
Family SES Is Associated with the Gut Microbiome in Infants and Children

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Demographics and Family SES
2.2. SNP Microarray and Polygenic Scores (PGS)
2.3. Stool Sample Collection and Handling
2.4. DNA Extraction and Sequencing of Metagenomes
2.5. Analyzing Metagenomes
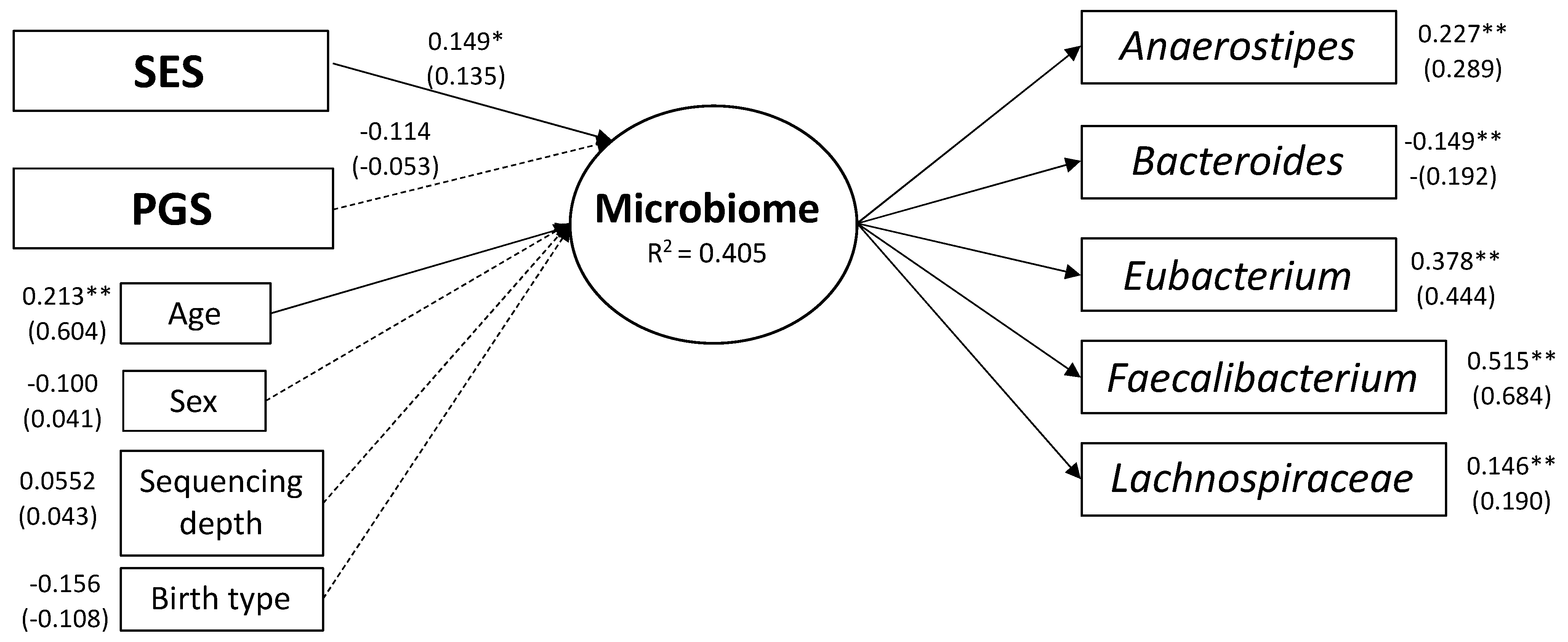
2.6. Structural Equation Modeling
