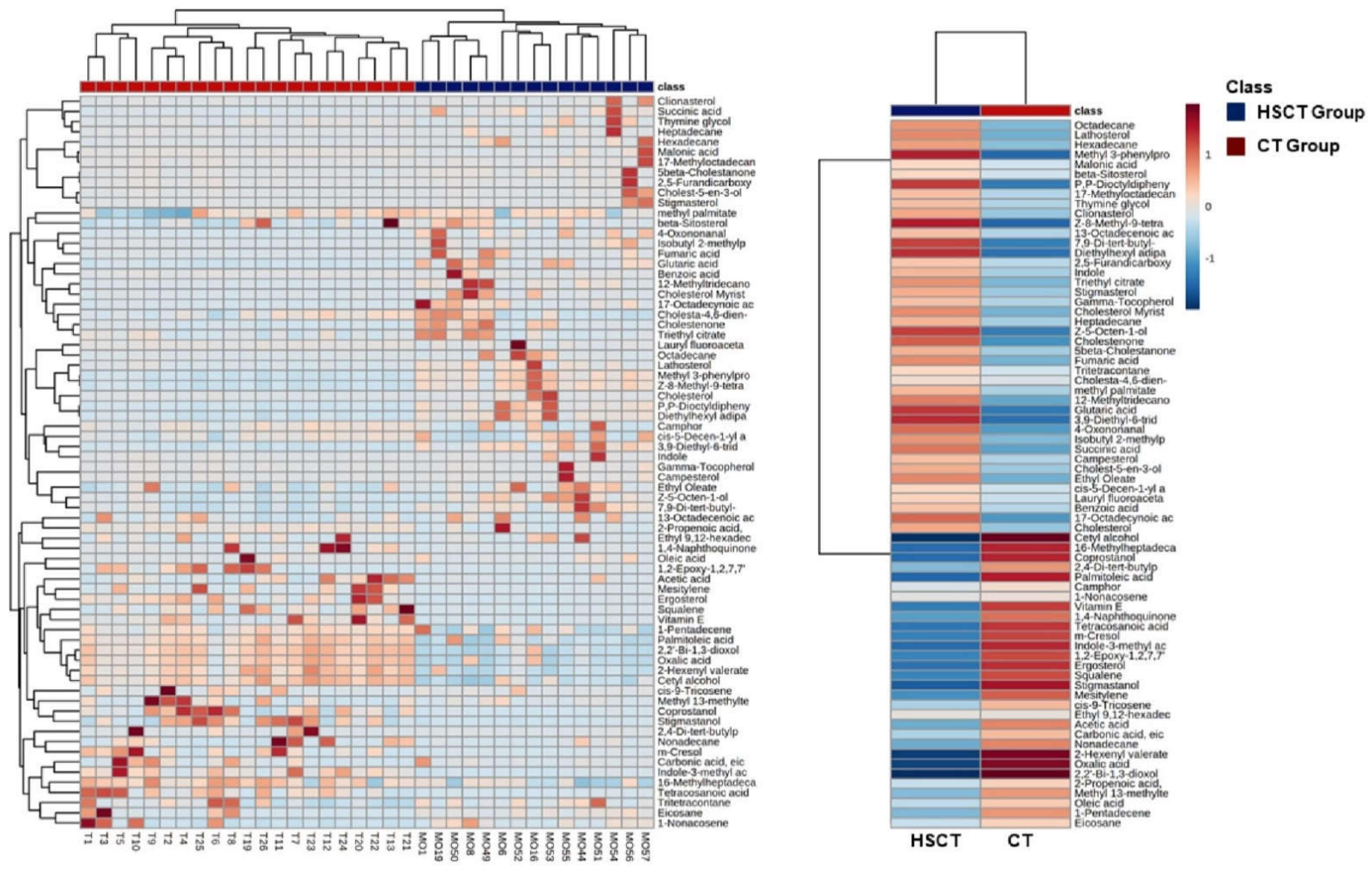
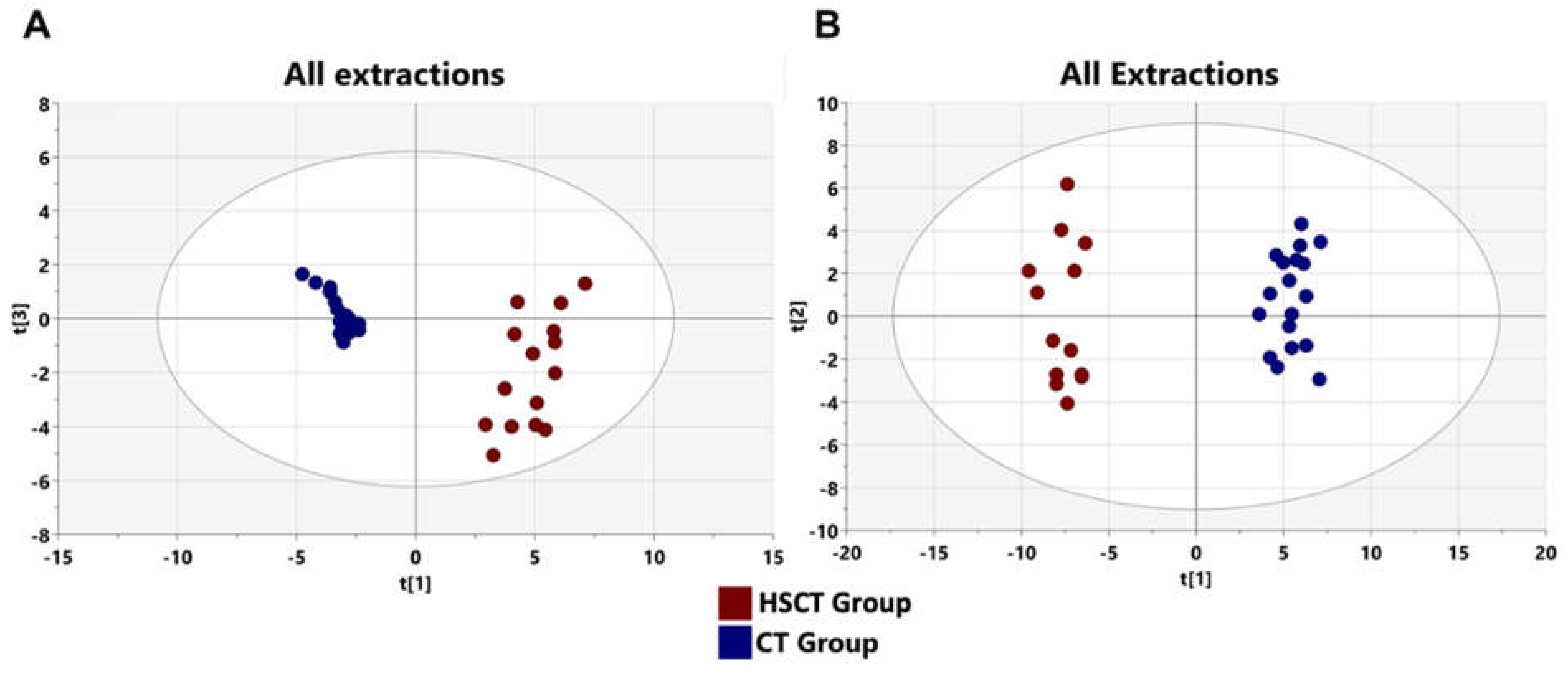
3.4. Metabolomics Profiling of Patients after allo-HSCT
3.5. Alteration of Patients’ Metabolome Compared to Healthy Subjects
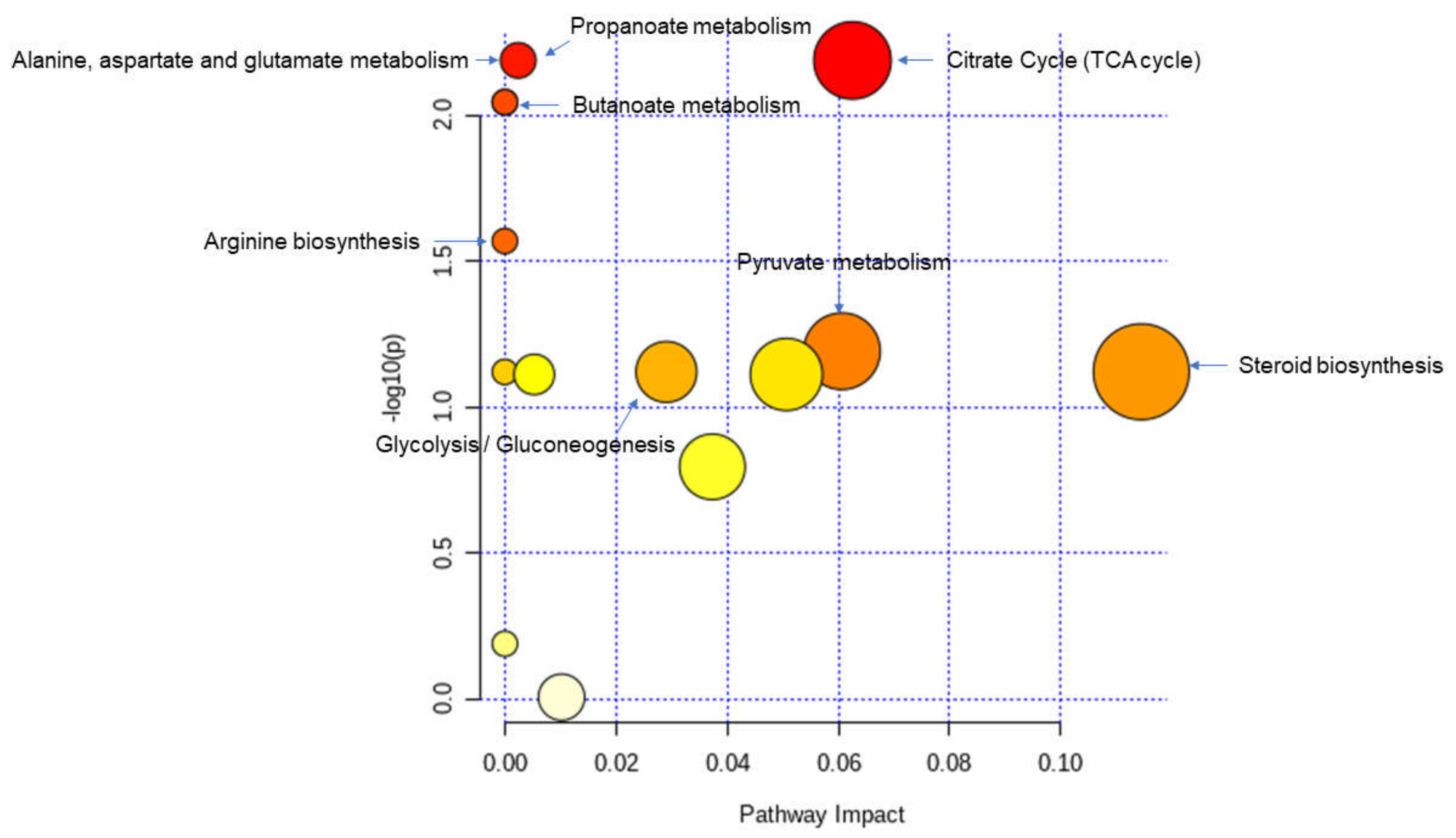
