
Microbial Shift in the Enteric Bacteriome of Coral Reef Fish Following Climate-Driven Regime Shifts

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
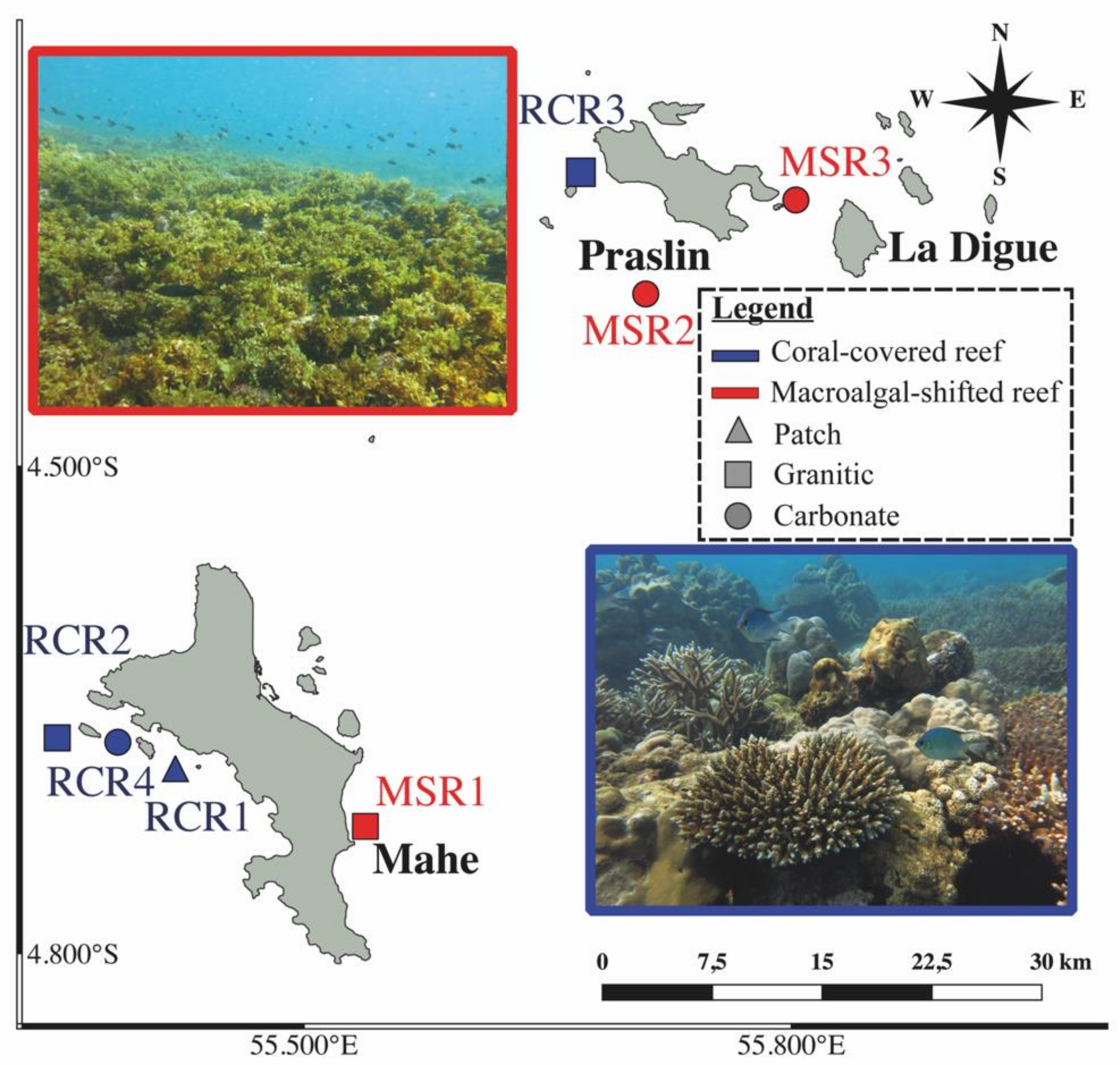
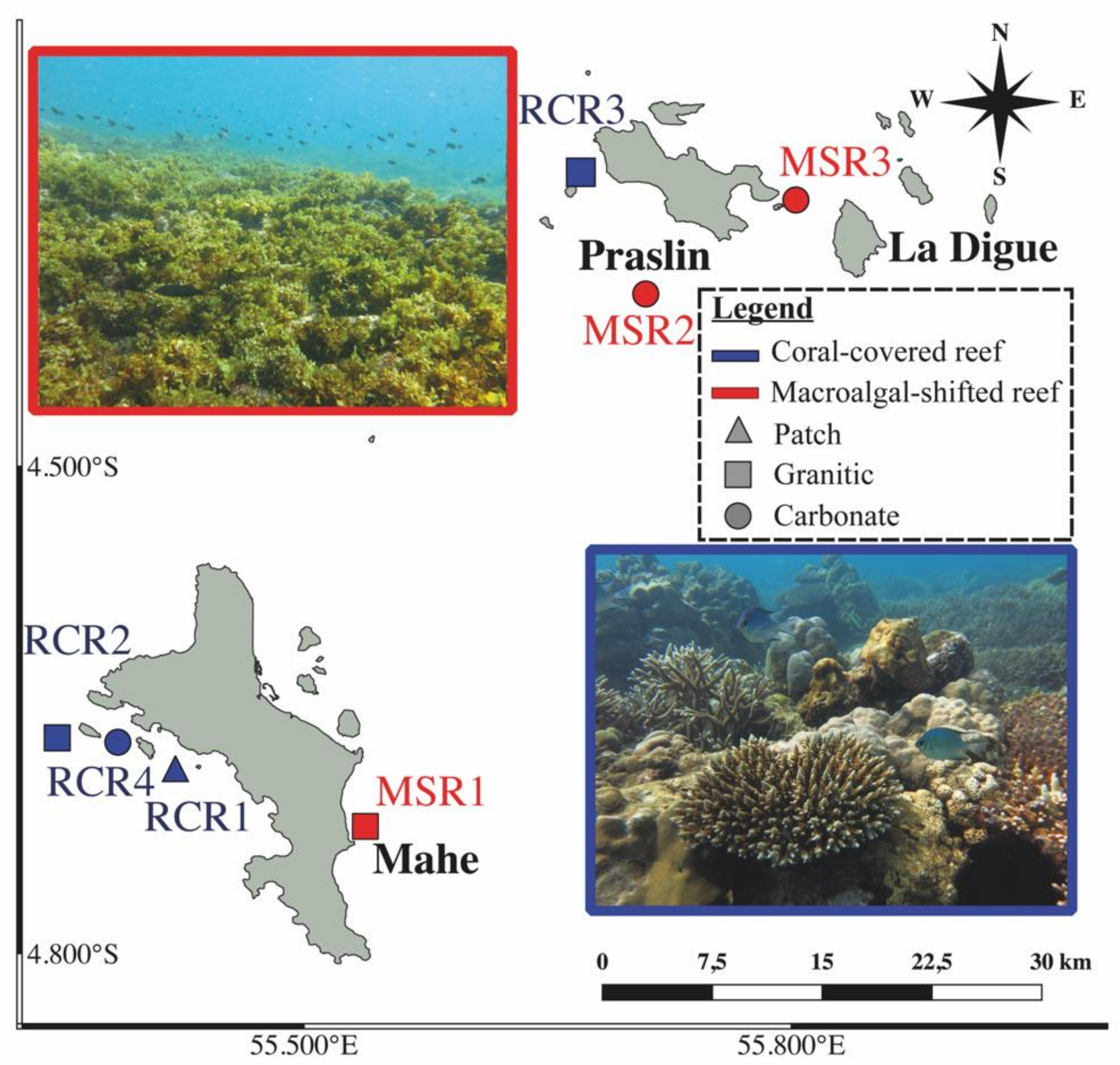
2.1. Study Area and Sample Collection
2.2. Fish Identification and Diet Type Definition
2.3. DNA Extraction and 16S rDNA Gene Amplification
2.4. Sequence Processing
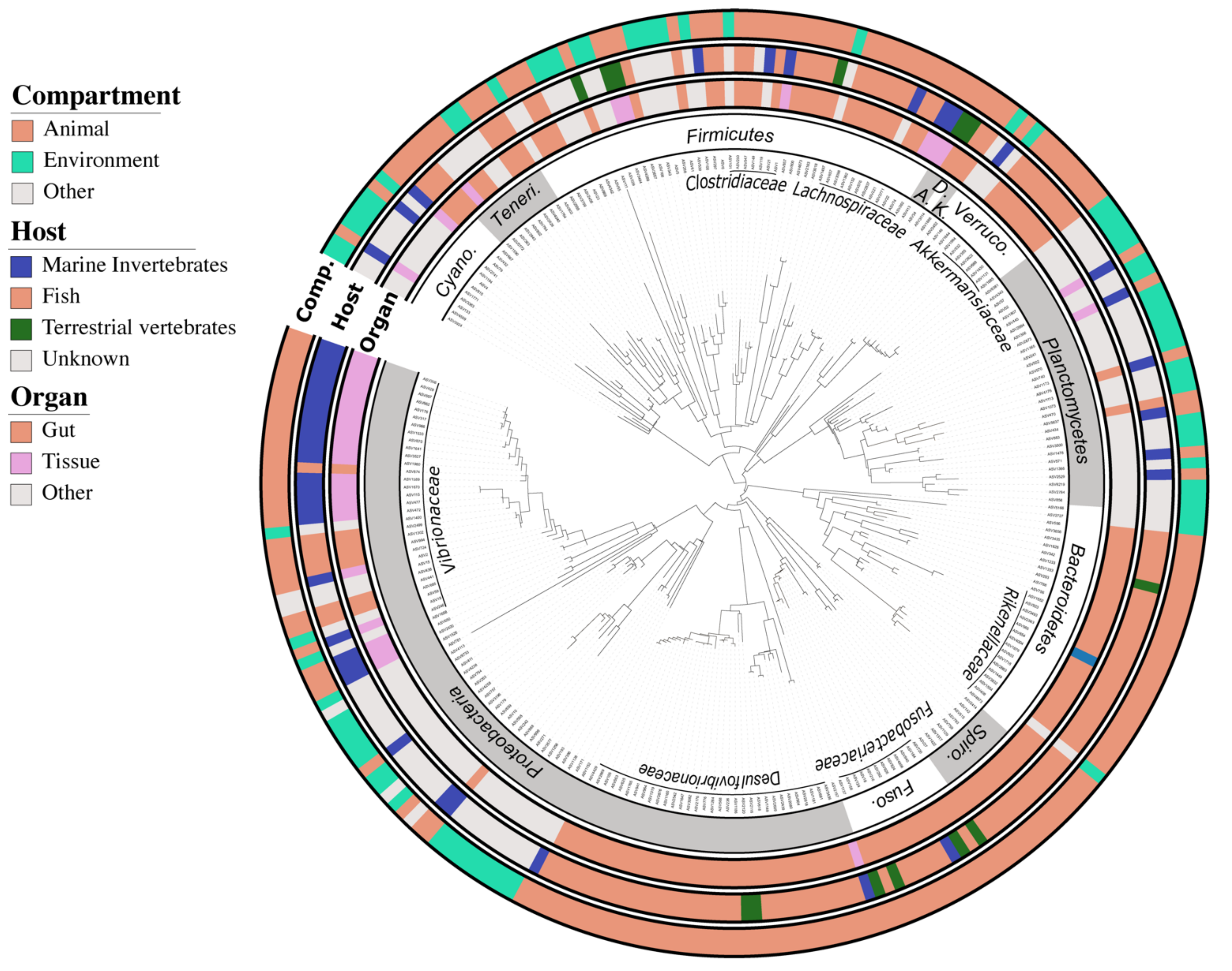
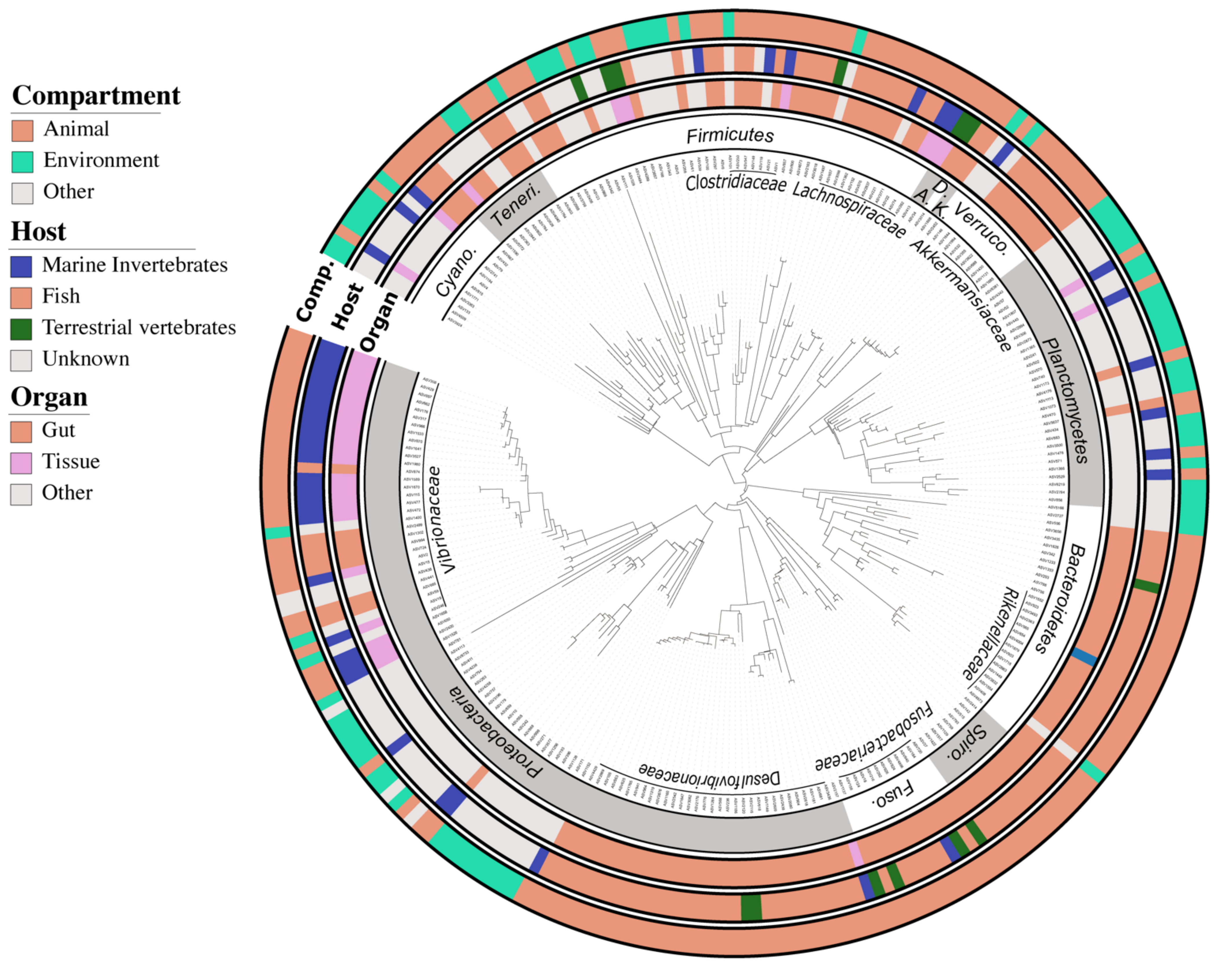
2.5. Defining the Core Bacteriome of Reef Organisms
2.6. Inference of ASVs Habitat Preference
2.7. Computation of Alpha and Beta-diversity of Bacteriomes
2.8. Functional Diversity Predictions of Bacteriome
2.9. Statistical Tests
3. Results
3.1. Sampling Size and Composition of Fish Catch between Reef Conditions
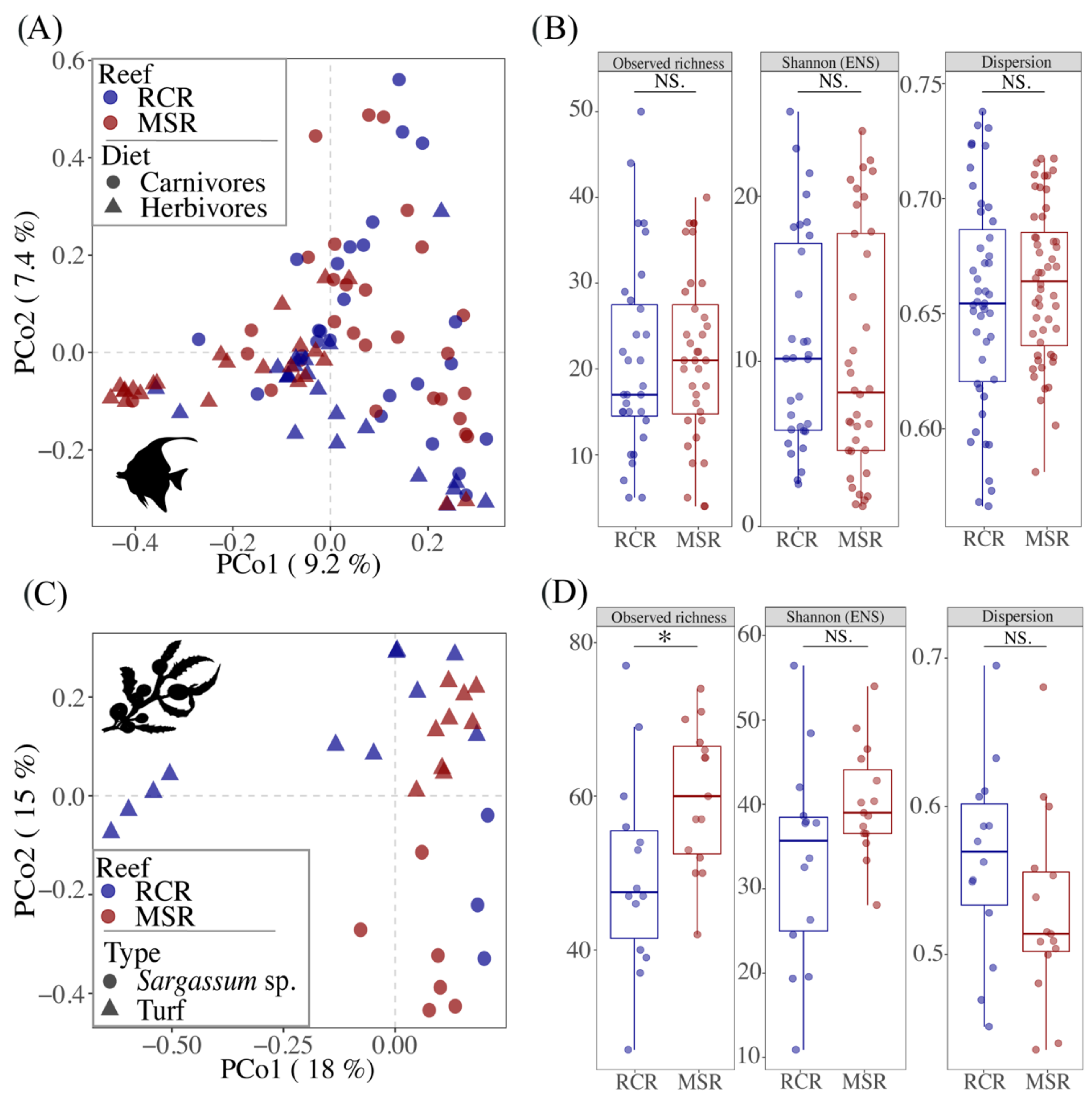
3.2. Composition and Diversity of the Fish Core Gut Bacteriome
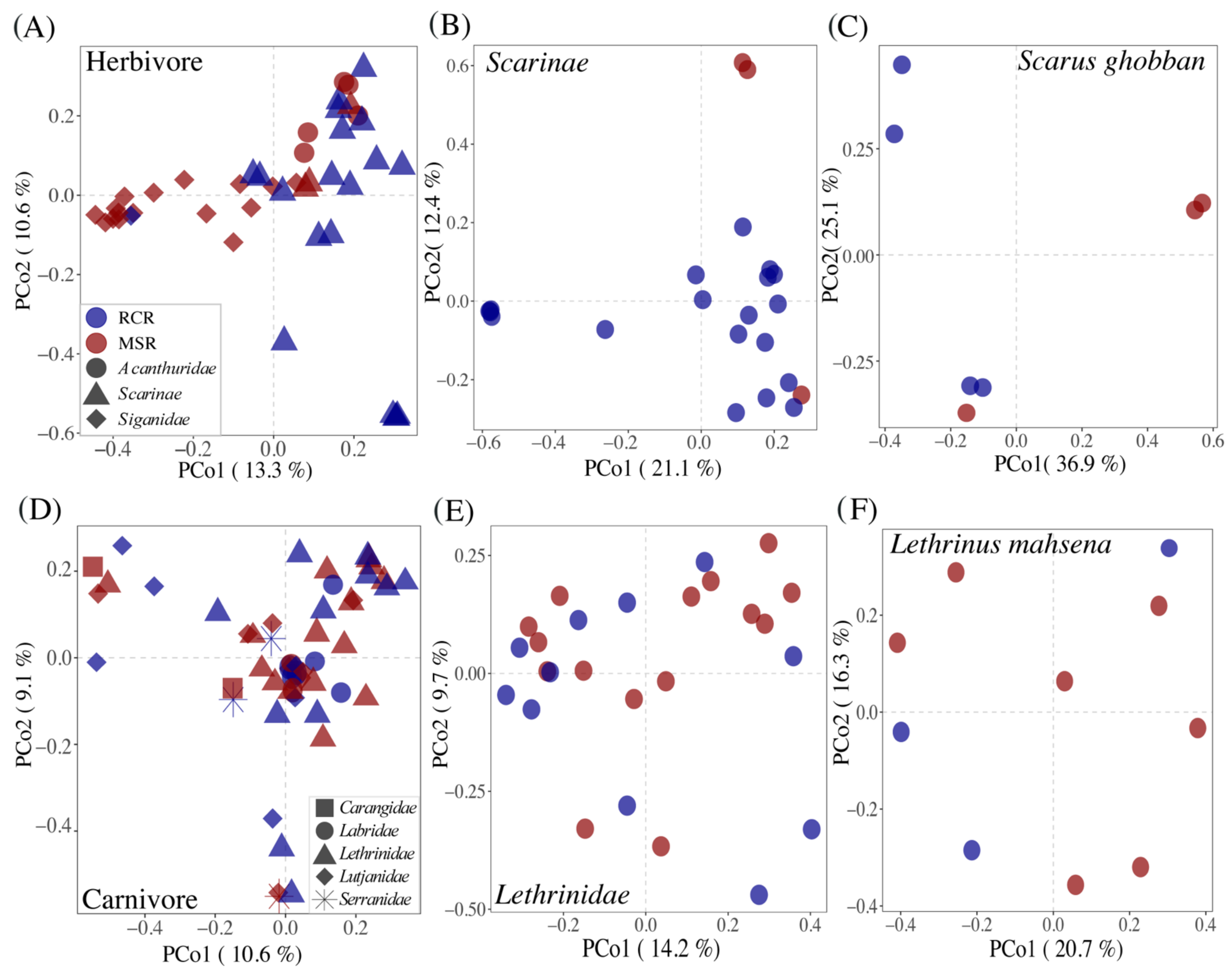
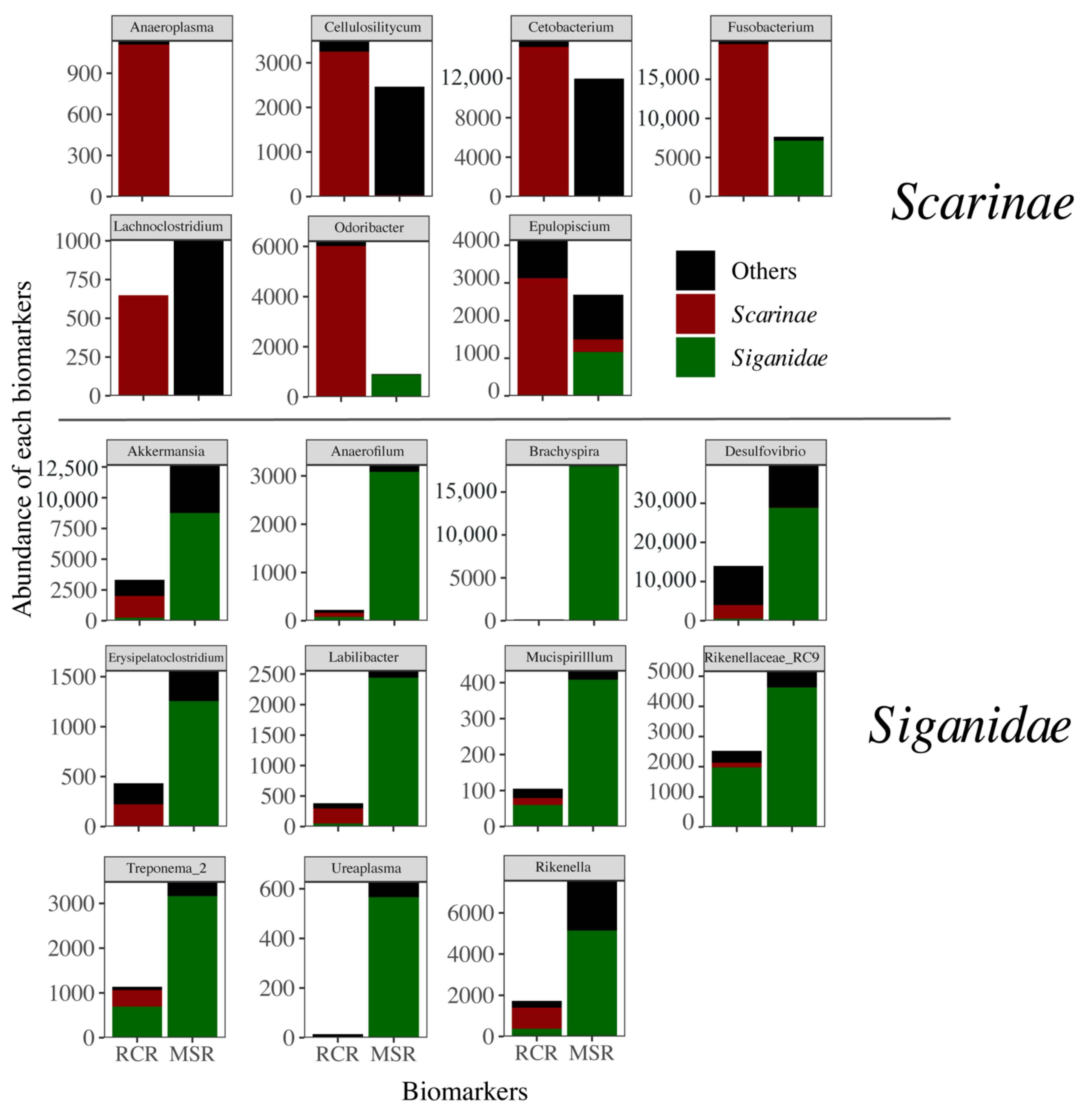
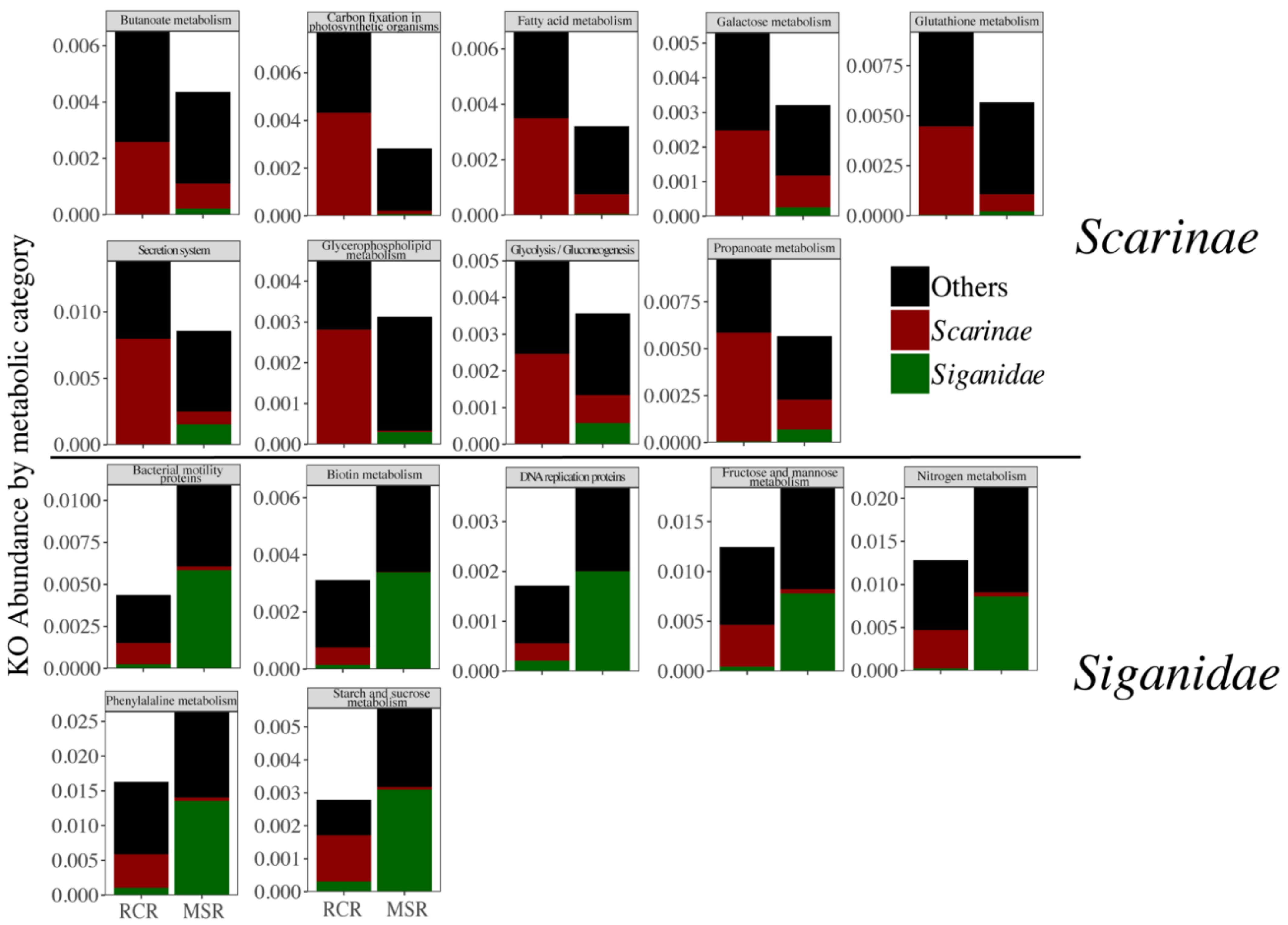
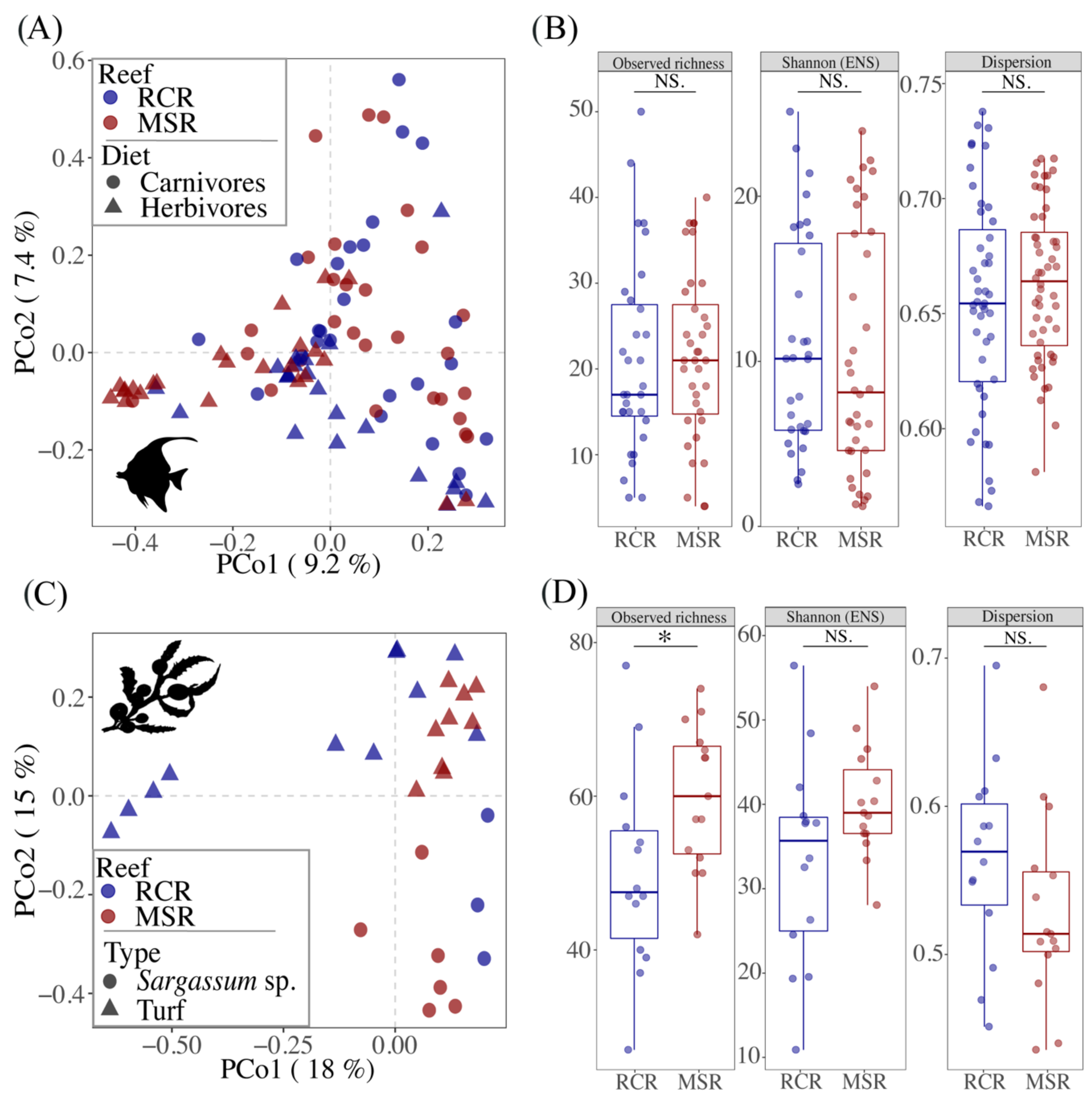
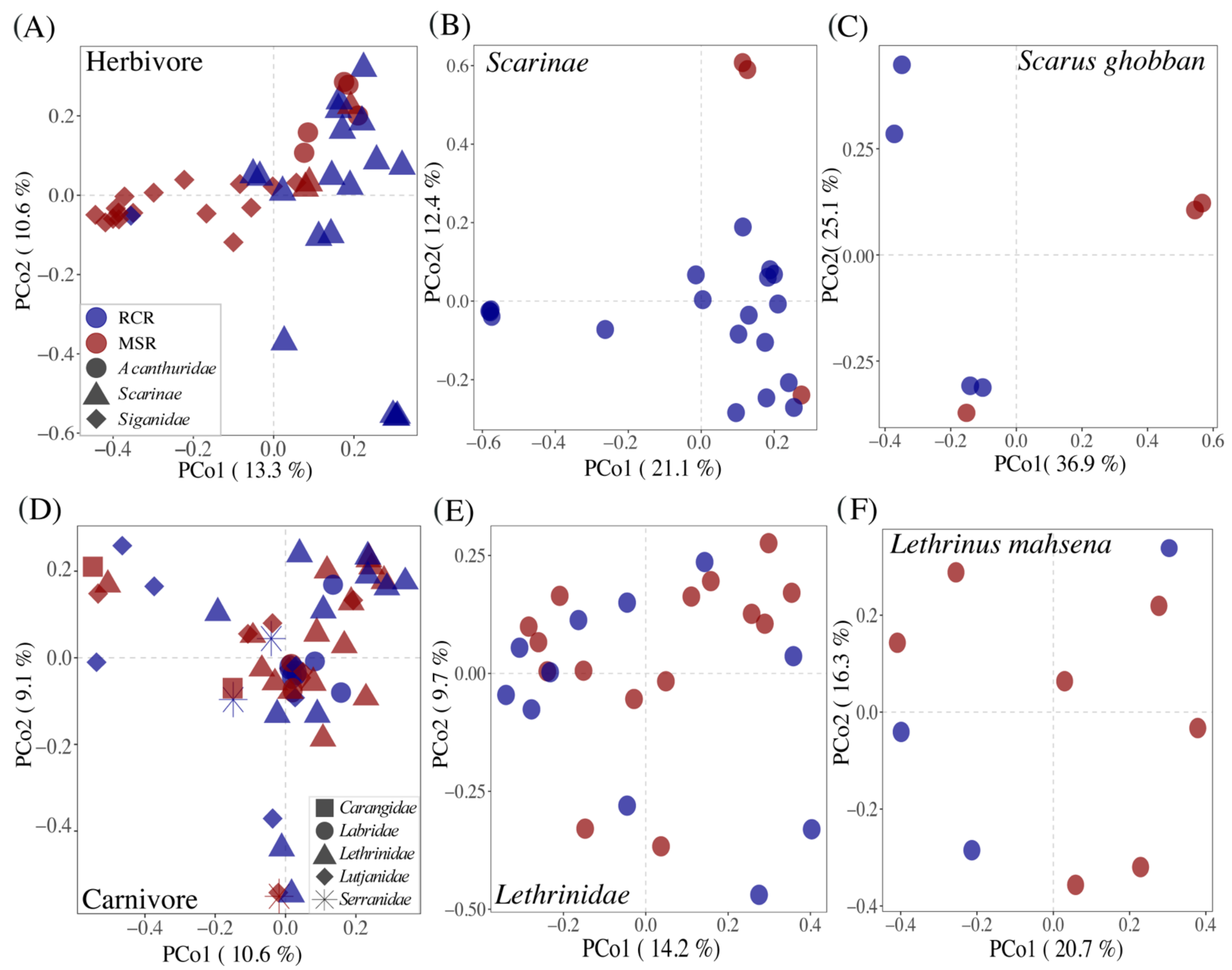
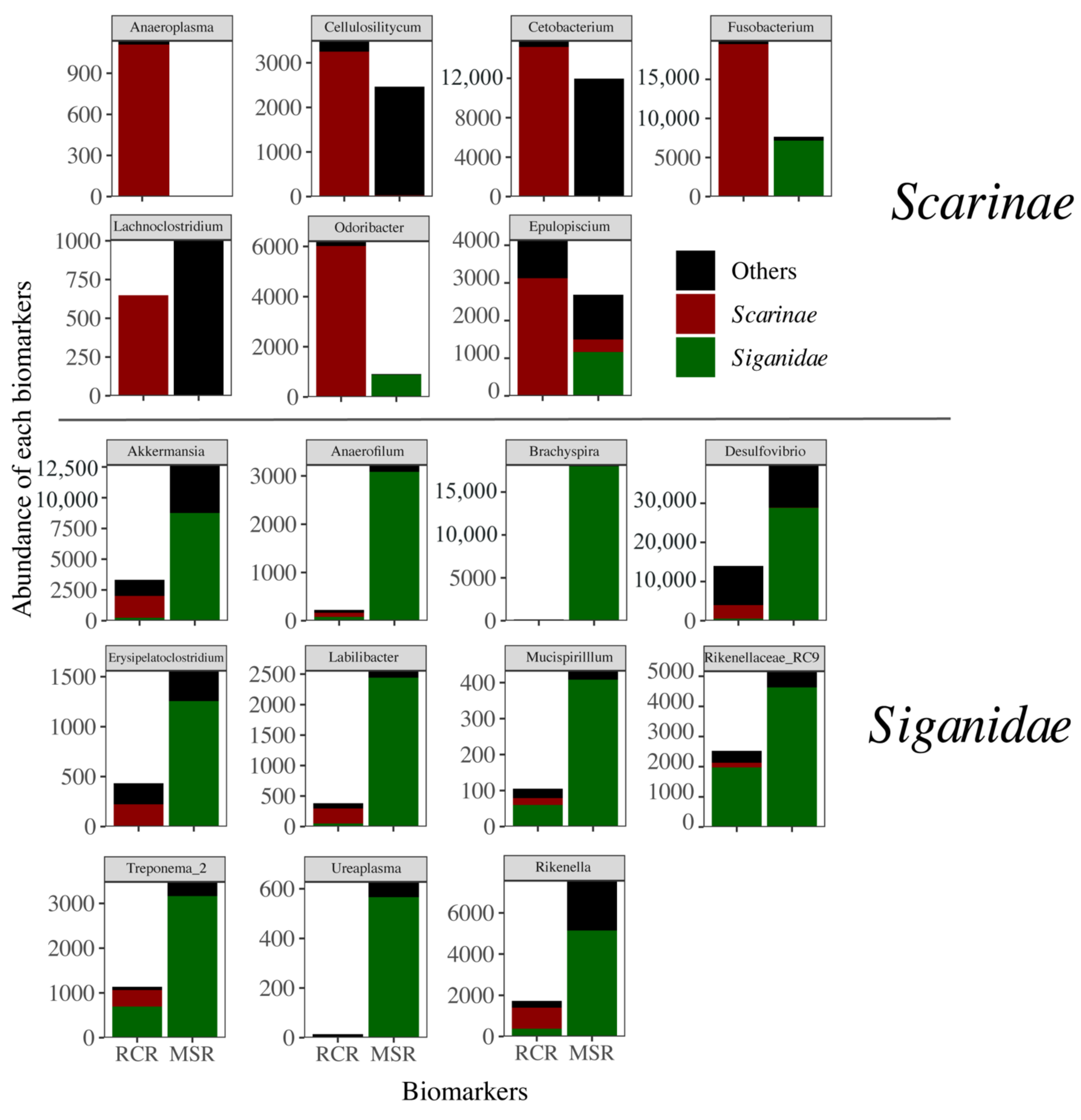
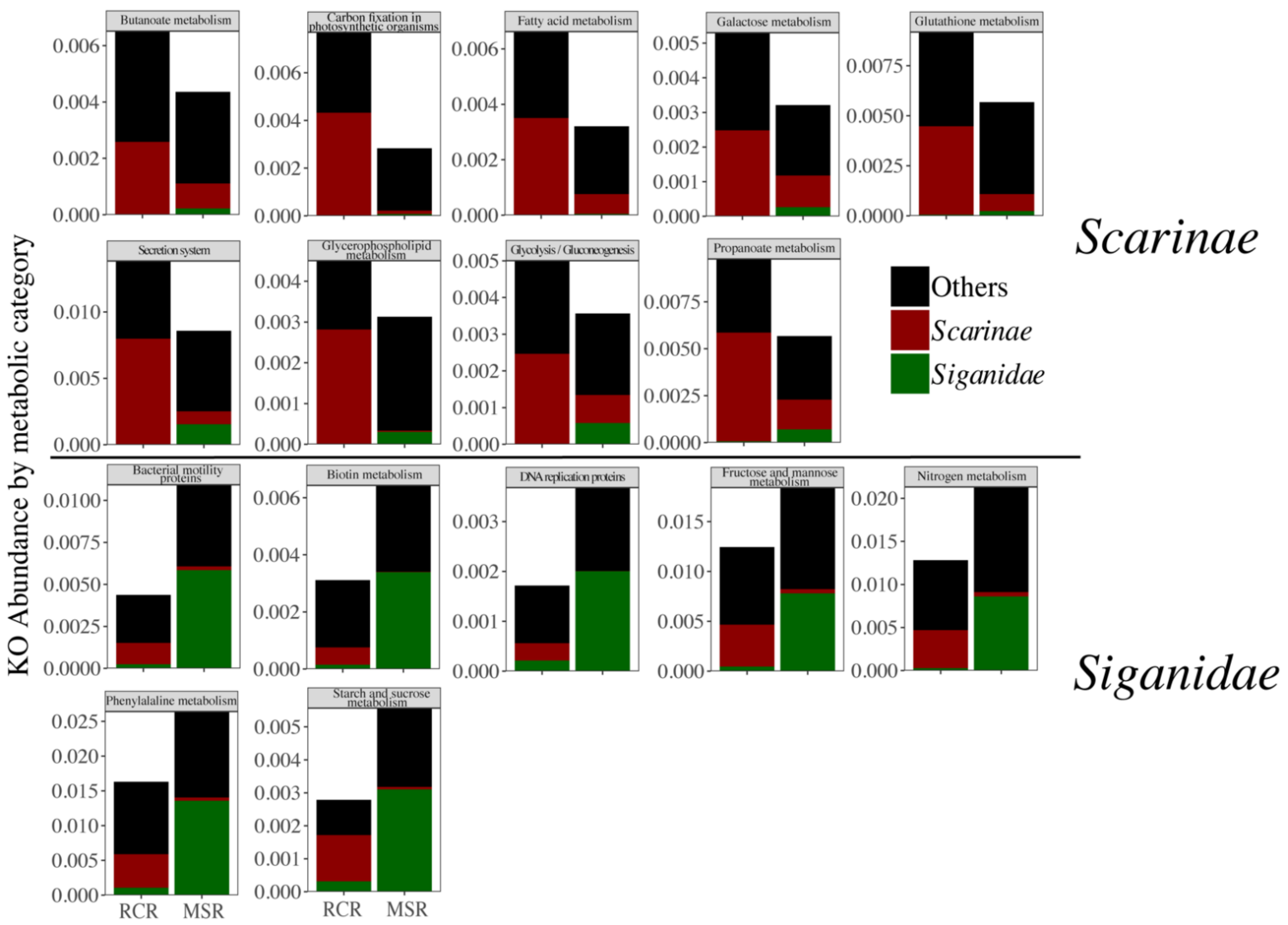
3.3. Alteration of the Coral Reef Significantly Disrupts Herbivore but Not Carnivore Bacteriomes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hughes, T.P. Climate Change, Human Impacts, and the Resilience of Coral Reefs. Science 2003, 301, 929–933. [Google Scholar] [CrossRef] [Green Version]
- Jackson, J.B.C. Ecological extinction and evolution in the brave new ocean. Proc. Natl. Acad. Sci. USA 2008, 105, 11458–11465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodhead, A.J.; Hicks, C.C.; Norström, A.V.; Williams, G.J.; Graham, N.A.J. Coral reef ecosystem services in the Anthropocene. Funct. Ecol. 2019, 33, 1023–1034. [Google Scholar] [CrossRef] [Green Version]
- McWilliam, M.; Pratchett, M.S.; Hoogenboom, M.O.; Hughes, T.P. Deficits in functional trait diversity following recovery on coral reefs. Proc. R. Soc. B Biol. Sci. 2020, 287, 20192628. [Google Scholar] [CrossRef] [Green Version]
