
Descriptive Study of Gut Microbiota in Infected and Colonized Subjects by Clostridiodes difficile

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design and Subject Inclusion
2.2. Metataxonomic Determination of the Composition of the Gut Microbiota
2.3. Bioinformatic Analysis of the Amplicon Sequences of the Bacterial 16S rDNA Gene and Taxonomic Assignment
2.4. Expression of Results
3. Results
3.1. Alpha Diversity in Study Groups
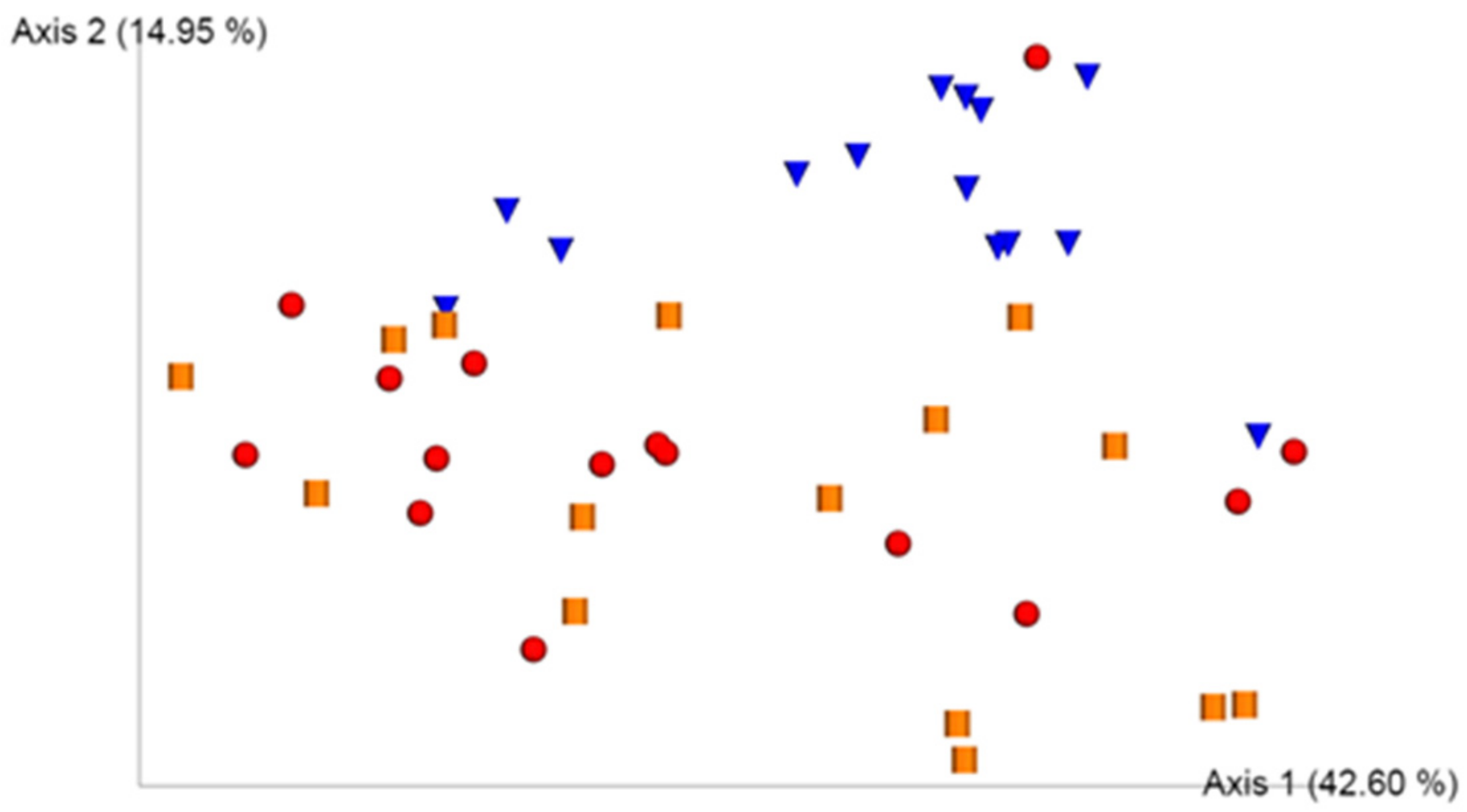
3.2. UniFrac Model
3.3. Composition Analyses of Gut Microbiota
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lawson, P.A.; Citron, D.M.; Tyrrell, K.L.; Finegold, S.M. Reclassification of Clostridium difficile as Clostridioides difficile (Hall and O’Toole 1935) Prévot 1938. Anaerobe 2016, 40, 95–99. [Google Scholar] [CrossRef]
- McDonald, L.C.; Owings, M.; Jernigan, D.B. Clostridium difficile Infection in Patients Discharged from US Short-stay Hospitals, 1996–2003. Emerg. Infect. Dis. 2006, 12, 409–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loo, V.G.; Bourgault, A.-M.; Poirier, L.; Lamothe, F.; Michaud, S.; Turgeon, N.; Toye, B.; Beaudoin, A.; Frost, E.; Gilca, R.; et al. Host and Pathogen Factors for Clostridium difficile Infection and Colonization. N. Engl. J. Med. 2011, 365, 1693–1703. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekaran, R.; Lacy, D.B. The role of toxins in Clostridium difficile infection. FEMS Microbiol. Rev. 2017, 41, 723–750. [Google Scholar] [CrossRef] [Green Version]
- Crobach, M.; Planche, T.; Eckert, C.; Barbut, F.; Terveer, E.; Dekkers, O.; Wilcox, M.; Kuijper, E. European Society of Clinical Microbiology and Infectious Diseases: Update of the diagnostic guidance document for Clostridium difficile infection. Clin. Microbiol. Infect. 2016, 22, S63–S81. [Google Scholar] [CrossRef] [Green Version]
- Crobach, M.J.T.; Vernon, J.J.; Loo, V.G.; Kong, L.Y.; Péchiné, S.; Wilcox, M.H.; Kuijper, E.J. Understanding Clostridium difficile Colonization. Clin. Microbiol. Rev. 2018, 31, e00021-17. [Google Scholar] [CrossRef] [Green Version]
- Pépin, J.; Valiquette, L.; Alary, M.-E.; Villemure, P.; Pelletier, A.; Forget, K.; Pépin, K.; Chouinard, D. Clostridium difficile-associated diarrhea in a region of Quebec from 1991 to 2003: A changing pattern of disease severity. Can. Med. Assoc. J. 2004, 171, 466–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Britton, R.A.; Young, V.B. Interaction between the intestinal microbiota and host in Clostridium difficile colonization resistance. Trends Microbiol. 2012, 20, 313–319. [Google Scholar] [CrossRef] [Green Version]
