
Recovering Escherichia coli Plasmids in the Absence of Long-Read Sequencing Data

 and
and
Abstract
:1. Introduction
2. Materials and Methods
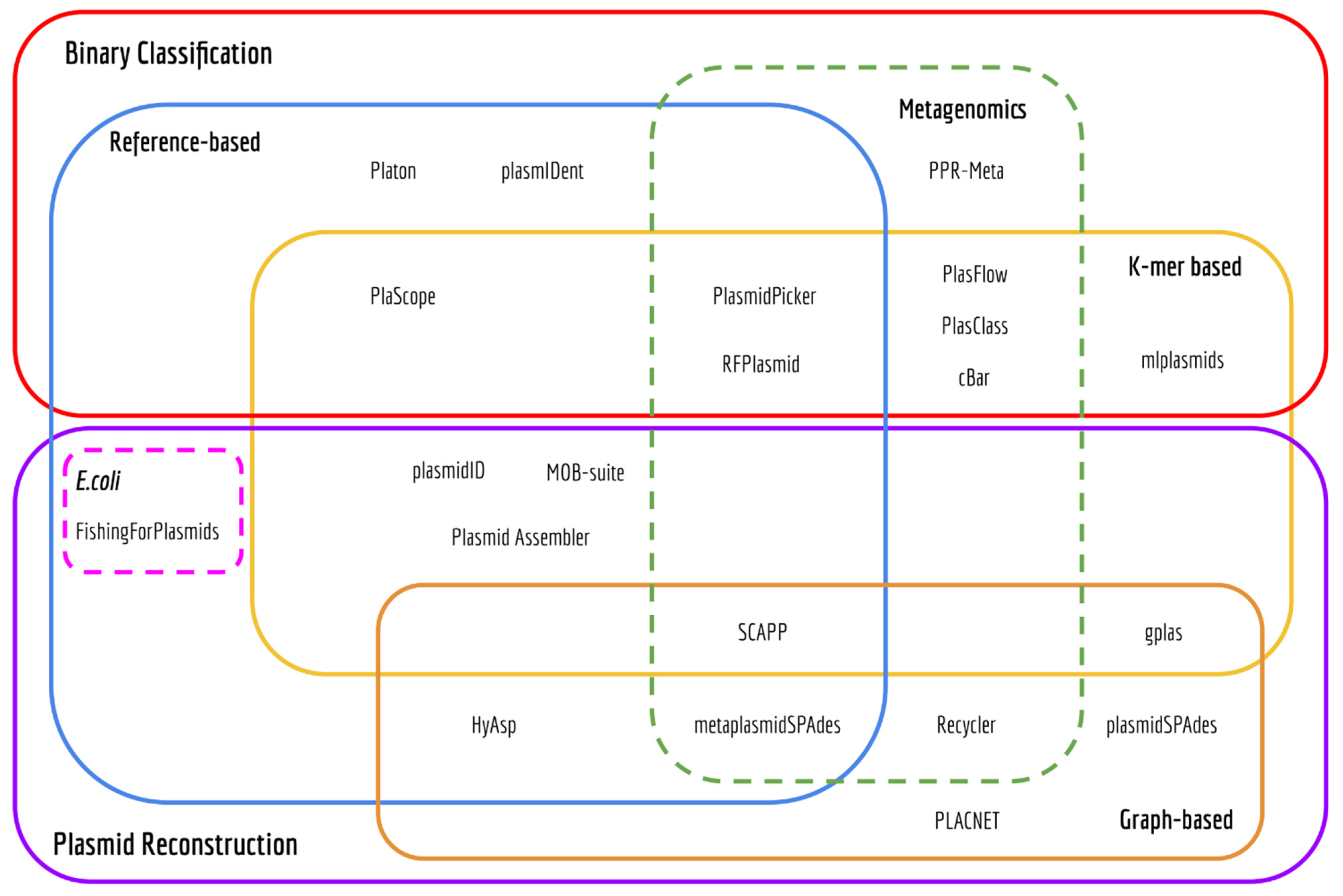
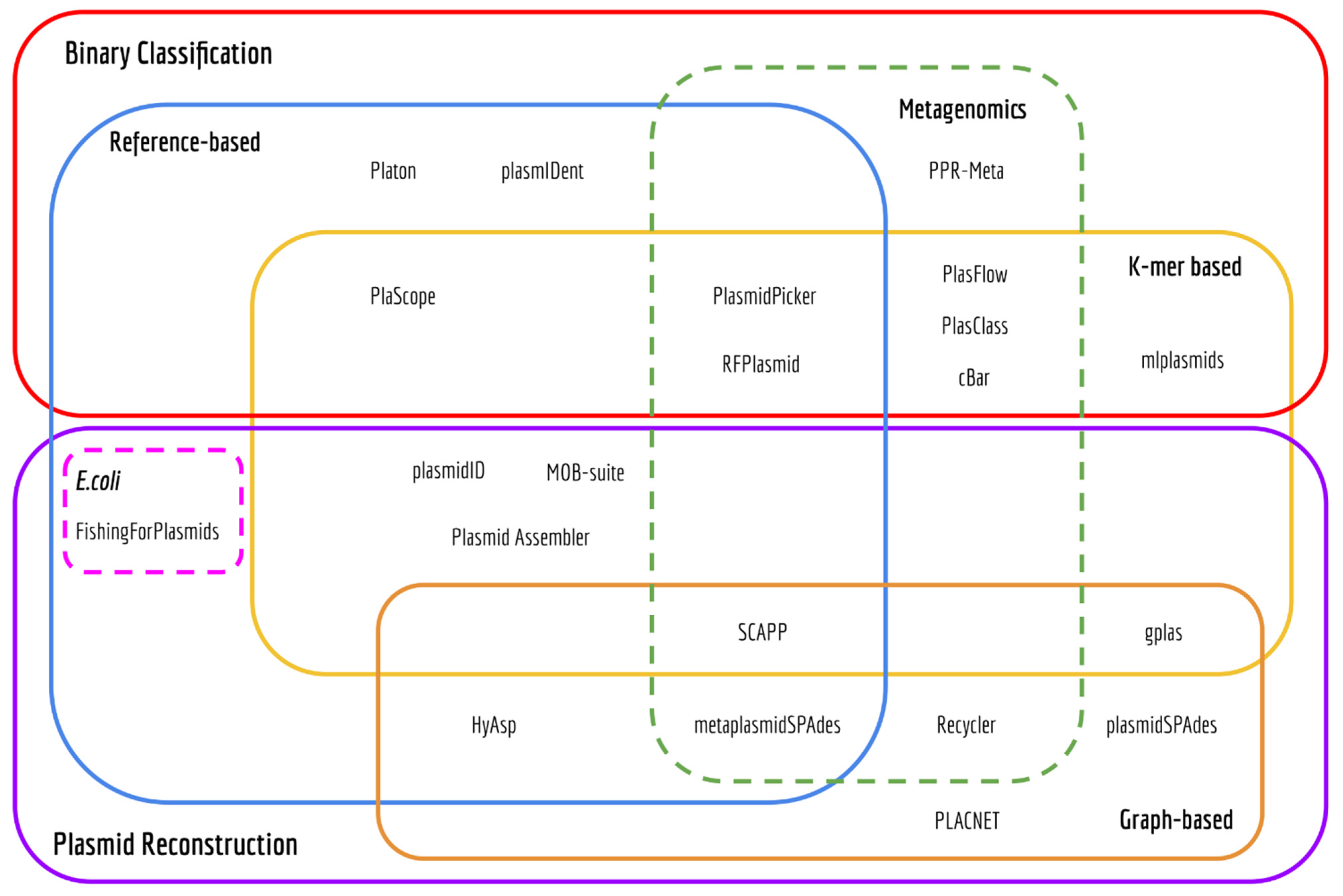
2.1. Review of Plasmid Prediction Tools
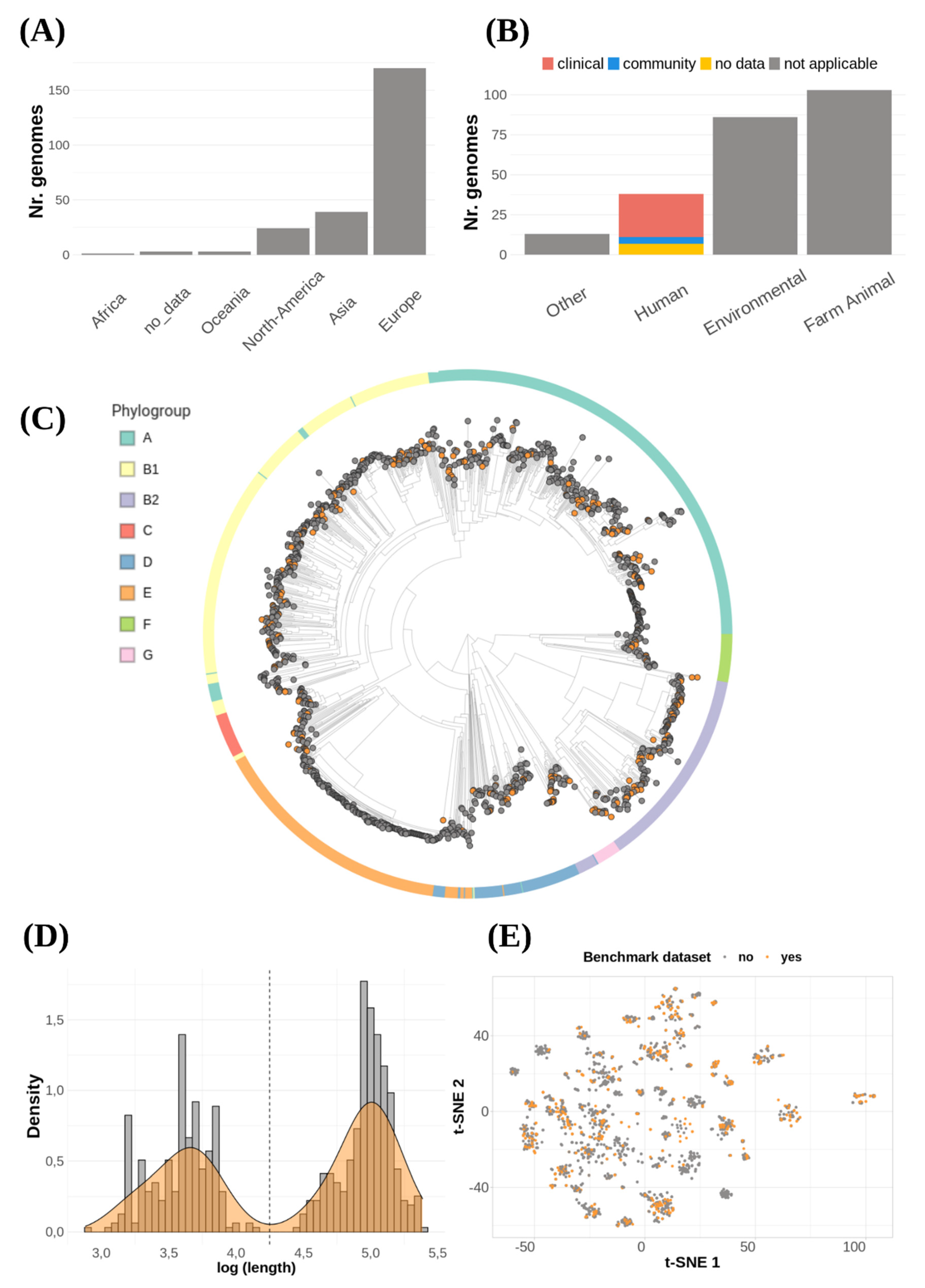
2.2. Retrieving E. coli Complete Genomes and Metadata from NCBI Database
2.3. Phylogenetic Analysis
2.4. Benchmark Data Set Selection
2.5. Evaluating Plasmid Diversity in Benchmarking Data
2.6. Plasmid Predictions
2.7. Analysis of the Plasmid Bins Composition
2.8. Evaluating Maximum Theoretical Recall for Each Reference Plasmid
2.9. Antibiotic Resistance Gene (ARG) Prediction
2.10. Evaluating Reconstruction of ARG Plasmids
3. Results
3.1. Computational Methods to Predict the Plasmidome or Distinct Plasmids
3.1.1. Binary Classification Tools
3.1.2. Plasmid Reconstruction Tools
3.2. The Benchmark Data Set Represents the Diversity of Sequenced Plasmids
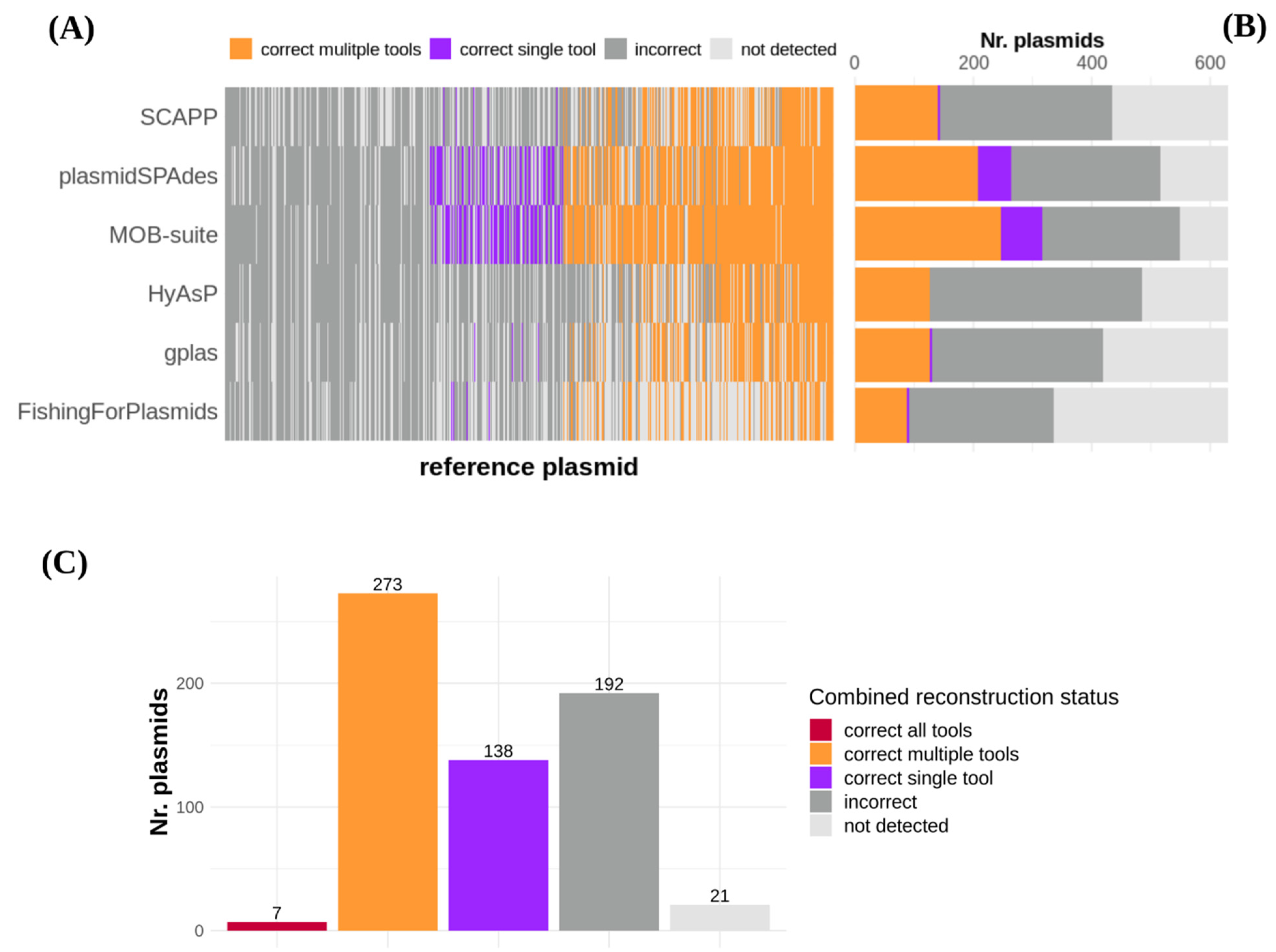
3.3. A Third of All Plasmids Could Not Be Correctly Reconstructed by Any of the Tools
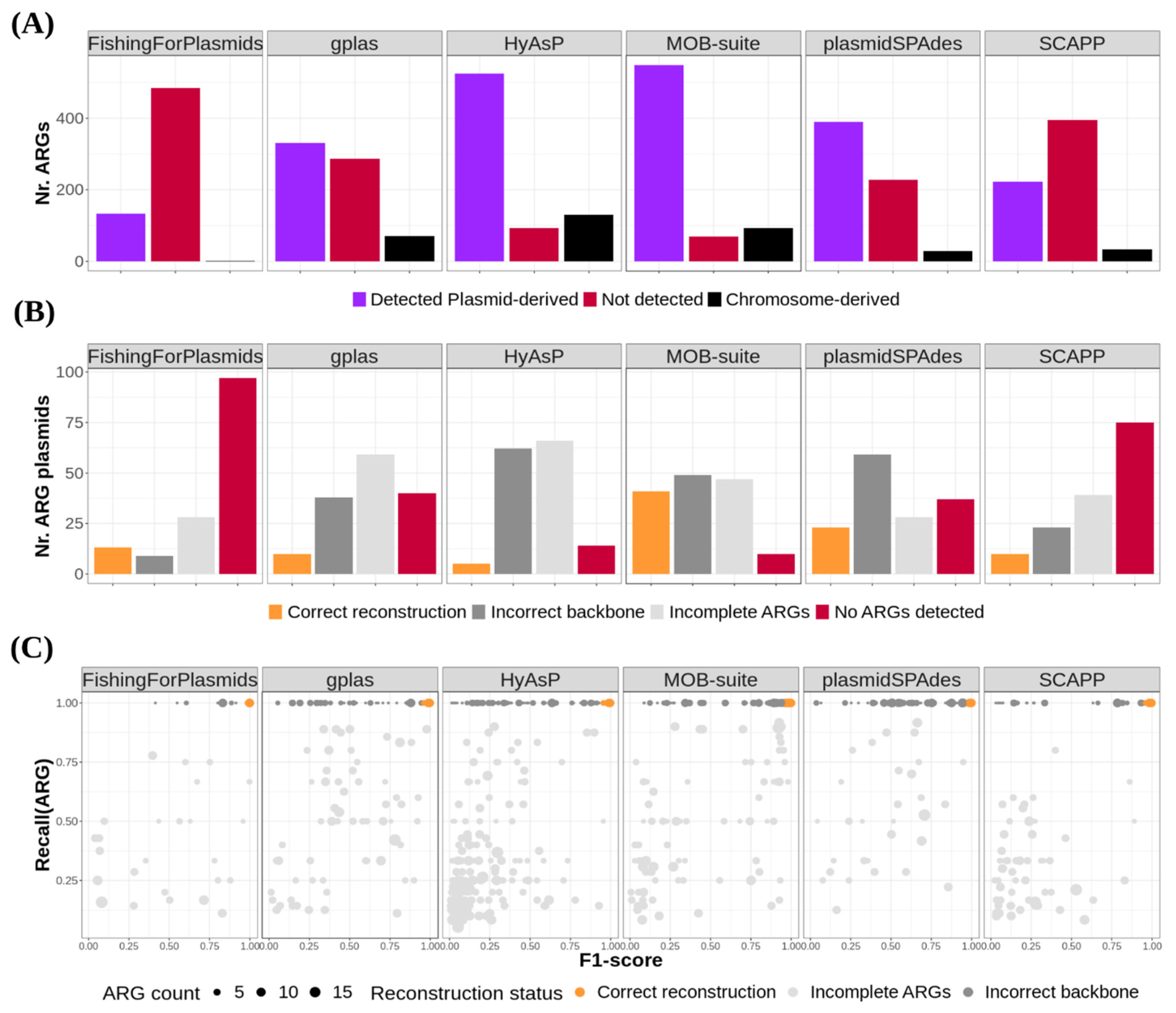
3.4. Plasmids Carrying Antibiotic Resistance Genes Were Difficult to Reconstruct for All Tools
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- The Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Ochoa, T.J.; Barletta, F.; Contreras, C.; Mercado, E. New insights into the epidemiology of enteropathogenic Escherichia coli infection. Trans. R. Soc. Trop. Med. Hyg. 2008, 102, 852–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biran, D.; Ron, E.Z. Extraintestinal Pathogenic Escherichia coli. In Current Topics in Microbiology and Immunology; Springer: Cham, Switzerland, 2018; Volume 416, pp. 149–161. [Google Scholar]
- European Centre for Disease Prevention and Control. Healthcare-associated infections acquired in intensive care units. In ECDC Annual Epidemiological Report for 2017; ECDC: Stockholm, Sweden, 2019. [Google Scholar]
- Laupland, K.B.; Church, D.L. Population-Based Epidemiology and Microbiology of Community-Onset Bloodstream Infections. Clin. Microbiol. Rev. 2014, 27, 647–664. [Google Scholar] [CrossRef] [Green Version]
- Denamur, E.; Clermont, O.; Bonacorsi, S.; Gordon, D. The population genetics of pathogenic Escherichia coli. Nat. Rev. Genet. 2021, 19, 37–54. [Google Scholar] [CrossRef] [PubMed]