- Mumby, P.J. The impact of exploiting grazers (Scaridae) on the dynamics of Caribbean coral reefs. Ecol. Appl. 2006, 16, 747–769. [Google Scholar] [CrossRef] [Green Version]
- Graham, N.A.J.; Jennings, S.; MacNeil, M.A.; Mouillot, D.; Wilson, S.K. Predicting climate-driven regime shifts versus rebound potential in coral reefs. Nature 2015, 518, 94–97. [Google Scholar] [CrossRef]
- Norström, A.; Nyström, M.; Lokrantz, J.; Folke, C. Alternative states on coral reefs: Beyond coral–macroalgal phase shifts. Mar. Ecol. Prog. Ser. 2009, 376, 295–306. [Google Scholar] [CrossRef]
- Cheal, A.J.; MacNeil, M.A.; Cripps, E.; Emslie, M.J.; Jonker, M.; Schaffelke, B.; Sweatman, H. Coral-macroalgal phase shifts or reef resilience: Links with diversity and functional roles of herbivorous fishes on the Great Barrier Reef. Coral Reefs 2010, 29, 1005–1015. [Google Scholar] [CrossRef]
- McCook, L.; Jompa, J.; Diaz-Pulido, G. Competition between corals and algae on coral reefs: A review of evidence and mechanisms. Coral Reefs 2001, 19, 400–417. [Google Scholar] [CrossRef]
- Hughes, T.P.; Rodrigues, M.J.; Bellwood, D.R.; Ceccarelli, D.; Hoegh-Guldberg, O.; McCook, L.; Moltschaniwskyj, N.; Pratchett, M.S.; Steneck, R.S.; Willis, B. Phase Shifts, Herbivory, and the Resilience of Coral Reefs to Climate Change. Curr. Biol. 2007, 17, 360–365. [Google Scholar] [CrossRef] [Green Version]
- Morrow, K.M.; Bromhall, K.; Motti, C.A.; Munn, C.B.; Bourne, D.G. Allelochemicals Produced by Brown Macroalgae of the Lobophora Genus Are Active against Coral Larvae and Associated Bacteria, Supporting Pathogenic Shifts to Vibrio Dominance. Appl. Environ. Microbiol. 2017, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrow, K.M.; Liles, M.R.; Paul, V.J.; Moss, A.G.; Chadwick, N.E. Bacterial shifts associated with coral-macroalgal competition in the Caribbean Sea. Mar. Ecol. Prog. Ser. 2013, 488, 103–117. [Google Scholar] [CrossRef]
- Burkepile, D.E.; Hay, M.E. Herbivore vs. nutrient control of marine primary producers: Context-dependent effects. Ecology 2006, 87, 3128–3139. [Google Scholar] [CrossRef] [Green Version]