- Weingarden, A.R.; Chen, C.; Bobr, A.; Yao, D.; Lu, Y.; Nelson, V.M.; Sadowsky, M.; Khoruts, A. Microbiota transplantation restores normal fecal bile acid composition in recurrent Clostridium difficile infection. Am. J. Physiol. Liver Physiol. 2014, 306, G310–G319. [Google Scholar] [CrossRef] [Green Version]
- Thanissery, R.; Winston, J.A.; Theriot, C.M. Inhibition of spore germination, growth, and toxin activity of clinically relevant C. difficile strains by gut microbiota derived secondary bile acids. Anaerobe 2017, 45, 86–100. [Google Scholar] [CrossRef]
- Solbach, P.; Chhatwal, P.; Woltemate, S.; Tacconelli, E.; Buhl, M.; Gerhard, M.; Thoeringer, C.K.; Vehreschild, M.J.G.T.; Jazmati, N.; Rupp, J.; et al. BaiCD gene cluster abundance is negatively correlated with Clostridium difficile infection. PLoS ONE 2018, 13, e0196977. [Google Scholar] [CrossRef] [Green Version]
- Buffie, C.G.; Bucci, V.; Stein, R.R.; McKenney, P.T.; Ling, L.; Gobourne, A.; No, D.; Liu, H.; Kinnebrew, M.; Viale, A.; et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nat. Cell. Biol. 2015, 517, 205–208. [Google Scholar] [CrossRef] [Green Version]
- Antharam, V.C.; Li, E.C.; Ishmael, A.; Sharma, A.; Mai, V.; Rand, K.H.; Wang, G.P. Intestinal Dysbiosis and Depletion of Butyrogenic Bacteria in Clostridium difficile Infection and Nosocomial Diarrhea. J. Clin. Microbiol. 2013, 51, 2884–2892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milani, C.; Ticinesi, A.; Gerritsen, J.; Nouvenne, A.; Lugli, G.A.; Mancabelli, L.; Turroni, F.; Duranti, S.; Mangifesta, M.; Viappiani, A.; et al. Gut microbiota composition and Clostridium difficile infection in hospitalized elderly individuals: A metagenomic study. Sci. Rep. 2016, 6, 25945. [Google Scholar] [CrossRef] [PubMed]
- McDonald, L.C.; Gerding, D.N.; Johnson, S.; Bakken, J.S.; Carroll, K.C.; Coffin, S.E.; Dubberke, E.R.; Garey, K.W.; Gould, C.V.; Kelly, C.; et al. Clinical Practice Guidelines for Clostridium difficile Infection in Adults and Children: 2017 Update by the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA). Clin. Infect. Dis. 2018, 66, e1–e48. [Google Scholar] [CrossRef]
- Furuya-Kanamori, L.; Marquess, J.; Yakob, L.; Riley, T.V.; Paterson, D.L.; Foster, N.F.; Huber, C.A.; Clements, A.C.A. Asymptomatic Clostridium difficile colonization: Epidemiology and clinical implications. BMC. Infect. Dis. 2015, 15, 516. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2012, 41, e1. [Google Scholar] [CrossRef]