- Cassini, A.; Högberg, L.D.; Plachouras, D.; Quattrocchi, A.; Hoxha, A.; Simonsen, G.S.; Colomb-Cotinat, M.; Kretzschmar, M.E.; Devleesschauwer, B.; Cecchini, M.; et al. Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European Economic Area in 2015: A population-level modelling analysis. Lancet Infect. Dis. 2019, 19, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Stephens, C.; Arismendi, T.; Wright, M.; Hartman, A.; Gonzalez, A.; Gill, M.; Pandori, M.; Hess, D. F Plasmids Are the Major Carriers of Antibiotic Resistance Genes in Human-Associated Commensal Escherichia coli. mSphere 2020, 5. [Google Scholar] [CrossRef]
- Matamoros, S.; Van Hattem, J.M.; Arcilla, M.S.; Willemse, N.; Melles, D.C.; Penders, J.; Vinh, T.N.; Hoa, N.T.; Bootsma, M.C.J.; Van Genderen, P.J.; et al. Global phylogenetic analysis of Escherichia coli and plasmids carrying the mcr-1 gene indicates bacterial diversity but plasmid restriction. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Kaper, J.B. Pathogenic Escherichia coli. Int. J. Med. Microbiol. 2005, 295, 355–356. [Google Scholar] [CrossRef]
- Arredondo-Alonso, S.; Willems, R.; van Schaik, W.; Schürch, A.C. On the (im)possibility of reconstructing plasmids from whole-genome short-read sequencing data. Microb. Genom. 2017, 3, e000128. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.; Gorrie, C.; Holt, K. Completing bacterial genome assemblies with multiplex MinION sequencing. Microb. Genom. 2017, 3, e000132. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carattoli, A.; Zankari, E.; García-Fernández, A.; Larsen, M.V.; Lund, O.; Villa, L.; Aarestrup, F.; Hasman, H. In Silico Detection and Typing of Plasmids using PlasmidFinder and Plasmid Multilocus Sequence Typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ten Doesschate, T.; Abbott, I.J.; Willems, R.J.L.; Top, J.; Rogers, M.R.; Bonten, M.M.; Paganelli, F.L. In vivo acquisition of fosfomycin resistance in Escherichia coli by fosA transmission from commensal flora. J. Antimicrob. Chemother. 2019, 74, 3630–3632. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.M.; Eng, W.W.H.; Dhanoa, A. First genomic insights into carbapenem-resistant Klebsiella pneumoniae from Malaysia. J. Glob. Antimicrob. Resist. 2020, 20, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Van Driessche, L.; Vanneste, K.; Bogaerts, B.; De Keersmaecker, S.C.; Roosens, N.H.; Haesebrouck, F.; De Cremer, L.; Deprez, P.; Pardon, B.; Boyen, F. Isolation of Drug-Resistant Gallibacterium anatis from Calves with Unresponsive Bronchopneumonia, Belgium. Emerg. Infect. Dis. 2020, 26, 721–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.K.; Shin, H.; Han, D.; Hur, H.G.; Uno, T. Metagenomic analysis reveals the prevalence and persistence of antibiotic- and heavy met-al-resistance genes in wastewater treatment plant. J. Microbiol. 2018, 56, 408–415. [Google Scholar] [CrossRef]