- Nyström, M.; Graham, N.A.J.; Lokrantz, J.; Norström, A.V. Capturing the cornerstones of coral reef resilience: Linking theory to practice. Coral Reefs 2008, 27, 795–809. [Google Scholar] [CrossRef]
- Hughes, T.P.; Barnes, M.L.; Bellwood, D.R.; Cinner, J.E.; Cumming, G.S.; Jackson, J.B.C.; Kleypas, J.; van de Leemput, I.A.; Lough, J.M.; Morrison, T.H.; et al. Coral reefs in the Anthropocene. Nature 2017, 546, 82–90. [Google Scholar] [CrossRef]
- Bellwood, D.R.; Pratchett, M.S.; Morrison, T.H.; Gurney, G.G.; Hughes, T.P.; Álvarez-Romero, J.G.; Day, J.C.; Grantham, R.; Grech, A.; Hoey, A.S.; et al. Coral reef conservation in the Anthropocene: Confronting spatial mismatches and prioritizing functions. Biol. Conserv. 2019, 236, 604–615. [Google Scholar] [CrossRef]
- Bellwood, D.R.; Hughes, T.P.; Folke, C.; Nyström, M. Confronting the coral reef crisis. Nature 2004, 429, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Chiarello, M.; Auguet, J.C.; Bettarel, Y.; Bouvier, C.; Claverie, T.; Graham, N.A.J.; Rieuvilleneuve, F.; Sucré, E.; Bouvier, T.; Villéger, S. Skin microbiome of coral reef fish is highly variable and driven by host phylogeny and diet. Microbiome 2018, 6, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Chiarello, M.; Auguet, J.-C.; Graham, N.A.J.; Claverie, T.; Sucré, E.; Bouvier, C.; Rieuvilleneuve, F.; Restrepo-Ortiz, C.X.; Bettarel, Y.; Villéger, S.; et al. Exceptional but vulnerable microbial diversity in coral reef animal surface microbiomes. Proc. R. Soc. B Biol. Sci. 2020, 287, 20200642. [Google Scholar] [CrossRef] [PubMed]
- Miyake, S.; Ngugi, D.K.; Stingl, U. Diet strongly influences the gut microbiota of surgeonfishes. Mol. Ecol. 2015, 24, 656–672. [Google Scholar] [CrossRef] [PubMed]
- Pratte, Z.A.; Besson, M.; Hollman, R.D.; Stewarta, F.J. The gills of reef fish support a distinct microbiome influenced by hostspecific factors. Appl. Environ. Microbiol. 2018, 84, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Scott, J.J.; Adam, T.C.; Duran, A.; Burkepile, D.E.; Rasher, D.B. Intestinal microbes: An axis of functional diversity among large marine consumers. Proc. R. Soc. B Biol. Sci. 2020, 287, 20192367. [Google Scholar] [CrossRef] [Green Version]