- Chang, J.Y.; Antonopoulos, D.A.; Kalra, A.; Tonelli, A.; Khalife, W.T.; Schmidt, T.; Young, V. Decreased Diversity of the Fecal Microbiome in Recurrent Clostridium difficile–Associated Diarrhea. J. Infect Dis. 2008, 197, 435–438. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Dong, D.; Jiang, C.; Li, Z.; Wang, X.; Peng, Y. Insight into alteration of gut microbiota in Clostridium difficile infection and asymptomatic C. difficile colonization. Anaerobe 2015, 34, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Khanna, S.; Pardi, D.S. Clinical implications of antibiotic impact on gastrointestinal microbiota and Clostridium difficile infection. Exper.t Rev. Gastroenterol. Hepato. 2016, 10, 1145–1152. [Google Scholar] [CrossRef]
- Lee, Y.J.; Arguello, E.S.; Jenq, R.R.; Littmann, E.; Kim, G.J.; Miller, L.C.; Ling, L.; Figueroa, C.; Robilotti, E.; Perales, M.-A.; et al. Protective Factors in the Intestinal Microbiome Against Clostridium difficile Infection in Recipients of Allogeneic Hematopoietic Stem Cell Transplantation. J. Infect. Dis. 2017, 215, 1117–1123. [Google Scholar] [CrossRef] [Green Version]
- Kriss, M.; Hazleton, K.; Nusbacher, N.M.; Martin, C.G.; Lozupone, C.A. Low diversity gut microbiota dysbiosis: Drivers, functional implications and recovery. Curr. Opin. Microbiol. 2018, 44, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Buonomo, E.L.; Petri, W.A. The microbiota and immune response during Clostridium difficile infection. Anaerobe 2016, 41, 79–84. [Google Scholar] [CrossRef] [Green Version]
- Doerner, K.C.; Takamine, F.; LaVoie, C.P.; Mallonee, D.H.; Hylemon, P.B. Assessment of fecal bacteria with bile acid 7 alpha-dehydroxylating activity for the presence of bai-like genes. Appl. Environ. Microbiol. 1997, 63, 1185–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivera-Chávez, F.; Lopez, C.A.; Bäumler, A.J. Oxygen as a driver of gut dysbiosis. Free. Radic. Biol. Med. 2017, 105, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Kolling, G.L.; Wu, M.; Warren, C.A.; Durmaz, E.; Klaenhammer, T.R.; Guerrant, R.L. Lactic acid production by Streptococcus thermophilus alters Clostridium difficile infection and in vitro Toxin A production. Gut Microbes 2012, 3, 523–529. [Google Scholar] [CrossRef] [Green Version]
- Sangster, W.; Hegarty, J.; Schieffer, K.; Wright, J.R.; Hackman, J.; Toole, D.R.; Lamendella, R.; Stewart, D.B. Bacterial and Fungal Microbiota Changes Distinguish C. difficile Infection from Other Forms of Diarrhea: Results of a Prospective Inpatient Study. Front. Microbiol. 2016, 7, 789. [Google Scholar] [CrossRef]
- Han, S.-H.; Yi, J.; Kim, J.-H.; Lee, S.; Moon, H.-W. Composition of gut microbiota in patients with toxigenic Clostridioides (Clostridium) difficile: Comparison between subgroups according to clinical criteria and toxin gene load. PLoS ONE 2019, 14, e0212626. [Google Scholar] [CrossRef] [Green Version]