- Arredondo-Alonso, S.; Top, J.; McNally, A.; Puranen, S.; Pesonen, M.; Pensar, J.; Marttinen, P.; Braat, J.C.; Rogers, M.R.C.; van Schaik, W.; et al. Plasmids Shaped the Recent Emergence of the Major Nosocomial Pathogen Enterococcus faecium. mBio 2020, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arredondo-Alonso, S.; Top, J.; Corander, J.; Willems, R.J.; Schürch, A.C. Mode and dynamics of vanA-type vancomycin-resistance dissemination in Dutch hospitals. Genome Med. 2020, 13, 1–8. [Google Scholar]
- Kans, J. Entrez Direct: E-utilities on the Unix Command Line. In Entrez Programming Utilities Help [Internet]; 2013 Apr 23 [Updated 2021 Jul 16]; National Center for Biotechnology Information: Bethesda, MD, USA, 2010. [Google Scholar]
- Beghain, J.; Bridier-Nahmias, A.; Le Nagard, H.; Denamur, E.; Clermont, O. ClermonTyping: An easy-to-use and accurate in silico method for Escherichia genus strain phylotyping. Microb. Genom. 2018, 4, e000192. [Google Scholar] [CrossRef]
- Lees, J.A.; Harris, S.R.; Tonkin-Hill, G.; Gladstone, R.A.; Lo, S.W.; Weiser, J.N.; Corander, J.; Bentley, S.D.; Croucher, N.J. Fast and flexible bacterial genomic epidemiology with PopPUNK. Genome Res. 2019, 29, 304–316. [Google Scholar] [CrossRef] [Green Version]
- Argimón, S.; Abudahab, K.; Goater, R.J.E.; Fedosejev, A.; Bhai, J.; Glasner, C.; Feil, E.J.; Holden, M.T.G.; Yeats, C.A.; Grundmann, H.; et al. Microreact: Visualizing and sharing data for genomic epidemiology and phylo-geography. Microb. Genom. 2016, 2, e000093. [Google Scholar]
- Walk, S.T.; Alm, E.W.; Gordon, D.M.; Ram, J.L.; Toranzos, G.A.; Tiedje, J.M.; Whittam, T.S. Cryptic Lineages of the Genus Escherichia. Appl. Environ. Microbiol. 2009, 75, 6534–6544. [Google Scholar] [CrossRef] [Green Version]
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes De Novo Assembler. Curr. Protoc. Bioinform. 2020, 70, e102. [Google Scholar] [CrossRef] [PubMed]
- Royer, G.; Decousser, J.W.; Branger, C.; Dubois, M.; Médigue, C.; Denamur, E.; Vallenet, D. PlaScope: A targeted approach to assess the plasmidome from genome assemblies at the species level. Microb. Genom. 2018, 4. [Google Scholar] [CrossRef] [PubMed]
- Schwengers, O.; Barth, P.; Falgenhauer, L.; Hain, T.; Chakraborty, T.; Goesmann, A. Platon: Identification and characterization of bacterial plasmid contigs in short-read draft assemblies exploiting protein sequence-based replicon distribution scores. Microb. Genom. 2020, 6, e000398. [Google Scholar] [CrossRef]