- Fischbach, M.A.; Segre, J.A. Signaling in Host-Associated Microbial Communities. Cell 2016, 164, 1288–1300. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhou, L.; Yu, Y.; Ni, J.; Xu, W.; Yan, Q. Composition of Gut Microbiota in the Gibel Carp (Carassius auratus gibelio) Varies with Host Development. Microb. Ecol. 2017, 74, 239–249. [Google Scholar] [CrossRef]
- Piazzon, M.C.; Calduch-Giner, J.A.; Fouz, B.; Estensoro, I.; Simó-Mirabet, P.; Puyalto, M.; Karalazos, V.; Palenzuela, O.; Sitjà-Bobadilla, A.; Pérez-Sánchez, J. Under control: How a dietary additive can restore the gut microbiome and proteomic profile, and improve disease resilience in a marine teleostean fish fed vegetable diets. Microbiome 2017, 5, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngugi, D.K.; Miyake, S.; Cahill, M.; Vinu, M.; Hackmann, T.J.; Blom, J.; Tietbohl, M.D.; Berumen, M.L.; Stingl, U. Genomic diversification of giant enteric symbionts reflects host dietary lifestyles. Proc. Natl. Acad. Sci. USA 2017, 114, E7592–E7601. [Google Scholar] [CrossRef] [Green Version]
- Butt, R.L.; Volkoff, H. Gut microbiota and energy homeostasis in fish. Front. Endocrinol. (Lausanne) 2019, 10, 6–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullam, K.E.; Essinger, S.D.; Lozupone, C.A.; O’Connor, M.P.; Rosen, G.L.; Knight, R.; Kilham, S.S.; Russell, J.A. Environmental and ecological factors that shape the gut bacterial communities of fish: A meta-analysis. Mol. Ecol. 2012, 21, 3363–3378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Bruijn, I.; Liu, Y.; Wiegertjes, G.F.; Raaijmakers, J.M. Exploring fish microbial communities to mitigate emerging diseases in aquaculture. FEMS Microbiol. Ecol. 2018, 94, 1–12. [Google Scholar] [CrossRef]
- Clements, K.D.; Angert, E.R.; Montgomery, W.L.; Choat, J.H. Intestinal microbiota in fishes: What’s known and what’s not. Mol. Ecol. 2014, 23, 1891–1898. [Google Scholar] [CrossRef] [PubMed]