- Planche, T.D.; Davies, K.A.; Coen, P.G.; Finney, J.M.; Monahan, I.M.; Morris, K.A.; O’Connor, L.; Oakley, S.J.; Pope, C.F.; Wren, M.W.; et al. Differences in outcome according to Clostridium difficile testing method: A prospective multicentre diagnostic validation study of C difficile infection. Lancet Infect. Dis. 2013, 13, 936–945. [Google Scholar] [CrossRef] [Green Version]
- Rupnik, M. Heterogeneity of large clostridial toxins: Importance of Clostridium difficile toxinotypes. FEMS Microbiol. Rev. 2008, 32, 541–555. [Google Scholar] [CrossRef] [Green Version]
- Deng, H.; Yang, S.; Zhang, Y.; Qian, K.; Zhang, Z.; Liu, Y.; Wang, Y.; Bai, Y.; Fan, H.; Zhao, X.; et al. Bacteroides fragilis Prevents Clostridium difficile Infection in a Mouse Model by Restoring Gut Barrier and Microbiome Regulation. Front. Microbiol. 2018, 9, 2976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, B.; Xu, W.; Santini, P.A.; Polydorides, A.D.; Chiu, A.; Estrella, J.; Shan, M.; Chadburn, A.; Villanacci, V.; Plebani, A.; et al. Intestinal Bacteria Trigger T Cell-Independent Immunoglobulin A2 Class Switching by Inducing Epithelial-Cell Secretion of the Cytokine APRIL. Immunity 2007, 26, 812–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wexler, A.G.; Goodman, A.L. An insider’s perspective: Bacteroides as a window into the microbiome. Nat. Microbiol. 2017, 2, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Gu, S.; Chen, Y.; Zhang, X.; Lu, H.; Lv, T.; Shen, P.; Lv, L.; Zheng, B.; Jiang, X.; Li, L. Identification of key taxa that favor intestinal colonization of Clostridium difficile in an adult Chinese population. Microbes Infect. 2016, 18, 30–38. [Google Scholar] [CrossRef]
- Kaper, J.B.; Nataro, J.P.; Mobley, H. Pathogenic Escherichia coli. Nat. Rev. Genet. 2004, 2, 123–140. [Google Scholar] [CrossRef]
- Boto-Ordóñez, M.; Urpi-Sarda, M.; Queipo-Ortuño, M.I.; Tulipani, S.; Tinahones, F.J.; Andres-Lacueva, C. High levels of Bifidobacteria are associated with increased levels of anthocyanin microbial metabolites: A randomized clinical trial. Food Funct. 2014, 5, 1932–1938. [Google Scholar] [CrossRef] [Green Version]
- Hidalgo-Cantabrana, C.; Delgado, S.; Ruiz, L.; Ruas-Madiedo, P.; Sánchez, B.; Margolles, A. Bifidobacteria and Their Health-Promoting Effects. Microbiol. Spectr. 2017, 5, 5. [Google Scholar] [CrossRef]
- Valdés-Varela, L.; Hernández-Barranco, A.M.; Ruas-Madiedo, P.; Gueimonde, M. Effect of Bifidobacterium upon Clostridium difficile Growth and Toxicity When Co-cultured in Different Prebiotic Substrates. Front. Microbiol. 2016, 7, 738. [Google Scholar] [CrossRef]
- Wei, Y.; Yang, F.; Wu, Q.; Gao, J.; Liu, W.; Liu, C.; Guo, X.-K.; Suwal, S.; Kou, Y.; Zhang, B.; et al. Protective Effects of Bifidobacterial Strains Against Toxigenic Clostridium difficile. Front. Microbiol. 2018, 9, 888. [Google Scholar] [CrossRef]