3.6. Identification of Potential Biomarkers of Allogenic Hematopoietic Stem Cell Transplant Status and Biological Explanation
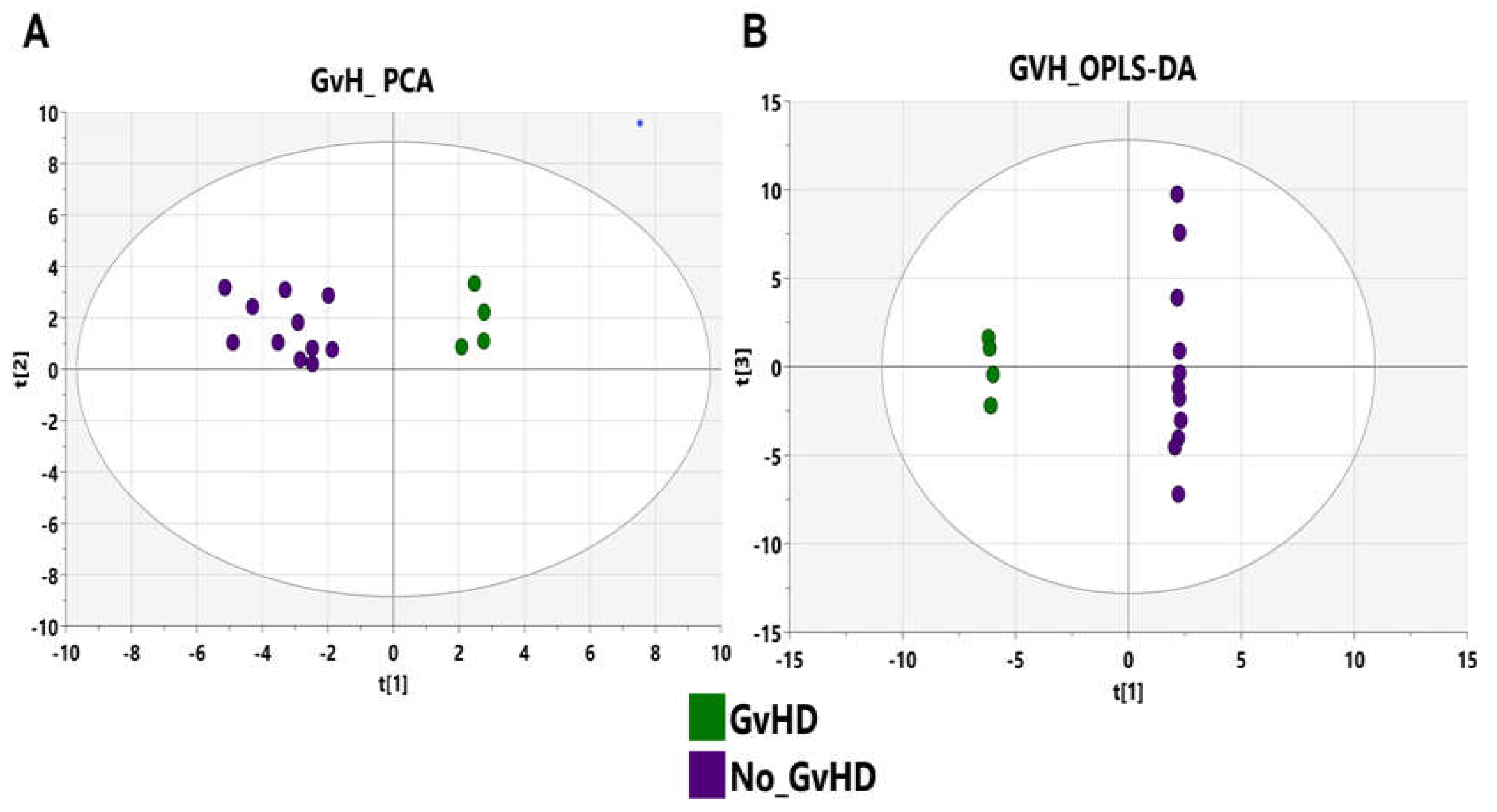
3.7. GvHD Onset Is Characterized by Specific Metabolomic Changes
4. Discussion
Metabolomics Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Khaddour, K.; Hana, C.K.; Mewawalla, P. Hematopoietic stem cell transplantation. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. Available online: http://www.ncbi.nlm.nih.gov/books/NBK536951/ (accessed on 20 May 2021).