- Neave, M.J.; Apprill, A.; Aeby, G.; Miyake, S.; Voolstra, C.R. Microbial Communities of Red Sea Coral Reefs; Springer International Publishing: Cham, Switzerland, 2019; pp. 53–68. [Google Scholar] [CrossRef]
- Amato, K.R.; Yeoman, C.J.; Kent, A.; Righini, N.; Carbonero, F.; Estrada, A.; Rex Gaskins, H.; Stumpf, R.M.; Yildirim, S.; Torralba, M.; et al. Habitat degradation impacts black howler monkey (Alouatta pigra) gastrointestinal microbiomes. ISME J. 2013, 7, 1344–1353. [Google Scholar] [CrossRef]
- Kohl, K.D.; Amaya, J.; Passement, C.A.; Dearing, M.D.; Mccue, M.D. Unique and shared responses of the gut microbiota to prolonged fasting: A comparative study across five classes of vertebrate hosts. FEMS Microbiol. Ecol. 2014, 90, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Borbón-García, A.; Reyes, A.; Vives-Flórez, M.; Caballero, S. Captivity shapes the gut microbiota of Andean bears: Insights into health surveillance. Front. Microbiol. 2017, 8, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clayton, J.B.; Vangay, P.; Huang, H.; Ward, T.; Hillmann, B.M.; Al-Ghalith, G.A.; Travis, D.A.; Long, H.T.; Van Tuan, B.; Van Minh, V.; et al. Captivity humanizes the primate microbiome. Proc. Natl. Acad. Sci. USA 2016, 113, 10376–10381. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, E.; Mykytczuk, N.; Schulte-Hostedde, A.I. Effects of the captive and wild environment on diversity of the gut microbiome of deer mice (Peromyscus maniculatus). ISME J. 2019, 13, 1293–1305. [Google Scholar] [CrossRef]
- Gudka, M.; Obura, D.; Mbugua, J.; Ahamada, S.; Kloiber, U.; Holter, T. Participatory reporting of the 2016 bleaching event in the Western Indian Ocean. Coral Reefs 2020, 39. [Google Scholar] [CrossRef]
- Faure, G.; Guillaume, M.; Payri, C.; Thomassin, B.; Vanpraet, M.; Vasseur, P. Massive bleaching and death of corals in the Mayotte Reef ecosystem (SW Indian-Ocean). Comptes rendus l’académie des Sci. Série III-Sciences la vie-Life Sci. 1984, 299, 637–642. [Google Scholar]
- Wilkinson, C.; Lindén, O.; Cesar, H.; Hodgson, G.; Rubens, J.; Strong, A.E. Ecological and socioeconomic impacts of 1998 coral mortality in the Indian Ocean: An ENSO impact and a warning of future change? Ambio 1999, 28, 188–196. [Google Scholar]