- Yun, B.; Song, M.; Park, D.J.; Oh, S. Beneficial Effect of Bifidobacterium longum ATCC 15707 on Survival Rate of Clostridium difficile Infection in Mice. Korean J. Food Sci. Anim. Resour. 2017, 37, 368–375. [Google Scholar] [CrossRef] [Green Version]
- Derrien, M.; Vaughan, E.E.; Plugge, C.M.; De Vos, W.M. Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int. J. Syst. Evol. Microbiol. 2004, 54, 1469–1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, K.M.; Ferreyra, J.A.; Higginbottom, S.K.; Lynch, J.; Kashyap, P.C.; Gopinath, S.; Naidu, N.; Choudhury, B.; Weimer, B.; Monack, D.M.; et al. Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nat. Cell Biol. 2013, 502, 96–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
| Clinical and Demographic Characteristics | Group CDI | Group P | Group CTRL |
|---|---|---|---|
| Sex, number (%) | |||
| Men | 4 (26%) | 10 (66%) | 7 (46%) |
| Women | 11 (74%) | 5 (34%) | 8 (54%) |
| Age (mean ± SD) | 69 ± 19 | 51 ± 26 | 44 ± 12 |
| Antibiotics last three months, number (%) | 14 (93%) | 8 (53%) | - |
| Cephalosporins | 5 (33%) | 2 (13%) | - |
| Fluorquinolones | 4 (27%) | 2 (13%) | - |
| Β-Lactamics | 5 (33%) | 3 (20%) | - |
| Others | 5 (33%) | 4 (27%) | - |
| Without antibiotics | 1 (7%) | 2 (13%) | - |
| Unknown | 0 (0%) | 4 (27%) | - |
| Strain Type, number (%) | |||
| Toxigenic | 15 (100%) | 8 (53%) | - |
| Non-toxigenic | 0 (0%) | 7 (47%) | - |
| Comorbidities | |||
| Hepatic disease | 1 (7%) | 0 (0%) | - |
| Crohn’s disease | 1 (7%) | 0 (0%) | - |
| Malignant blood disease | 2 (13%) | 0 (0%) | - |
| Other intestinal disease | 3 (20%) | 0 (0%) | - |
| Other comorbidity | 13 (87%) | 6 (40%) | - |
| Previous CD, number (%) | 2 (13%) | 0 (0%) | - |
| Origin, number (%) | |||
| Hospital | 7 (47%) | 2 (13%) | - |
| Community | 8 (53%) | 13 (87%) | - |
| Resolution, number (%) | |||
| Complete | 11 (73%) | - | - |
| Exitus letalis | 3 (20%) | - | - |
| Recurrence | 1 (7%) | - | - |
| Group 1 | Group 2 | Index | Mean Group 1 | Mean Group 2 | p-Values Wilcoxon Test |
|---|---|---|---|---|---|
| CDI | CTRL | Shannon | 2.0 | 2.8 | 0.0002 |
| CDI | P | Shannon | 2.0 | 1.9 | 0.3724 |
| CTRL | P | Shannon | 2.8 | 1.9 | 0.0003 |
| CDI | CTRL | Simpson | 0.7 | 0.9 | 0.0006 |
| CDI | P | Simpson | 0.7 | 0.7 | 0.3091 |
| CTRL | P | Simpson | 0.9 | 0.7 | 0.0003 |
| CDI | CTRL | ACE | 47.2 | 113.1 | <0.0001 |
| CDI | P | ACE | 47.2 | 51.6 | 0.9339 |
| CTRL | P | ACE | 113.1 | 51.6 | <0.0001 |
| CDI | CTRL | CHAO1 | 47.2 | 113.1 | <0.0001 |
| CDI | P | CHAO1 | 47.2 | 51.6 | 0.9339 |
| CTRL | P | CHAO1 | 113.1 | 51.6 | <0.0001 |
| Family | Genus | Group CDI | Group P | Group CTRL | CDI versus CTRL | P versus CTRL | CDI versus P |
|---|---|---|---|---|---|---|---|