- Zama, D.; Bossù, G.; Leardini, D.; Muratore, E.; Biagi, E.; Prete, A.; Pession, A.; Masetti, R. Insights into the role of intestinal microbiota in hematopoietic stem-cell transplantation. Ther. Adv. Hematol. 2020, 11, 2040620719896961. [Google Scholar] [CrossRef] [Green Version]
- Pichereau, C.; Lengliné, E.; Valade, S.; Michonneau, D.; Ghrenassia, E.; Lemiale, V.; Socié, G.; Azoulay, E. Trajectories of acute graft-versus-host disease and mortality in critically ill allogeneic-hematopoietic stem cell recipients: The Allo-GRRR-OH score. Bone Marrow Transpl. 2020, 55, 1966–1974. [Google Scholar] [CrossRef]
- Peled, J.U.; Gomes, A.L.C.; Devlin, S.M.; Littmann, E.R.; Taur, Y.; Sung, A.D.; Weber, D.; Hashimoto, D.; Slingerland, A.E.; Maloy, M.; et al. Microbiota as predictor of mortality in allogeneic hematopoietic-cell transplantation. N. Engl. J. Med. 2020, 382, 822–834. [Google Scholar] [CrossRef]
- Hong, T.; Wang, R.; Wang, X.; Yang, S.; Wang, W.; Gao, Q.; Zhang, X. Interplay between the intestinal microbiota and acute graft-versus-host disease: Experimental evidence and clinical significance. Front. Immunol. 2021, 12, 644982. [Google Scholar] [CrossRef]
- Fiorenza, S.; Turtle, C.J. Associations between the gut microbiota, immune reconstitution, and outcomes of allogeneic hematopoietic stem cell transplantation. Immunometabolism 2021, 3, e210004. [Google Scholar] [CrossRef]
- Noor, F.; Kaysen, A.; Wilmes, P.; Schneider, J.G. The gut microbiota and hematopoietic stem cell transplantation: Challenges and potentials. J. Innate Immun. 2019, 11, 405–415. [Google Scholar] [CrossRef]
- Zama, D.; Biagi, E.; Masetti, R.; Gasperini, P.; Prete, A.; Candela, M.; Brigidi, P.; Pession, A. Gut microbiota and hematopoietic stem cell transplantation: Where do we stand? Bone Marrow Transplant. 2017, 52, 7–14. [Google Scholar] [CrossRef] [Green Version]
- Malard, F.; Gasc, C.; Plantamura, E.; Doré, J. High gastrointestinal microbial diversity and clinical outcome in graft-versus-host disease patients. Bone Marrow Transpl. 2018, 53, 1493–1497. [Google Scholar] [CrossRef]
- Henig, I.; Yehudai-Ofir, D.; Zuckerman, T. The clinical role of the gut microbiome and fecal microbiota transplantation in allogeneic stem cell transplantation. Haematologica 2020, 106, 933. [Google Scholar] [CrossRef]
- Biagi, E.; Zama, D.; Rampelli, S.; Turroni, S.; Brigidi, P.; Consolandi, C.; Severgnini, M.; Picotti, E.; Gasperini, P.; Merli, P.; et al. Early gut microbiota signature of aGvHD in children given allogeneic hematopoietic cell transplantation for hematological disorders. BMC Med. Genom. 2019, 12, 1–11. [Google Scholar] [CrossRef]
- Devaux, C.A.; Million, M.; Raoult, D. The Butyrogenic and Lactic Bacteria of the Gut Microbiota Determine the Outcome of Allogenic Hematopoietic Cell Transplant. Front. Microbiol. 2020, 11, 1642. [Google Scholar] [CrossRef] [PubMed]
- Jenq, R.R.; Taur, Y.; Devlin, S.M.; Ponce, D.M.; Goldberg, J.D.; Ahr, K.F.; Littmann, E.R.; Ling, L.; Gobourne, A.C.; Miller, L.C.; et al. Intestinal Blautia is associated with reduced death from graft-versus-host disease. Biol. Blood Marrow Transplant. 2015, 21, 1373–1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michonneau, D.; Latis, E.; Curis, E.; Dubouchet, L.; Ramamoorthy, S.; Ingram, B.; de Latour, R.P.; Robin, M.; de Fontbrune, F.S.; Chevret, S.; et al. Metabolomics analysis of human acute graft-versus-host disease reveals changes in host and microbiota-derived metabolites. Nat. Commun. 2019, 10, 5695. [Google Scholar] [CrossRef] [Green Version]
- Romick-Rosendale, L.E.; Haslam, D.B.; Lane, A.; Denson, L.; Lake, K.; Wilkey, A.; Watanabe, M.; Bauer, S.; Litts, B.; Luebbering, N.; et al. Antibiotic Exposure and Reduced Short Chain Fatty Acid Production after Hematopoietic Stem Cell Transplant. Biol Blood Marrow Transplant. 2018, 24, 2418–2424. [Google Scholar] [CrossRef] [Green Version]
- Koper, J.E.; Loonen, L.M.; Wells, J.M.; Troise, A.D.; Capuano, E.; Fogliano, V. Polyphenols and tryptophan metabolites activate the aryl hydrocarbon receptor in an in vitro model of colonic fermentation. Mol. Nutr. Food Res. 2019, 63, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. SEARCH_16S: A new algorithm for identifying 16S ribosomal RNA genes in contigs and chromosomes. bioRxiv 2017, 124131. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Edgar, R.C. UNOISE2: Improved error-correction for Illumina 16S and ITS amplicon sequencing. bioRxiv 2016, 081257. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. SINTAX: A simple non-Bayesian taxonomy classifier for 16S and ITS sequences. bioRxiv 2016, 074161. [Google Scholar] [CrossRef] [Green Version]
- Ijaz, D.U.Z. Umerijaz/Microbiomeseq. Published: 14 August 2020 [Online]. 2020. Available online: https://github.com/umerijaz/microbiomeSeq (accessed on 14 August 2020).
- Nordstokke, D.; Stelnicki, A.M. Pairwise comparisons. In Encyclopedia of Quality of Life and Well-Being Research; Michalos, A.C., Ed.; Springer: Dordrecht, The Netherlands, 2014; pp. 4575–4576. [Google Scholar] [CrossRef]
- Hollander, M.; Wolfe, D.A.; Chicken, E. Nonparametric Statistical Methods, 1st ed.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2015. [Google Scholar] [CrossRef]
- Anderson, M.J. Permutational multivariate analysis of variance (PERMANOVA). Wiley Statsref: Stat. Ref. Online 2014, 1–15. Available online: https://onlinelibrary.wiley.com/doi/abs/10.1002/9781118445112.stat07841. (accessed on 15 November 2017).