- Arredondo-Alonso, S.; Rogers, M.R.C.; Braat, J.C.; Verschuuren, T.D.; Top, J.; Corander, J.; Willems, R.J.L.; Schürch, A.C. mlplasmids: A user-friendly tool to predict plasmid- and chromosome-derived sequences for single species. Microb. Genom. 2018, 4, e000224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krawczyk, P.S.; Lipinski, L.; Dziembowski, A. PlasFlow: Predicting plasmid sequences in metagenomic data using genome signatures. Nucleic Acids Res. 2018, 46, e35. [Google Scholar] [CrossRef] [Green Version]
- Pellow, D.; Mizrahi, I.; Shamir, R. PlasClass improves plasmid sequence classification. PLoS Comput. Biol. 2020, 16, e1007781. [Google Scholar] [CrossRef] [Green Version]
- van Bloois, L.V.D.G.; Wagenaar, J.A.; Zomer, A.L. RFPlasmid: Predicting plasmid sequences from short read assembly data using machine learning. bioRxiv 2020. [Google Scholar] [CrossRef]
- Fang, Z.; Tan, J.; Wu, S.; Li, M.; Xu, C.; Xie, Z.; Zhu, H. PPR-Meta: A tool for identifying phages and plasmids from metagenomic fragments using deep learning. GigaScience 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Antipov, D.; Hartwick, N.; Shen, M.; Rayko, M.; Lapidus, A.; Pevzner, P.A. plasmidSPAdes: Assembling plasmids from whole genome sequencing data. Bioinformatics 2016, 32, 3380–3387. [Google Scholar] [CrossRef] [Green Version]
- Rozov, R.; Brown Kav, A.; Bogumil, D.; Shterzer, N.; Halperin, E.; Mizrahi, I.; Shamir, R. Recycler: An algorithm for detecting plasmids from de novo assembly graphs. Bioinformatics 2017, 33, 475–482. [Google Scholar]
- Arredondo-Alonso, S.; Bootsma, M.; Hein, Y.; Rogers, M.R.C.; Corander, J.; Willems, R.J.L.; Schürch, A.C. gplas: A comprehensive tool for plasmid analysis using short-read graphs. Bioinformatics 2020, 36, 3874–3876. [Google Scholar] [CrossRef] [PubMed]
- Robertson, J.; Nash, J.H.E. MOB-suite: Software tools for clustering, reconstruction and typing of plasmids from draft assemblies. Microb. Genom. 2018, 4, e000206. [Google Scholar] [CrossRef]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carattoli, A.; Hasman, H. PlasmidFinder and In Silico pMLST: Identification and Typing of Plasmid Replicons in Whole-Genome Sequencing (WGS). Methods Mol. Biol. 2020, 2075, 285–294. [Google Scholar] [PubMed]
- Pellow, D.; Zorea, A.; Probst, M.; Furman, O.; Segal, A.; Mizrahi, I.; Shamir, R. SCAPP: An algorithm for improved plasmid assembly in metagenomes. Microbiome 2021, 9, 1–12. [Google Scholar] [CrossRef]
- Goswami, C.; Fox, S.; Holden, M.T.; Connor, M.; Leanord, A.; Evans, T.J. Origin, maintenance and spread of antibiotic resistance genes within plasmids and chromosomes of bloodstream isolates of Escherichia coli. Microb. Genom. 2020, 6, mgen000353. [Google Scholar] [CrossRef]