- Wilson, S.K.; Robinson, J.P.W.; Chong-Seng, K.; Robinson, J.; Graham, N.A.J. Boom and bust of keystone structure on coral reefs. Coral Reefs 2019, 38, 625–635. [Google Scholar] [CrossRef]
- Graham, N.A.J.; Wilson, S.K.; Jennings, S.; Polunin, N.V.C.; Bijoux, J.P.; Robinson, J. Dynamic fragility of oceanic coral reef ecosystems. Proc. Natl. Acad. Sci. 2006, 103, 8425–8429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, J.P.W.; Wilson, S.K.; Jennings, S.; Graham, N.A.J. Thermal stress induces persistently altered coral reef fish assemblages. Glob. Chang. Biol. 2019, 25, 2739–2750. [Google Scholar] [CrossRef]
- Clements, K.D.; Pasch, I.B.Y.; Moran, D.; Turner, S.J. Clostridia dominate 16S rRNA gene libraries prepared from the hindgut of temperate marine herbivorous fishes. Mar. Biol. 2007, 150, 1431–1440. [Google Scholar] [CrossRef]
- Taquet, M.; Delinguer, A. Poissons de l’Océan Indien et de la Mer Rouge; Quae: Versailles, 2007; ISBN 978-2-7592-0045-0. [Google Scholar]
- Mouillot, D.; Villéger, S.; Parravicini, V.; Kulbicki, M.; Arias-González, J.E.; Bender, M.; Chabanet, P.; Floeter, S.R.; Friedlander, A.; Vigliola, L.; et al. Functional over-redundancy and high functional vulnerability in global fish faunas on tropical reefs. Proc. Natl. Acad. Sci. USA 2014, 111, 13757–13762. [Google Scholar] [CrossRef] [Green Version]
- Clements, K.D.; German, D.P.; Piché, J.; Tribollet, A.; Choat, J.H. Integrating ecological roles and trophic diversification on coral reefs: Multiple lines of evidence identify parrotfishes as microphages. Biol. J. Linn. Soc. 2017, 120, 729–751. [Google Scholar] [CrossRef]
- McArdle, B.H.; Anderson, M.J. Fitting multivariate models to community data: A comment on distance-based redundancy analysis. Ecology 2001, 82, 290–297. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Michael, F.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Peter, S.; et al. vegan: Community Ecology Package. 2019. Available online: inecol.mx (accessed on 9 August 2021).