- Allen, B.; Kon, M.; Bar-Yam, Y. A new phylogenetic diversity measure generalizing the Shannon index and its application to phyllostomid bats. Am. Nat. 2009, 174, 236–243. [Google Scholar] [CrossRef] [Green Version]
- Marcon, E.; Hérault, B. entropart: An R package to measure and partition diversity. J. Stat. Softw. 2015, 67, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Steele, J.A.; Countway, P.D.; Xia, L.; Vigil, P.D.; Beman, J.M.; Kim, D.Y.; Chow, C.T.; Sachdeva, R.; Jones, A.C.; Schwalbach, M.S.; et al. Marine bacterial, archaeal and protistan association networks reveal ecological linkages. ISME J. 2011, 5, 1414–1425. [Google Scholar] [CrossRef]
- Zhou, J.; Deng, Y.; Luo, F.; He, Z.; Yang, Y. Phylogenetic molecular ecological network of soil microbial communities in response to elevated CO2. mBio 2011, 2, e00122-11. [Google Scholar] [CrossRef] [Green Version]
- Newman, M.E. The structure and function of complex networks. SIAM Rev. 2003, 45, 167–256. [Google Scholar] [CrossRef] [Green Version]
- Egestad, B.; Pettersson, P.; Sjövall, J.; Rafter, J.; Hyvönen, K.; Gustafsson, J.Å. Studies on the chromatographic fractionation of metabolites of benzo [a] pyrene in faeces and urine from germfree and conventional rats. Biomed. Chromatogr. 1987, 2, 120–134. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yin, Y.; Chen, X.; Chen, C.; Xia, Y.; Qi, H.; Baker, P.N.; Zhang, H.; Han, T.-L. Evaluating different extraction solvents for GC-MS based metabolomic analysis of the fecal metabolome of adult and baby giant pandas. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, O. Metabolomics by gas chromatography–mass spectrometry: Combined targeted and untargeted profiling. Curr. Protoc. Mol. Biol. 2016, 114, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Alfaro, A.C.; Nguyen, T.V.; Young, T.; Lulijwa, R. An integrated omics approach to investigate summer mortality of New Zealand Greenshell™ mussels. Metabolomics 2020, 16, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Broadhurst, D.I.; Wilson, M.; Wishart, D.S. Translational biomarker discovery in clinical metabolomics: An introductory tutorial. Metabolomics 2013, 9, 280–299. [Google Scholar] [CrossRef] [Green Version]
- Chong, J.; Wishart, D.S.; Xia, J. Using MetaboAnalyst 4.0 for comprehensive and integrative metabolomics data analysis. Curr. Protoc. Bioinform. 2019, 68, e86. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Wishart, D.S. MetPA: A web-based metabolomics tool for pathway analysis and visualization. Bioinformatics 2010, 26, 2342–2344. [Google Scholar] [CrossRef] [Green Version]
- Galloway-Peña, J.R.; Peterson, C.B.; Malik, F.; Sahasrabhojane, P.V.; Shah, D.P.; Brumlow, C.E.; Carlin, L.G.; Chemaly, R.F.; Im, J.S.; Rondon, G.; et al. Fecal microbiome, metabolites, and stem cell transplant outcomes: A single-center pilot study. In Open Forum Infectious Diseases; Oxford University Press: Oxford, UK, 2019; Volume 6, p. ofz173. [Google Scholar] [CrossRef]
- Taur, Y.; Xavier, J.B.; Lipuma, L.; Ubeda, C.; Goldberg, J.; Gobourne, A.; Lee, Y.J.; Dubin, K.A.; Socci, N.D.; Viale, A.; et al. Intestinal domination and the risk of bacteremia in patients undergoing allogeneic hematopoietic stem cell transplantation. Clin. Infect. Dis. 2012, 55, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Harris, B.; Morjaria, S.M.; Littmann, E.R.; Geyer, A.I.; Stover, D.E.; Barker, J.N.; Giralt, S.A.; Taur, Y.; Pamer, E.G. Gut microbiota predict pulmonary infiltrates after allogeneic hematopoietic cell transplantation. Am. J. Respir. Crit. Care Med. 2016, 194, 450–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ubeda, C.; Bucci, V.; Caballero, S.; Djukovic, A.; Toussaint, N.C.; Equinda, M.; Lipuma, L.; Ling, L.; Gobourne, A.; No, D.; et al. Intestinal microbiota containing Barnesiella species cures vancomycin-resistant Enterococcus faecium colonization. Infect. Immun. 2013, 81, 965–973. [Google Scholar] [CrossRef] [Green Version]
- Tamburini, F.B.; Andermann, T.M.; Tkachenko, E.; Senchyna, F.; Banaei, N.; Bhatt, A.S. Precision identification of diverse bloodstream pathogens in the gut microbiome. Nat. Med. 2018, 24, 1809–1814. [Google Scholar] [CrossRef]
- Poutsiaka, D.D.; Price, L.L.; Ucuzian, A.; Chan, G.W.; Miller, K.B.; Snydman, D.R. Blood stream infection after hematopoietic stem cell transplantation is associated with increased mortality. Bone Marrow Transplant. 2007, 40, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Ghimire, S.; Weber, D.; Mavin, E.; Dickinson, A.M.; Holler, E. Pathophysiology of GvHD and other HSCT-related major complications. Front. Immunol. 2017, 8, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathewson, N.; Reddy, P. The microbiome and graft versus host disease. Curr. Stem Cell Rep. 2015, 1, 39–47. Available online: https://scholar.google.com/scholar_lookup?journal=Current+Stem+Cell+Reports&title=The+Microbiome+and+Graft+Versus+Host+Disease&author=N+Mathewson&author=P+Reddy&volume=1&publication_year=2015&pages=39-47& (accessed on 25 July 2021). [CrossRef] [Green Version]