- Peter, S.; Bosio, M.; Gross, C.; Bezdan, D.; Gutierrez, J.; Oberhettinger, P.; Liese, J.; Vogel, W.; Dörfel, D.; Berger, L.; et al. Tracking of Antibiotic Resistance Transfer and Rapid Plasmid Evolution in a Hospital Setting by Nanopore Sequencing. mSphere 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Tang, N.; Chen, D.; Sun, K.; Lan, R.; Zhang, W.; Zhou, H.; Yuan, M.; Chen, X.; Zhao, X.; et al. A Nosocomial Respiratory Infection Outbreak of Carbapenem-Resistant ST131 With Multi-ple Transmissible Carrying Plasmids. Front. Microbiol. 2020, 11, 2068. [Google Scholar] [CrossRef]
- Paramita, R.I.; Nelwan, E.J.; Fadilah, F.; Renesteen, E.; Puspandari, N.; Erlina, L. Genome-based characterization of Escherichia coli causing bloodstream infection through next-generation sequencing. PLoS ONE 2020, 15, e0244358. [Google Scholar] [CrossRef]
- Mughini-Gras, L.; Dorado-García, A.; van Duijkeren, E.; van den Bunt, G.; Dierikx, C.M.; Bonten, M.J.M.; Bootsma, M.C.J.; Schmitt, H.; Hald, T.; Evers, E.G.; et al. Attributable sources of community-acquired carriage of Escherich-ia coli containing β-lactam antibiotic resistance genes: A population-based modelling study. Lancet Planet Health 2019, 3, e357–e369. [Google Scholar] [CrossRef] [Green Version]
- Ludden, C.; Raven, K.E.; Jamrozy, D.; Gouliouris, T.; Blane, B.; Coll, F.; de Goffau, M.; Naydenova, P.; Horner, C.; Hernandez-Garcia, J.; et al. One Health Genomic Surveillance of Escherichia coli Demonstrates Distinct Lineages and Mobile Genetic Elements in Isolates from Humans versus Livestock. mBio 2019, 10, e02693-18. [Google Scholar] [CrossRef] [Green Version]
- Hendriksen, R.S.; Bortolaia, V.; Tate, H.; Tyson, G.H.; Aarestrup, F.; McDermott, P.F. Using Genomics to Track Global Antimicrobial Resistance. Front. Public Health 2019, 7, 242. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Beltrán, J.; DelaFuente, J.; León-Sampedro, R.; MacLean, R.C.; Millán, Á.S. Beyond horizontal gene transfer: The role of plasmids in bacterial evolution. Nat. Rev. Genet. 2021, 19, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Che, Y.; Yang, Y.; Xu, X.; Břinda, K.; Polz, M.F.; Hanage, W.P.; Zhang, T. Conjugative plasmids interact with insertion sequences to shape the horizontal transfer of antimicrobial resistance genes. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- He, S.; Hickman, A.B.; Varani, A.M.; Siguier, P.; Chandler, M.; Dekker, J.P.; Dyda, F. Insertion Sequence IS 26 Reorganizes Plasmids in Clinically Isolated Multidrug-Resistant Bacteria by Replicative Transposition. mBio 2015, 6, e00762-15. [Google Scholar] [CrossRef] [Green Version]
- Vandecraen, J.; Chandler, M.; Aertsen, A.; Van Houdt, R. The impact of insertion sequences on bacterial genome plasticity and adapta-bility. Crit. Rev. Microbiol. 2017, 43, 709–730. [Google Scholar] [CrossRef] [PubMed]