| 38.5533 | 39.1305 | 66.8691 | 0.0006 | 0.0016 | 1.0000 | ||
| Lachnospiraceae | 11.7971 | 12.8014 | 18.7516 | 0.0771 | 0.1213 | 0.8035 | |
| Lachnospiraceae | Agathobacter | 0.0374 | 0.1254 | 6.2873 | <0.0001 | <0.0001 | 1.0000 |
| Lachnospiraceae | Roseburia | 0.1196 | 0.1654 | 2.7259 | <0.0001 | 0.0001 | 0.6545 |
| Ruminococcaceae | 9.3575 | 6.3853 | 31.4165 | <0.0001 | <0.0001 | 0.1150 | |
| Ruminococcaceae | Faecalibaterium | 3.2948 | 0.9126 | 11.2857 | 0.0013 | <0.0001 | 0.1299 |
| Ruminococcaceae | Ruminococcus | 0.1022 | 0.0560 | 4.5458 | 0.0009 | 0.0002 | 0.3894 |
| Ruminococcaceae | Subdoligranolum | 0.3407 | 0.2808 | 0.2808 | 0.0001 | <0.0001 | 0.4092 |
| Peptostreptococcaceae | 0.5298 | 0.4781 | 0.0600 | 0.0134 | 0.0242 | 0.5755 | |
| Peptostreptococcaceae | Clostridiodes | 0.3668 | 0.3668 | 0.0000 | <0.0001 | <0.0001 | 0.1568 |
| Enterococcaceae | 0.6853 | 0.4993 | 0.0000 | 0.0009 | 0.0021 | 1.0000 | |
| Enterococcaceae | Enterococcus | 0.6853 | 0.4993 | 0.0000 | 0.0009 | 0.0021 | 1.0000 |
| Streptococcaceae | 2.3442 | 3.5860 | 0.7527 | 0.0701 | 0.3481 | 0.1300 | |
| Streptococcaceae | Streptococcus | 2.3431 | 3.5834 | 0.7448 | 0.0701 | 0.3481 | 0.1408 |
| Lactobacillaceae | 1.0287 | 0.2269 | 0.0111 | 0.3307 | 0.1475 | 0.8470 | |
| Lactobacillaceae | Lactobacillus | 1.0017 | 0.2256 | 0.0111 | 0.3307 | 0.2624 | 1.0000 |
| Veillonellaceae | 3.4787 | 12.1379 | 10.2983 | 0.2216 | 0.7765 | 0.6185 | |
| Veillonellaceae | Veillonella | 2.9121 | 4.5646 | 0.0824 | 0.1075 | 0.3898 | 0.8342 |
| Family | Genus | Group CDI | Group P | Group CTRL | CDI versus CTRL | P versus CTRL | CDI versus P |
|---|---|---|---|---|---|---|---|
| Lachnospiraceae | Blautia | 0.5904 | 1.5385 | 0.6994 | 0.1831 | 0.1829 | 0.7395 |
| Lachnospiraceae | Eubacterium ventriosum | 0.0000 | 0.0032 | 0.7224 | <0.0001 | 0.0001 | 0.3506 |
| Lachnospiraceae | Eubacterium eligens | 0.0063 | 0.1217 | 0.3189 | 0.0260 | 0.1126 | 0.5383 |
| Lachnospiraceae | Eubacterium xylanophilum | 0.0000 | 0.0010 | 0.1578 | 0.0001 | 0.0003 | 1.0000 |
| Lachnospiraceae | Eubacterium ruminantium | 0.0000 | 0.0000 | 0.0420 | 0.1498 | 0.1498 | 1.0000 |
| Lachnospiraceae | Eubacterium fissicatena | 0.0082 | 0.0485 | 0.0053 | 0.5155 | 0.6324 | 0.2879 |
| Lachnospiraceae | Eubacterium hallii | 0.0011 | 0.0056 | 0.0040 | 0.0740 | 0.2220 | 0.5772 |
| Lachnospiraceae | Eubacterium oxidoreducens | 0.0000 | 0.0000 | 0.0001 | 0.3340 | 0.3340 | 1.0000 |
| Eubacteriaceae | Eubacterium | 0.0011 | 0.0118 | 0.0003 | 0.5634 | 0.0790 | 0.2401 |
| Clostridiales family XIII | Eubacterium brachy | 0.0000 | 0.0051 | 0.3334 | 0.0004 | 0.0036 | 0.3506 |
| Clostridiales family XIII | Eubacterium nodatum | 0.0239 | 0.1099 | 0.0196 | 0.0261 | 0.1174 | 0.0040 |