- Docampo, M.D.; Auletta, J.J.; Jenq, R.R. Emerging influence of the intestinal microbiota during allogeneic hematopoietic cell transplantation: Control the gut and the body will follow. Biol. Blood Marrow Transplant. 2015, 21, 1360–1366. [Google Scholar] [CrossRef] [Green Version]
- Hanash, A.M.; Dudakov, J.A.; Hua, G.; O’Connor, M.H.; Young, L.F.; Singer, N.V.; West, M.L.; Jenq, R.R.; Holland, A.M.; Kappel, L.W.; et al. Interleukin-22 protects intestinal stem cells from immune-mediated tissue damage and regulates sensitivity to graft versus host disease. Immunity 2012, 37, 339–350. [Google Scholar] [CrossRef] [Green Version]
- Hubbard-Lucey, V.M.; Shono, Y.; Maurer, K.; West, M.L.; Singer, N.V.; Ziegler, C.G.K.; Lezcano, C.; Motta, A.C.F.; Schmid, K.; Levi, S.M.; et al. Autophagy gene Atg16L1 prevents lethal T cell alloreactivity mediated by dendritic cells. Immunity 2014, 41, 579–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, D.; Oefner, P.J.; Hiergeist, A.; Koestler, J.; Gessner, A.; Weber, M.; Hahn, J.; Wolff, D.; Stämmler, F.; Spang, R.; et al. Low urinary indoxyl sulfate levels early after transplantation reflect a disrupted microbiome and are associated with poor outcome. Blood J. Am. Soc. Hematol. 2015, 126, 1723–1728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heimesaat, M.M.; Nogai, A.; Bereswill, S.; Plickert, R.; Fischer, A.; Loddenkemper, C.; Steinhoff, U.; Tchaptchet, S.; Thiel, E.; Freudenberg, M.A.; et al. MyD88/TLR9 mediated immunopathology and gut microbiota dynamics in a novel murine model of intestinal graft-versus-host disease. Gut 2010, 59, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Jenq, R.R.; Ubeda, C.; Taur, Y.; Menezes, C.C.; Khanin, R.; Dudakov, J.A.; Liu, C.; West, M.L.; Singer, N.V.; Equinda, M.J.; et al. Regulation of intestinal inflammation by microbiota following allogeneic bone marrow transplantation. J. Exp. Med. 2012, 209, 903–911. [Google Scholar] [CrossRef]
- Kaakoush, N.O.; Day, A.S.; Huinao, K.D.; Leach, S.T.; Lemberg, D.A.; Dowd, S.E.; Mitchell, H.M. Microbial dysbiosis in pediatric patients with Crohn’s disease. J. Clin. Microbiol. 2012, 50, 3258–3266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biagi, E.; Zama, D.; Nastasi, C.; Consolandi, C.; Fiori, J.; Rampelli, S.; Turroni, S.; Centanni, M.; Severgnini, M.; Peano, C.; et al. Gut microbiota trajectory in pediatric patients undergoing hematopoietic SCT. Bone Marrow Transplant. 2015, 50, 992–998. [Google Scholar] [CrossRef] [Green Version]
- Kaysen, A.; Heintz-Buschart, A.; Muller, E.E.L.; Narayanasamy, S.; Wampach, L.; Laczny, C.C.; Graf, N.; Simon, A.; Franke, K.; Bittenbring, J.; et al. Integrated meta-omic analyses of the gastrointestinal tract microbiome in patients undergoing allogeneic hematopoietic stem cell transplantation. Transl. Res. 2017, 186, 79–94. [Google Scholar] [CrossRef] [Green Version]
- Vital, M.; Howe, A.C.; Tiedje, J.M. Revealing the bacterial butyrate synthesis pathways by analyzing (meta) genomic data. mBio 2014, 5, e00889-14. [Google Scholar] [CrossRef] [Green Version]
- Venegas, D.P.; De la Fuente, M.K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short chain fatty acids (SCFAs)-mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef] [Green Version]
- Han, L.; Jin, H.; Zhou, L.; Zhang, X.; Fan, Z.; Dai, M.; Lin, Q.; Huang, F.; Xuan, L.; Zhang, H.; et al. Intestinal microbiota at engraftment influence acute graft-versus-host disease via the Treg/Th17 balance in allo-HSCT recipients. Front. Immunol. 2018, 9, 669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shono, Y.; Docampo, M.D.; Peled, J.U.; Perobelli, S.U.; Velardi, E.; Tsai, J.J.; Slingerland, A.E.; Smith, O.M.; Young, L.F.; Gupta, J.; et al. Increased GVHD-related mortality with broad-spectrum antibiotic use after allogeneic hematopoietic stem cell transplantation in human patients and mice. Sci. Transl. Med. 2016, 8, 339ra71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taur, Y.; Jenq, R.R.; Perales, M.-A.; Littmann, E.R.; Morjaria, S.; Ling, L.; No, D.; Gobourne, A.; Viale, A.; Dahi, P.B.; et al. The effects of intestinal tract bacterial diversity on mortality following allogeneic hematopoietic stem cell transplantation. Blood J. Am. Soc. Hematol. 2014, 124, 1174–1182. [Google Scholar] [CrossRef] [PubMed]
- Ilett, E.E.; Jørgensen, M.; Noguera-Julian, M.; Nørgaard, J.C.; Daugaard, G.; Helleberg, M.; Paredes, R.; Murray, D.D.; Lundgren, J.; MacPherson, C.; et al. Associations of the gut microbiome and clinical factors with acute GVHD in allogeneic HSCT recipients. Blood Adv. 2020, 4, 5797–5809. [Google Scholar] [CrossRef]
- Golob, J.L.; Pergam, S.A.; Srinivasan, S.; Fiedler, T.L.; Liu, C.; Garcia, K.; Mielcarek, M.; Ko, D.; Aker, S.; Marquis, S.; et al. Stool microbiota at neutrophil recovery is predictive for severe acute graft vs host disease after hematopoietic cell transplantation. Clin. Infect. Dis. 2017, 65, 1984–1991. [Google Scholar] [CrossRef]