- Shaw, L.P.; Chau, K.K.; Kavanagh, J.; AbuOun, M.; Stubberfield, E.; Gweon, H.S.; Barker, L.; Rodger, G.; Bowes, M.J.; Hubbard, A.; et al. Niche and local geography shape the pangenome of wastewater- and livestock-associated Enterobacteriaceae. Sci. Adv. 2021, 7, eabe3868. [Google Scholar] [CrossRef] [PubMed]
- Pesesky, M.W.; Tilley, R.; Beck, D. Mosaic plasmids are abundant and unevenly distributed across prokaryotic taxa. Plasmid 2019, 102, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Jesus, T.F.; Ribeiro-Gonçalves, B.; Silva, D.N.; Bortolaia, V.; Ramirez, M.; Carriço, J.A. Plasmid ATLAS: Plasmid visual analytics and identification in high-throughput sequencing data. Nucleic Acids Res. 2019, 47, D188–D194. [Google Scholar] [CrossRef]
- Bosi, E.; Fani, R.; Fondi, M. The mosaicism of plasmids revealed by atypical genes detection and analysis. BMC Genom. 2011, 12, 403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arredondo-Alonso, S.; Pöntinen, A.K.; Cléon, F.; Gladstone, R.A.; Schurch, A.C.; Johnsen, P.J.; Samuelsen, O.; Corander, J. A high-throughput multiplexing and selection strategy to complete bac-terial genomes. BioRxiv 2021, 6, 448320. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Wyres, K.L.; Holt, K.E. Recovery of small plasmid sequences via Oxford Nanopore sequencing. BioRxiv 2021. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HyAsP | MOB-Suite | gplas | plasmidSPAdes | SCAPP | FishingFor Plasmids | |
|---|---|---|---|---|---|---|
| Computational Performance | ||||||
| Memory Usage (GB) | 299.2 | 202.82 | 150.36 | 442.03 | 435.23 | 36.57 |
| CPU-Time (hr) | 46.57 | 46.62 | 83.64 | 321.07 | 70.96 | 10.6 |
| Nr. of plasmid predictions | ||||||
| Nr. total predicted plasmids (bins) | 2590 | 738 | 550 | 642 | 986 | 377 |
| Nr. correct predictions of plasmid absence (%) | 2 (10.53) | 13 (68.42) | 17 (89.47) | 9 (47.37) | 17 (89.47) | 18 (94.74) |
| Plasmids reconstruction All Plasmid (n = 631) | ||||||
| Nr. correctly reconstructed plasmids (%) | 127 (20.13) | 317 (50.24) | 130 (20.6) | 263 (41.68) | 152 (24.09) | 92 (14.58) |
| Nr. small plasmids (%) | 82 (30.04) | 151 (55.31) | 87 (31.87) | 168 (61.54) | 98 (35.9) | 14 (5.13) |
| Nr. large plasmids (%) | 45 (12.57) | 166 (46.37) | 43 (12.01) | 95 (26.54) | 54 (15.08) | 78 (21.79) |
| Nr. incorrectly reconstructed plasmids (%) | 358 (56.74) | 231 (36.61) | 289 (45.8) | 252 (39.94) | 291 (46.12) | 243 (38.51) |
| Nr. small plasmids (%) | 53 (19.41) | 50 (18.32) | 17 (6.23) | 47 (17.22) | 59 (21.61) | 14 (5.13) |