- Parada, A.E.; Needham, D.M.; Fuhrman, J.A. Every base matters: Assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 2016, 18, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- R Core Team R. A language and environment for statistical computing. 2020. Available online: r-project.org (accessed on 9 August 2021).
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Glassman, S.I.; Martiny, J.B.H. Broadscale Ecological Patterns Are Robust to Use of Exact. mSphere 2018, 3, e00148-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, 590–596. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.; Loman, N.J.; Walker, A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014, 12, 87. [Google Scholar] [CrossRef] [Green Version]
- Cariveau, D.P.; Elijah Powell, J.; Koch, H.; Winfree, R.; Moran, N.A. Variation in gut microbial communities and its association with pathogen infection in wild bumble bees (Bombus). ISME J. 2014, 8, 2369–2379. [Google Scholar] [CrossRef]
- Madhusoodanan, J. News Feature: Do hosts and their microbes evolve as a unit? Proc. Natl. Acad. Sci. USA 2019, 116, 14391–14394. [Google Scholar] [CrossRef] [Green Version]
- Berg, R. The indigenous gastrointestinal microflora. Trends Microbiol. 1996, 4, 430–435. [Google Scholar] [CrossRef]
- Martinson, V.G.; Danforth, B.N.; Minckley, R.L.; Rueppell, O.; Tingek, S.; Moran, N.A. A simple and distinctive microbiota associated with honey bees and bumble bees. Mol. Ecol. 2011, 20, 619–628. [Google Scholar] [CrossRef]
- Koch, H.; Abrol, D.P.; Li, J.; Schmid-Hempel, P. Diversity and evolutionary patterns of bacterial gut associates of corbiculate bees. Mol. Ecol. 2013, 22, 2028–2044. [Google Scholar] [CrossRef]
- Magurran, A.E.; Henderson, P.A. Explaining the excess of rare species in natural species abundance distributions. Nature 2003, 422, 714–716. [Google Scholar] [CrossRef] [PubMed]
- Krebs, C.J. Ecological Methodology; Pearson; Addison-Wesley Educational Publishers: Menlo Park, 1999; ISBN 9780321021731. [Google Scholar]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwig, W.; Strunk, O.; Westram, R.; Richter, L.; Meier, H.; Yadhukumar, A.; Buchner, A.; Lai, T.; Steppi, S.; Jacob, G.; et al. ARB: A software environment for sequence data. Nucleic Acids Res. 2004, 32, 1363–1371. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef] [PubMed]
- Chao, A.; Lee, S.-M.; Chen, T.-C. A generalized Good’s nonparametric coverage estimator. Chin. J. Math. 1988, 16, 189–199. [Google Scholar]
- Jost, L. Entropy and diversity. Oikos 2006, 113, 363–375. [Google Scholar] [CrossRef]
- Wemheuer, F.; Taylor, J.A.; Daniel, R.; Johnston, E.; Meinicke, P.; Thomas, T.; Wemheuer, B. Tax4Fun2: Prediction of habitat-specific functional profiles and functional redundancy based on 16S rRNA gene sequences. Environ. Microbiome 2020, 15, 11. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Chong-Seng, K.M.; Mannering, T.D.; Pratchett, M.S.; Bellwood, D.R.; Graham, N.A.J. The influence of coral reef benthic condition on associated fish assemblages. PLoS ONE 2012, 7, e42167. [Google Scholar] [CrossRef]
- Graham, N.A.J.; Robinson, J.P.W.; Smith, S.E.; Govinden, R.; Gendron, G.; Wilson, S.K. Changing role of coral reef marine reserves in a warming climate. Nat. Commun. 2020, 11, 2000. [Google Scholar] [CrossRef] [PubMed]
- Silveira, C.B.; Cavalcanti, G.S.; Walter, J.M.; Silva-Lima, A.W.; Dinsdale, E.A.; Bourne, D.G.; Thompson, C.C.; Thompson, F.L. Microbial processes driving coral reef organic carbon flow. FEMS Microbiol. Rev. 2017, 41, 575–595. [Google Scholar] [CrossRef] [Green Version]
- McDole, T.; Nulton, J.; Barott, K.L.; Felts, B.; Hand, C.; Hatay, M.; Lee, H.; Nadon, M.O.; Nosrat, B.; Salamon, P.; et al. Assessing Coral Reefs on a Pacific-Wide Scale Using the Microbialization Score. PLoS ONE 2012, 7, e43233. [Google Scholar] [CrossRef]
- Meirelles, P.M.; Amado-Filho, G.M.; Pereira-Filho, G.H.; Pinheiro, H.T.; De Moura, R.L.; Joyeux, J.C.; Mazzei, E.F.; Bastos, A.C.; Edwards, R.A.; Dinsdale, E.; et al. Baseline assessment of mesophotic reefs of the Vitória-Trindade Seamount Chain based on water quality, microbial diversity, benthic cover and fish biomass data. PLoS ONE 2015, 10, e0130084. [Google Scholar] [CrossRef]
- Haas, A.F.; Fairoz, M.F.M.; Kelly, L.W.; Nelson, C.E.; Dinsdale, E.A.; Edwards, R.A.; Giles, S.; Hatay, M.; Hisakawa, N.; Knowles, B.; et al. Global microbialization of coral reefs. Nat. Microbiol. 2016, 1, 16042. [Google Scholar] [CrossRef]
- Roach, T.N.F.; Abieri, M.L.; George, E.E.; Knowles, B.; Naliboff, D.S.; Smurthwaite, C.A.; Kelly, L.W.; Haas, A.F.; Rohwer, F.L. Microbial bioenergetics of coral-algal interactions. PeerJ 2017, 2017, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, A.F.; Nelson, C.E.; Kelly, L.W.; Carlson, C.A.; Rohwer, F.; Leichter, J.J.; Wyatt, A.; Smith, J.E. Effects of coral reef benthic primary producers on dissolved organic carbon and microbial activity. PLoS ONE 2011, 6, e27973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vega Thurber, R.; Burkepile, D.E.; Correa, A.M.S.; Thurber, A.R.; Shantz, A.A.; Welsh, R.; Pritchard, C.; Rosales, S. Macroalgae Decrease Growth and Alter Microbial Community Structure of the Reef-Building Coral, Porites astreoides. PLoS ONE 2012, 7, e44246. [Google Scholar] [CrossRef] [Green Version]
- Munday, P.L.; Jones, G.P.; Pratchett, M.S.; Williams, A.J. Climate change and the future for coral reef fishes. Fish. Fish. 2008, 9, 261–285. [Google Scholar] [CrossRef]
- Munday, P.; Jones, G.; Caley, M. Habitat specialisation and the distribution and abundance of coral-dwelling gobies. Mar. Ecol. Prog. Ser. 1997, 152, 227–239. [Google Scholar] [CrossRef] [Green Version]
- Gardiner, N.; Jones, G. Habitat specialisation and overlap in a guild of coral reef cardinalfishes (Apogonidae). Mar. Ecol. Prog. Ser. 2005, 305, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Pratchett, M.S. Dietary overlap among coral-feeding butterflyfishes (Chaetodontidae) at Lizard Island, northern Great Barrier Reef. Mar. Biol. 2005, 148, 373–382. [Google Scholar] [CrossRef]
- Bellwood, D.R.; Hughes, T.P.; Hoey, A.S. Sleeping Functional Group Drives Coral-Reef Recovery. Curr. Biol. 2006, 16, 2434–2439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feary, D.; Almany, G.; Jones, G.; McCormick, M. Coral degradation and the structure of tropical reef fish communities. Mar. Ecol. Prog. Ser. 2007, 333, 243–248. [Google Scholar] [CrossRef] [Green Version]
- Silveira, C.B.; Silva-Lima, A.W.; Francini-Filho, R.B.; Marques, J.S.M.; Almeida, M.G.; Thompson, C.C.; Rezende, C.E.; Paranhos, R.; Moura, R.L.; Salomon, P.S.; et al. Microbial and sponge loops modify fish production in phase-shifting coral reefs. Environ. Microbiol. 2015, 17, 3832–3846. [Google Scholar] [CrossRef] [PubMed]