- Louis, P.; Scott, K.P.; Duncan, S.H.; Flint, H.J. Understanding the effects of diet on bacterial metabolism in the large intestine. J. Appl. Microbiol. 2007, 102, 1197–1208. [Google Scholar] [CrossRef]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Tekahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef]
- Corrêa-Oliveira, R.; Fachi, J.L.; Vieira, A.; Sato, F.T.; Vinolo, M.A.R. Regulation of immune cell function by short-chain fatty acids. Clin. Transl. Immunol. 2016, 5, e73. [Google Scholar] [CrossRef]
- Donohoe, D.R.; Garge, N.; Zhang, X.; Sun, W.; O’Connell, T.M.; Bunger, M.K.; Bultman, S.J. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. 2011, 13, 517–526. [Google Scholar] [CrossRef] [Green Version]
- Fuhler, G.M.; Eppinga, H.; Peppelenbosch, M.P. Fumarates and cancer. Trends Mol. Med. 2015, 23, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Sciacovelli, M.; Frezza, C. Oncometabolites: Unconventional triggers of oncogenic signalling cascades. Free Radic. Biol. Med. 2016, 100, 175–181. [Google Scholar] [CrossRef] [Green Version]
- Engel, C.A.R.; Straathof, A.J.; Zijlmans, T.W.; van Gulik, W.M.; van der Wielen, L.A. Fumaric acid production by fermentation. Appl. Microbiol. Biotechnol. 2008, 78, 379–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aspuria, P.-J.P.; Lunt, S.Y.; Väremo, L.; Vergnes, L.; Gozo, M.; Beach, J.A.; Salumbides, B.; Reue, K.; Wiedemeyer, W.R.; Nielsen, J.; et al. Succinate dehydrogenase inhibition leads to epithelial-mesenchymal transition and reprogrammed carbon metabolism. Cancer Metab. 2014, 2, 1–15. [Google Scholar] [CrossRef]
- Mills, E.L.; Kelly, B.; O’Neill, L.A. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017, 18, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Bis, G.; Szlasa, W.; Sondaj, K.; Zendran, I.; Mielcarek-Siedziuk, M.; Barg, E. Lipid Complications after Hematopoietic Stem Cell Transplantation (HSCT) in Pediatric Patients. Nutrients 2020, 12, 2500. [Google Scholar] [CrossRef] [PubMed]
- Premstaller, M.; Perren, M.; Koçack, K.; Arranto, C.; Favre, G.; Lohri, A.; Gerull, S.; Passweg, J.R.; Halter, J.P.; Leuppi-Taegtmeyer, A.B.; et al. Dyslipidemia and lipid-lowering treatment in a hematopoietic stem cell transplant cohort: 25 years of follow-up data. J. Clin. Lipidol. 2018, 12, 464–480. [Google Scholar] [CrossRef] [PubMed]
- Marini, B.L.; Choi, S.W.; Byersdorfer, C.A.; Cronin, S.; Frame, D.G. Treatment of dyslipidemia in allogeneic hematopoietic stem cell transplant patients. Biol. Blood Marrow Transplant. 2015, 21, 809–820. [Google Scholar] [CrossRef] [Green Version]
- Kominsky, D.J.; Campbell, E.L.; Colgan, S.P. Metabolic shifts in immunity and inflammation. J. Immunol. 2010, 184, 4062–4068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calder, P.C.; Grimble, R.F. Polyunsaturated fatty acids, inflammation and immunity. Eur. J. Clin. Nutr. 2002, 56, S14–S19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezende, B.M.; Athayde, R.M.; Gonçalves, W.A.; Resende, C.B.; Bernardes, P.T.d.T.; Perez, D.A.; Esper, L.; Reis, A.C.; Rachid, M.A.; e Castor, M.G.M.; et al. Inhibition of 5-lipoxygenase alleviates graft-versus-host disease. J. Exp. Med. 2017, 214, 3399–3415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Q. Natural forms of vitamin E: Metabolism, antioxidant, and anti-inflammatory activities and their role in disease prevention and therapy. Free Radic. Biol. Med. 2014, 72, 76–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clemens, M.R.; Ladner, C.; Ehninger, G.; Einsele, H.; Renn, W.; Bühler, E.; Waller, H.D.; Gey, K.F. Plasma vitamin E and β-carotene concentrations during radiochemotherapy preceding bone marrow Transplantation. Am. J. Clin. Nutr. 1990, 51, 216–219. [Google Scholar] [CrossRef]
- Choi, Y.; Lee, S.; Kim, S.; Lee, J.; Ha, J.; Oh, H.; Lee, Y.; Kim, Y.; Yoon, Y. Vitamin E (α-tocopherol) consumption influences gut microbiota composition. Int. J. Food Sci. Nutr. 2020, 71, 221–225. [Google Scholar] [CrossRef]
- Buck, M.D.; O’sullivan, D.; Pearce, E.L. T cell metabolism drives immunity. J. Exp. Med. 2015, 212, 1345–1360. [Google Scholar] [CrossRef] [Green Version]
- Cameron, A.M.; Lawless, S.J.; Pearce, E.J. Metabolism and acetylation in innate immune cell function and fate. In Seminars in Immunology; Academic Press: Cambridge, MA, USA, 2016; Volume 28, pp. 408–416. [Google Scholar] [CrossRef]
- Filosa, S.; Fico, A.; Paglialunga, F.; Balestrieri, M.; Crooke, A.; Verde, P.; Abrescia, P.; Bautista, J.M.; Martini, G. Failure to increase glucose consumption through the pentose-phosphate pathway results in the death of glucose-6-phosphate dehydrogenase gene-deleted mouse embryonic stem cells subjected to oxidative stress. Biochem. J. 2003, 370, 935–943. [Google Scholar] [CrossRef]
- Nguyen, H.D.; Chatterjee, S.; Haarberg, K.M.K.; Wu, Y.; Bastian, D.; Heinrichs, J.; Fu, J.; Daenthanasanmak, A.; Schutt, S.; Shrestha, S.; et al. Metabolic reprogramming of alloantigen-activated T cells after hematopoietic cell transplantation. J. Clin. Investig. 2016, 126, 1337–1352. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.D.; Kuril, S.; Bastian, D.; Yu, X.Z. T-cell metabolism in hematopoietic cell transplantation. Front. Immunol. 2018, 9, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellor, A.L.; Munn, D.H. IDO expression by dendritic cells: Tolerance and tryptophan catabolism. Nat. Rev. Immunol. 2004, 4, 762–774. [Google Scholar] [CrossRef]