| Nr. large plasmids (%) | 305 (85.20) | 181 (50.56) | 272 (75.98) | 205 (57.26) | 232 (64.80) | 229 (63.97) |
| Nr. undetected plasmids (%) | 146 (23.14) | 83 (13.15) | 212 (33.6) | 116 (18.38) | 188 (29.79) | 296 (46.91) |
| Nr. small plasmids (%) | 138 (50.55) | 72 (26.37) | 169 (61.9) | 58 (21.25) | 116 (42.49) | 245 (89.74) |
| Nr. large plasmids (%) | 8 (2.23) | 11 (3.07) | 43 (12.01) | 58 (16.2) | 72 (20.11) | 51 (14.25) |
| F1-score (median-IQR) * | 0.12 (0.04–0.41) | 0.89 (0.3–0.98) | 0.59 (0.3–0.94) | 0.95 (0.49–0.99) | 0.18 (0.07−0.81) | 0.64 (0.29–0.93) |
| Small plasmids * | 0.98 (0.76–0.99) | 0.98 (0.94–0.99) | 0.99 (0.98–0.99) | 0.98 (0.96–0.99) | 0.96 (0.88–0.99) | 0.95 (0.7–0.98) |
| Large plasmids * | 0.11 (0.04–0.32) | 0.74 (0.17–0.97) | 0.49 (0.21–0.76) | 0.6 (0.31–0.97) | 0.12 (0.06–0.41) | 0.61 (0.28–0.91) |
| Recall (median-IQR) * | 0.07 (0.02–0.32) | 0.89 (0.21–0.99) | 0.5 (0.22–0.93) | 0.99 (0.88–1) | 0.13 (0.04–0.78) | 0.51 (0.18–0.93) |
| Small plasmids * | 1 (0.92–1) | 1 (0.96–1) | 1 (0.98−1) | 1 (1–1) | 0.99 (0.92–1) | 1 (0.96−1) |
| Large plasmids * | 0.06 (0.02–0.2) | 0.63 (0.12–0.96) | 0.4 (0.16–0.72) | 0.94 (0.36–0.99) | 0.07 (0.03–0.31) | 0.46 (0.16–0.84) |
| Precision (median-IQR) * | 0.87 (0.5–0.98) | 0.98 (0.68–1) | 0.97 (0.55–1) | 0.93 (0.41–0.98) | 0.8 (0.39–0.94) | 1 (1−1) |
| Small plasmids * | 0.96 (0.86–0.98) | 0.98 (0.95–0.99) | 0.98 (0.97–0.99) | 0.96 (0.92–0.98) | 0.95 (0.83–0.98) | 0.96 (0.65–0.97) |
| Large plasmids * | 0.84 (0.48–0.98) | 0.97 (0.53–1) | 0.93 (0.47–1) | 0.58 (0.33–0.99) | 0.75 (0.34–0.92) | 1 (1–1) |
| Chromosome contamination (Median-IQR) | 0.88 (0.59–0.99) | 0.1 (0.03–0.99) | 0.45 (0.11–1) | 0.75 (0.14–0.92) | 0.3 (0.09–0.66) | 1 (0.6–1) |
| Nr. bins with chromosome contamination (%) | 1340 (51.73) | 297 (40.2) | 197 (35.81) | 295 (45.95) | 249 (25.25) | 7 (1.86) |
| Nr. pure chromosome bins | 1251 | 65 | 70 | 20 | 1 | 4 |
| Plasmids reconstruction ARG-plasmids (n = 147) | ||||||
| ARGs in bins | ||||||
| Nr. plasmid-derived ARGs (%) | 525 (84.95) | 548 (88.67) | 331 (53.56) | 390 (63.11) | 223 (36.08) | 133 (21.52) |
| Nr. chromosome-derived ARGs | 130 | 92 | 71 | 29 | 34 | 1 |
| Reconstruction status | ||||||
| Nr. plasmids correctly reconstructed (%) | 5 (3.4) | 41 (27.89) | 10 (6.8) | 23 (15.65) | 10 (6.8) | 13 (8.84) |
| Nr. plasmids predicted with incorrect backbones (%) | 62 (42.18) | 49 (33.33) | 38 (25.85) | 59 (40.14) | 23 (15.65) | 9 (6.12) |
| Nr. plasmids predicted with incomplete ARG content (%) | 66 (44.9) | 47 (31.97) | 59 (40.14) | 28 (19.05) | 39 (26.53) | 28 (19.05) |
| Nr. plasmids with no ARGs predicted (%) | 14 (9.52) | 10 (6.8) | 40 (27.21) | 37 (25.17) | 75 (51.02) | 97 (65.99) |