| Ruminococcaceae | Eubacterium coprostanoligenes | 0.0939 | 0.1774 | 1.3705 | 0.0001 | 0.0001 | 0.9617 |
| Phylum | Family | Genus | Group CDI | Group P | Group CTRL | CDI versus CTRL | P versus CTRL | CDI versus P |
|---|---|---|---|---|---|---|---|---|
| Bacteroidetes | 39.3656 | 36.0149 | 19.5638 | 0.0275 | 0.0521 | 0.6783 | ||
| Bacteroidaceae | 26.8470 | 29.1252 | 11.0028 | 0.0307 | 0.0137 | 0.7400 | ||
| Bacteroidaceae | Bacteroides | 26.8470 | 29.1252 | 11.0028 | 0.0307 | 0.0137 | 0.7400 | |
| Protebacteria | 14.3918 | 20.5514 | 3.8165 | 0.0154 | 0.0636 | 0.8357 | ||
| Enterobacteriaceae | 10.6111 | 14.7078 | 3.0008 | 0.0521 | 0.3698 | 0.7399 | ||
| Enterobacteriaceae | Escherichia-Shigella | 9.1685 | 10.6014 | 2.5561 | 0.1620 | 0.2051 | 1.0000 | |
| Actinobacteria | 1.1694 | 2.7769 | 4.6633 | 0.0032 | 0.3261 | 0.0889 | ||
| Bifidobacteriaceae | 0.6280 | 2.0634 | 3.7001 | 0.0076 | 0.3698 | 0.1653 | ||
| Bifidobacteriaceae | Bifidobacterium | 0.6250 | 2.0620 | 3.6980 | 0.0071 | 0.3545 | 0.1564 | |
| Verrocomicrobia | 5.9703 | 1.2742 | 4.7534 | 0.1605 | 0.0516 | 0.8698 | ||
| Akkermansiaceae | 5.9703 | 1.2742 | 4.7534 | 0.1605 | 0.0516 | 0.8698 | ||
| Akkermansiaciae | Akkermansia | 5.9703 | 1.2742 | 4.7534 | 0.1605 | 0.0516 | 0.8698 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Pellicer, P.; Navarro-López, V.; González-Tamayo, R.; Llopis-Ruiz, C.; Núñez-Delegido, E.; Ruzafa-Costas, B.; Navarro-Moratalla, L.; Agüera-Santos, J. Descriptive Study of Gut Microbiota in Infected and Colonized Subjects by Clostridiodes difficile. Microorganisms 2021, 9, 1727. https://doi.org/10.3390/microorganisms9081727
Sánchez-Pellicer P, Navarro-López V, González-Tamayo R, Llopis-Ruiz C, Núñez-Delegido E, Ruzafa-Costas B, Navarro-Moratalla L, Agüera-Santos J. Descriptive Study of Gut Microbiota in Infected and Colonized Subjects by Clostridiodes difficile. Microorganisms. 2021; 9(8):1727. https://doi.org/10.3390/microorganisms9081727
Chicago/Turabian StyleSánchez-Pellicer, Pedro, Vicente Navarro-López, Ruth González-Tamayo, Coral Llopis-Ruiz, Eva Núñez-Delegido, Beatriz Ruzafa-Costas, Laura Navarro-Moratalla, and Juan Agüera-Santos. 2021. "Descriptive Study of Gut Microbiota in Infected and Colonized Subjects by Clostridiodes difficile" Microorganisms 9, no. 8: 1727. https://doi.org/10.3390/microorganisms9081727
APA StyleSánchez-Pellicer, P., Navarro-López, V., González-Tamayo, R., Llopis-Ruiz, C., Núñez-Delegido, E., Ruzafa-Costas, B., Navarro-Moratalla, L., & Agüera-Santos, J. (2021). Descriptive Study of Gut Microbiota in Infected and Colonized Subjects by Clostridiodes difficile. Microorganisms, 9(8), 1727. https://doi.org/10.3390/microorganisms9081727