- Alexander, B.E.; Liebrand, K.; Osinga, R.; van der Geest, H.G.; Admiraal, W.; Cleutjens, J.P.M.; Schutte, B.; Verheyen, F.; Ribes, M.; van Loon, E.; et al. Cell Turnover and Detritus Production in Marine Sponges from Tropical and Temperate Benthic Ecosystems. PLoS ONE 2014, 9, e109486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, B.; de Goeij, J.M.; Vermeij, M.J.A.; Mulders, Y.; van der Ent, E.; Ribes, M.; van Duyl, F.C. Natural Diet of Coral-Excavating Sponges Consists Mainly of Dissolved Organic Carbon (DOC). PLoS ONE 2014, 9, e90152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.E.; Shaw, M.; Edwards, R.A.; Obura, D.; Pantos, O.; Sala, E.; Sandin, S.A.; Smriga, S.; Hatay, M.; Rohwer, F.L. Indirect effects of algae on coral: Algae-mediated, microbe-induced coral mortality. Ecol. Lett. 2006, 9, 835–845. [Google Scholar] [CrossRef]
- Barott, K.L.; Rohwer, F.L. Unseen players shape benthic competition on coral reefs. Trends Microbiol. 2012, 20, 621–628. [Google Scholar] [CrossRef]
- Morrow, K.M.; Ritson-Williams, R.; Ross, C.; Liles, M.R.; Paul, V.J. Macroalgal Extracts Induce Bacterial Assemblage Shifts and Sublethal Tissue Stress in Caribbean Corals. PLoS ONE 2012, 7, e44859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beatty, D.; Clements, C.; Stewart, F.; Hay, M. Intergenerational effects of macroalgae on a reef coral: Major declines in larval survival but subtle changes in microbiomes. Mar. Ecol. Prog. Ser. 2018, 589, 97–114. [Google Scholar] [CrossRef]
- Nelson, C.E.; Goldberg, S.J.; Wegley Kelly, L.; Haas, A.F.; Smith, J.E.; Rohwer, F.; Carlson, C.A. Coral and macroalgal exudates vary in neutral sugar composition and differentially enrich reef bacterioplankton lineages. ISME J. 2013, 7, 962–979. [Google Scholar] [CrossRef]
- Meirelles, P.M.; Soares, A.C.; Oliveira, L.; Leomil, L.; Appolinario, L.R.; Francini-Filho, R.B.; De Moura, R.L.; De Barros Almeida, R.T.; Salomon, P.S.; Amado-Filho, G.M.; et al. Metagenomics of coral reefs under phase shift and high hydrodynamics. Front. Microbiol. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Aiken, D.E.; Waddy, S.L. Aquaculture. In Biology of the Lobster; Elsevier: Amsterdam, The Netherlands, 1995; pp. 153–175. [Google Scholar]
- Ghanbari, M.; Kneifel, W.; Domig, K.J. A new view of the fish gut microbiome: Advances from next-generation sequencing. Aquaculture 2015, 448, 464–475. [Google Scholar] [CrossRef]
- Egerton, S.; Culloty, S.; Whooley, J.; Stanton, C.; Ross, R.P. The gut microbiota of marine fish. Front. Microbiol. 2018, 9, 873. [Google Scholar] [CrossRef]
- Barelli, C.; Albanese, D.; Donati, C.; Pindo, M.; Dallago, C.; Rovero, F.; Cavalieri, D.; Michael Tuohy, K.; Christine Hauffe, H.; De Filippo, C. Habitat fragmentation is associated to gut microbiota diversity of an endangered primate: Implications for conservation. Sci. Rep. 2015, 5, 14862. [Google Scholar] [CrossRef] [Green Version]
- Hayden, B.; Palomares, M.L.D.; Smith, B.E.; Poelen, J.H. Biological and environmental drivers of trophic ecology in marine fishes-a global perspective. Sci. Rep. 2019, 9, 11415. [Google Scholar] [CrossRef] [PubMed]
- Green, A.L.; Maypa, A.P.; Almany, G.R.; Rhodes, K.L.; Weeks, R.; Abesamis, R.A.; Gleason, M.G.; Mumby, P.J.; White, A.T. Larval dispersal and movement patterns of coral reef fishes, and implications for marine reserve network design. Biol. Rev. 2015, 90, 1215–1247. [Google Scholar] [CrossRef]
- Zaneveld, J.R.; McMinds, R.; Thurber, R.V. Stress and stability: Applying the Anna Karenina principle to animal microbiomes. Nat. Microbiol. 2017, 2, 17121. [Google Scholar] [CrossRef] [PubMed]
- Van Soest, J.P. Nutritional ecology of the ruminant; Cornell University Press: Ithaca, NY, USA, 1994. [Google Scholar]
- Jones, J.; DiBattista, J.D.; Stat, M.; Bunce, M.; Boyce, M.C.; Fairclough, D.V.; Travers, M.J.; Huggett, M.J. The microbiome of the gastrointestinal tract of a range-shifting marine herbivorous fish. Front. Microbiol. 2018, 9, 2000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Wu, H.; Li, Z.; Li, Y.; Wang, S.; Zhu, D.; Wen, X.; Li, S. Effects of dietary supplementation of Ulva pertusa and non-starch polysaccharide enzymes on gut microbiota of Siganus canaliculatus. J. Oceanol. Limnol. 2018, 36, 438–449. [Google Scholar] [CrossRef]
- Miller, D.A.; Suen, G.; Bruce, D.; Copeland, A.; Cheng, J.-F.; Detter, C.; Goodwin, L.A.; Han, C.S.; Hauser, L.J.; Land, M.L.; et al. Complete Genome Sequence of the Cellulose-Degrading Bacterium Cellulosilyticum lentocellum. J. Bacteriol. 2011, 193, 2357–2358. [Google Scholar] [CrossRef] [Green Version]
- Mountfort, D.O.; Campbell, J.; Clements, K.D. Hindgut Fermentation in Three Species of Marine Herbivorous Fish. Appl. Environ. Microbiol. 2002, 68, 1374–1380. [Google Scholar] [CrossRef] [Green Version]
- Clements, K.D.; Choat, J.H. Fermentation in Tropical Marine Herbivorous Fishes. Physiol. Zool. 1995, 68, 355–378. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diet | Family | Genus | Species | Reef condition | |
|---|---|---|---|---|---|
| Algae (n = 29) | _ | _ | _ | R2 = 0.13 (***) | R2 = 0.07 (**) |
| Fish (n = 99) | R2 = 0.04 (***) | R2 = 0.16 (***) | R2 = 0.27 (***) | R2 = 0.46 (***) | R2 = 0.02 (***) |
| Herbivores (n = 44) | _ | R2 = 0.14 (***) | R2 = 0.24 (***) | R2 = 0.43 (***) | R2 = 0.07 (***) |
| Scarinae (n = 22) | _ | _ | R2 = 0.14 (**) | _ | R2 = 0.09 (**) |
| S.ghobban (n = 7) | _ | _ | _ | _ | R2 = 0.28 (NS.) |
| Siganidae (n = 17) | _ | _ | _ | R2 = 0.20 (**) | _ |
| Carnivores (n = 53) | _ | R2 = 0.10 (**) | R2 = 0.24 (**) | R2 = 0.44 (*) | R2 = 0.02 (NS.) |
| Lutjanidae (n = 14) | _ | _ | R2 = 0.08 (NS.) | R2 = 0.34 (NS.) | R2 = 0.10 (NS.) |
| A.virescens (n = 7) | _ | _ | _ | _ | R2 = 0.17 (NS.) |
| Lethrinidae (n = 26) | _ | _ | _ | R2 = 0.21 (NS.) | R2 = 0.05 (NS.) |
| L.mahsena (n = 10) | _ | _ | _ | _ | R2 = 0.12 (NS.) |
| L.enigmaticus (n = 6) | _ | _ | _ | _ | R2 = 0.17 (NS.) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheutin, M.-C.; Villéger, S.; Hicks, C.C.; Robinson, J.P.W.; Graham, N.A.J.; Marconnet, C.; Restrepo, C.X.O.; Bettarel, Y.; Bouvier, T.; Auguet, J.-C. Microbial Shift in the Enteric Bacteriome of Coral Reef Fish Following Climate-Driven Regime Shifts. Microorganisms 2021, 9, 1711. https://doi.org/10.3390/microorganisms9081711
Cheutin M-C, Villéger S, Hicks CC, Robinson JPW, Graham NAJ, Marconnet C, Restrepo CXO, Bettarel Y, Bouvier T, Auguet J-C. Microbial Shift in the Enteric Bacteriome of Coral Reef Fish Following Climate-Driven Regime Shifts. Microorganisms. 2021; 9(8):1711. https://doi.org/10.3390/microorganisms9081711
Chicago/Turabian StyleCheutin, Marie-Charlotte, Sébastien Villéger, Christina C. Hicks, James P. W. Robinson, Nicholas A. J. Graham, Clémence Marconnet, Claudia Ximena Ortiz Restrepo, Yvan Bettarel, Thierry Bouvier, and Jean-Christophe Auguet. 2021. "Microbial Shift in the Enteric Bacteriome of Coral Reef Fish Following Climate-Driven Regime Shifts" Microorganisms 9, no. 8: 1711. https://doi.org/10.3390/microorganisms9081711
APA StyleCheutin, M.-C., Villéger, S., Hicks, C. C., Robinson, J. P. W., Graham, N. A. J., Marconnet, C., Restrepo, C. X. O., Bettarel, Y., Bouvier, T., & Auguet, J.-C. (2021). Microbial Shift in the Enteric Bacteriome of Coral Reef Fish Following Climate-Driven Regime Shifts. Microorganisms, 9(8), 1711. https://doi.org/10.3390/microorganisms9081711