| Large ARG-plasmids reconstruction metrics (n = 143) | ||||||
| Recall (Median-IQR) * | 0.06 (0.02–0.16) | 0.38 (0.09–0.88) | 0.29 (0.14–0.62) | 0.87 (0.2–0.96) | 0.06 (0.03–0.17) | 0.35 (0.15–0.55) |
| Precision (Median-IQR) * | 0.84 (0.46–0.99) | 0.92 (0.42–1) | 0.86 (0.44–1) | 0.47 (0.31–0.92) | 0.71 (0.32–0.88) | 1 (1–1) |
| F1-score (Median-IQR) * | 0.1 (0.04–0.26) | 0.44 (0.13–0.9) | 0.41 (0.19–0.65) | 0.53 (0.24–0.73) | 0.1 (0.05–0.26) | 0.51 (0.25–0.69) |
| Nr. detected plasmids (%) | 141 (98.60) | 141 (98.60) | 135 (94.41) | 129 (90.21) | 113 (79.02) | 138 (96.50) |
| Plasmids reconstruction ESBL-plasmids (n = 60) | ||||||
| ESBL genes in bins | ||||||
| Nr. plasmid-derived (%) | 52 (86.67) | 57 (95) | 27 (45) | 40 (66.67) | 23 (38.33) | 11 (18.33) |
| Nr. chromosome-derived (%) | 10 | 8 | 7 | 2 | 2 | 0 |
| Reconstruction status | ||||||
| Nr. ESBL genes in correct plasmid backbone (%) | 0 (0) | 20 (33.33) | 4 (6.67) | 10 (16.67) | 5 (8.33) | 6 (10) |
| Nr. ESBL genes in incorrect plasmid backbone (%) | 52 (86.67) | 37 (61.67) | 23 (38.33) | 30 (50) | 18 (30) | 5 (8.33) |
| Reconstruction metrics | ||||||
| F1-score (Median-IQR) * | 0.29 (0.07–0.46) | 0.93 (0.72–0.97) | 0.69 (0.45–0.88) | 0.65 (0.51–0.95) | 0.27 (0.09–0.84) | 0.98 (0.71–0.99) |
| Recall (Median-IQR) * | 0.18 (0.04–0.31) | 0.89 (0.77–0.96) | 0.65 (0.36–0.84) | 0.96 (0.89–0.97) | 0.23 (0.05–0.84) | 0.95 (0.56–0.99) |
| Precision (Median-IQR) * | 0.91 (0.54–0.98) | 0.98 (0.93–1) | 0.97 (0.89–1) | 0.52 (0.38–0.95) | 0.85 (0.72–0.92) | 0.99 (1–1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paganini, J.A.; Plantinga, N.L.; Arredondo-Alonso, S.; Willems, R.J.L.; Schürch, A.C. Recovering Escherichia coli Plasmids in the Absence of Long-Read Sequencing Data. Microorganisms 2021, 9, 1613. https://doi.org/10.3390/microorganisms9081613
Paganini JA, Plantinga NL, Arredondo-Alonso S, Willems RJL, Schürch AC. Recovering Escherichia coli Plasmids in the Absence of Long-Read Sequencing Data. Microorganisms. 2021; 9(8):1613. https://doi.org/10.3390/microorganisms9081613
Chicago/Turabian StylePaganini, Julian A., Nienke L. Plantinga, Sergio Arredondo-Alonso, Rob J. L. Willems, and Anita C. Schürch. 2021. "Recovering Escherichia coli Plasmids in the Absence of Long-Read Sequencing Data" Microorganisms 9, no. 8: 1613. https://doi.org/10.3390/microorganisms9081613
APA StylePaganini, J. A., Plantinga, N. L., Arredondo-Alonso, S., Willems, R. J. L., & Schürch, A. C. (2021). Recovering Escherichia coli Plasmids in the Absence of Long-Read Sequencing Data. Microorganisms, 9(8), 1613. https://doi.org/10.3390/microorganisms9081613