- Nguyen, N.T.; Nakahama, T.; Le, D.H.; Van Son, L.; Chu, H.H.; Kishimoto, T. Aryl hydrocarbon receptor and kynurenine: Recent advances in autoimmune disease research. Front. Immunol. 2014, 5, 551. [Google Scholar] [CrossRef] [Green Version]
- Jasperson, L.K.; Bucher, C.; Panoskaltsis-Mortari, A.; Mellor, A.L.; Munn, D.H.; Blazar, B.R. Inducing the tryptophan catabolic pathway, indoleamine 2, 3-dioxygenase (IDO), for suppression of graft-versus-host disease (GVHD) lethality. Blood J. Am. Soc. Hematol. 2009, 114, 5062–5070. [Google Scholar] [CrossRef]
- Landfried, K.; Zhu, W.; Waldhier, M.C.; Schulz, U.; Ammer, J.; Holler, B.; Wolff, D.; Edinger, M.; Peter, K.; Kreutz, M.; et al. Tryptophan catabolism is associated with acute GVHD after human allogeneic stem cell transplantation and indicates activation of indoleamine 2, 3-dioxygenase. Blood J. Am. Soc. Hematol. 2011, 118, 6971–6974. [Google Scholar] [CrossRef] [Green Version]
- Ratajczak, P.; Jannin, A.; de Larour, R.P.; Koch, L.; Roche, B.; Munn, D.; Blazar, B.R.; Socié, G. IDO in human gut graft-versus-host disease. Biol. Blood Marrow Transplant. 2012, 18, 150–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jasperson, L.K.; Bucher, C.; Panoskaltsis-Mortari, A.; Taylor, P.A.; Mellor, A.L.; Munn, D.H.; Blazar, B.R. Indoleamine 2, 3-dioxygenase is a critical regulator of acute graft-versus-host disease lethality. Blood J. Am. Soc. Hematol. 2008, 111, 3257–3265. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Vázquez, C.; Quintana, F.J. Regulation of the immune response by the aryl hydrocarbon receptor. Immunity 2018, 48, 19–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ríos-Covián, D.; Ruas-Madiedo, P.; Margolles, A.; Gueimonde, M.; De Los Reyes-gavilán, C.G.; Salazar, N. Intestinal short chain fatty acids and their link with diet and human health. Front. Microbiol. 2016, 7, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, M.; Wrzosek, L.; Miquel, S.; Noordine, M.L.; Bouet, S.; Chevalier-Curt, M.J.; Robert, V.; Phillippe, C.; Bridonneau, C.; Cherbuy, C.; et al. 264 Bacteroides Thetaiotaomicron and Faecalibacterium prausnitzii Shape the Mucus Production and Mucin O-Glycosylation in Colon Epithelium. Gastroenterology 2013, 5, S-59. [Google Scholar] [CrossRef]
- Staels, B.; Fonseca, V.A. Bile acids and metabolic regulation: Mechanisms and clinical responses to bile acid sequestration. Diabetes Care 2009, 32, S237–S245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, S.-Y.; Ouyang, X.; Chen, Y.; Soroka, C.J.; Wang, J.; Mennone, A.; Wang, Y.; Mehal, W.Z.; Jain, D.; Boyer, J.L. Bile acids initiate cholestatic liver injury by triggering a hepatocyte-specific inflammatory response. JCI Insight 2017, 2, e90780. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Cai, S.Y.; Boyer, J.L. Mechanisms of bile acid mediated inflammation in the liver. Mol. Asp. Med. 2017, 56, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Xie, S.; Chi, Z.; Zhang, J.; Liu, Y.; Zhang, L.; Zheng, M.; Zhang, X.; Xia, D.; Ke, Y.; et al. Bile acids control inflammation and metabolic disorder through inhibition of NLRP3 inflammasome. Immunity 2016, 45, 802–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kouidhi, S.; Souai, N.; Zidi, O.; Mosbah, A.; Lakhal, A.; Ben Othmane, T.; Belloumi, D.; Ben Ayed, F.; Asimakis, E.; Stathopoulou, P.; et al. High Throughput Analysis Reveals Changes in Gut Microbiota and Specific Fecal Metabolomic Signature in Hematopoietic Stem Cell Transplant Patients. Microorganisms 2021, 9, 1845. https://doi.org/10.3390/microorganisms9091845
Kouidhi S, Souai N, Zidi O, Mosbah A, Lakhal A, Ben Othmane T, Belloumi D, Ben Ayed F, Asimakis E, Stathopoulou P, et al. High Throughput Analysis Reveals Changes in Gut Microbiota and Specific Fecal Metabolomic Signature in Hematopoietic Stem Cell Transplant Patients. Microorganisms. 2021; 9(9):1845. https://doi.org/10.3390/microorganisms9091845
Chicago/Turabian StyleKouidhi, Soumaya, Nessrine Souai, Oumaima Zidi, Amor Mosbah, Amel Lakhal, Tarek Ben Othmane, Dorra Belloumi, Farhat Ben Ayed, Elias Asimakis, Panagiota Stathopoulou, and et al. 2021. "High Throughput Analysis Reveals Changes in Gut Microbiota and Specific Fecal Metabolomic Signature in Hematopoietic Stem Cell Transplant Patients" Microorganisms 9, no. 9: 1845. https://doi.org/10.3390/microorganisms9091845
APA StyleKouidhi, S., Souai, N., Zidi, O., Mosbah, A., Lakhal, A., Ben Othmane, T., Belloumi, D., Ben Ayed, F., Asimakis, E., Stathopoulou, P., Cherif, A., & Tsiamis, G. (2021). High Throughput Analysis Reveals Changes in Gut Microbiota and Specific Fecal Metabolomic Signature in Hematopoietic Stem Cell Transplant Patients. Microorganisms, 9(9), 1845. https://doi.org/10.3390/microorganisms9091845