2.7. Microbiome Diversity
3. Results
3.1. Sample Characteristics
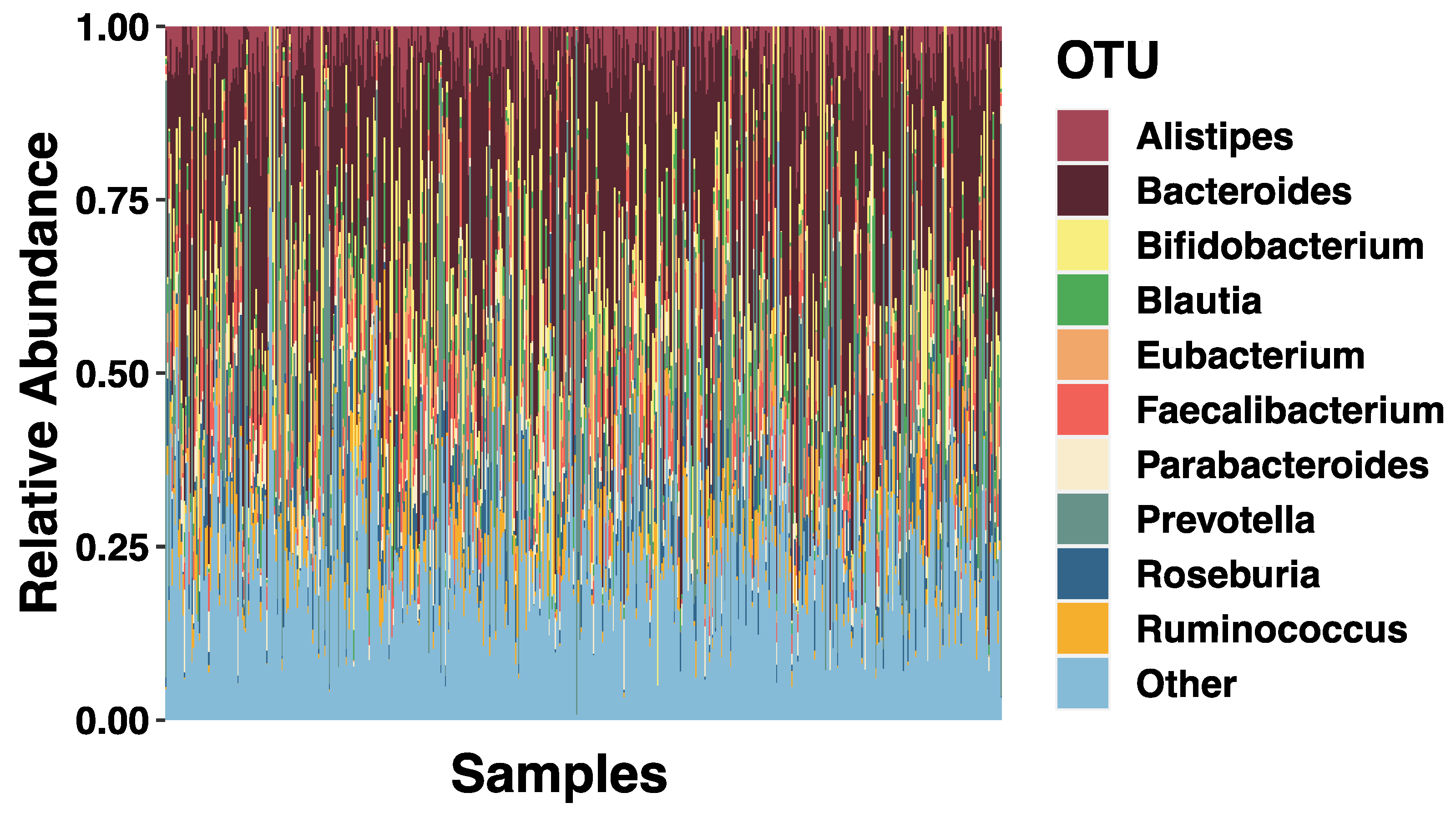
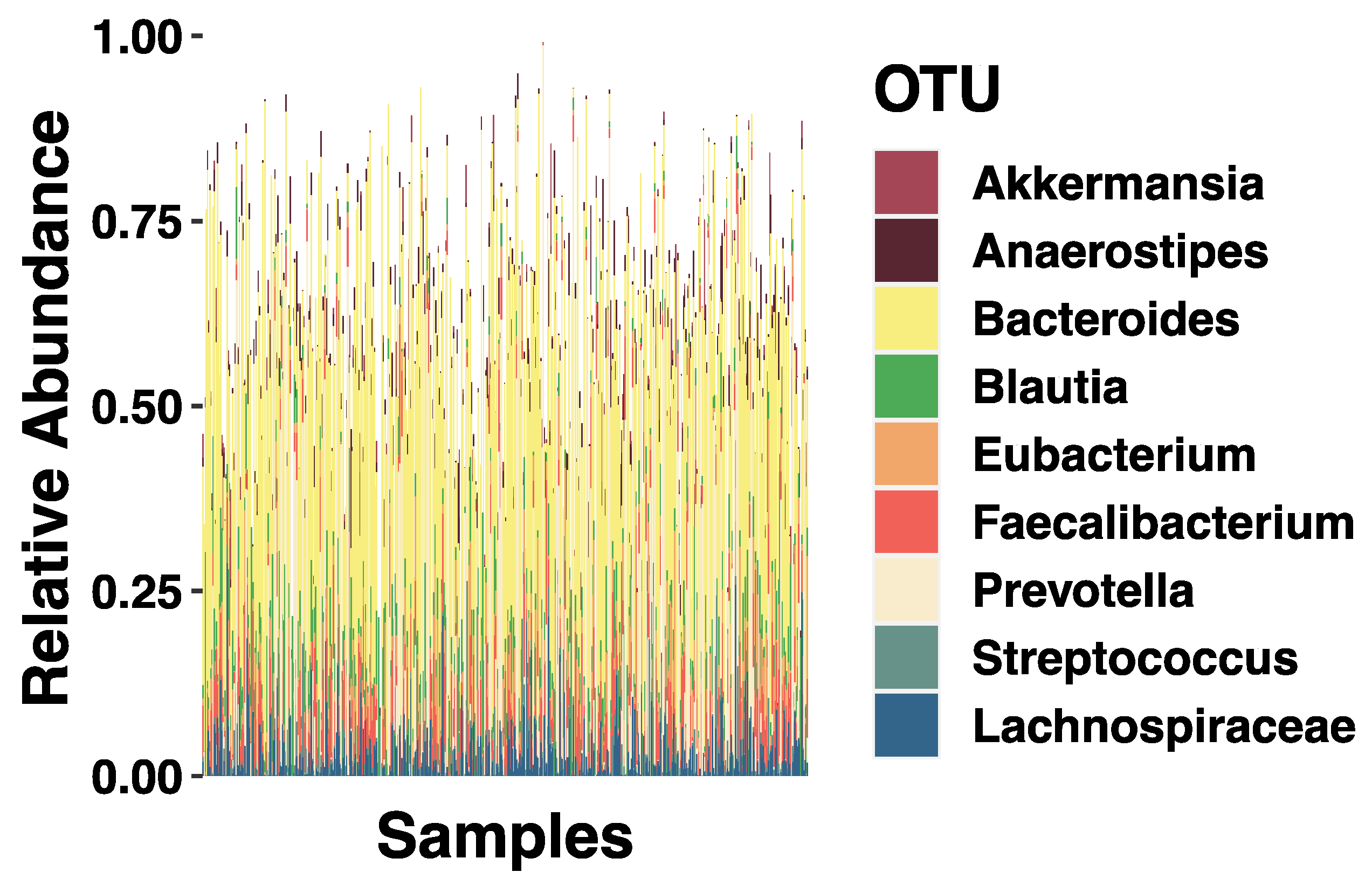
3.2. SES and Relative Abundance
3.3. Alpha Diversity
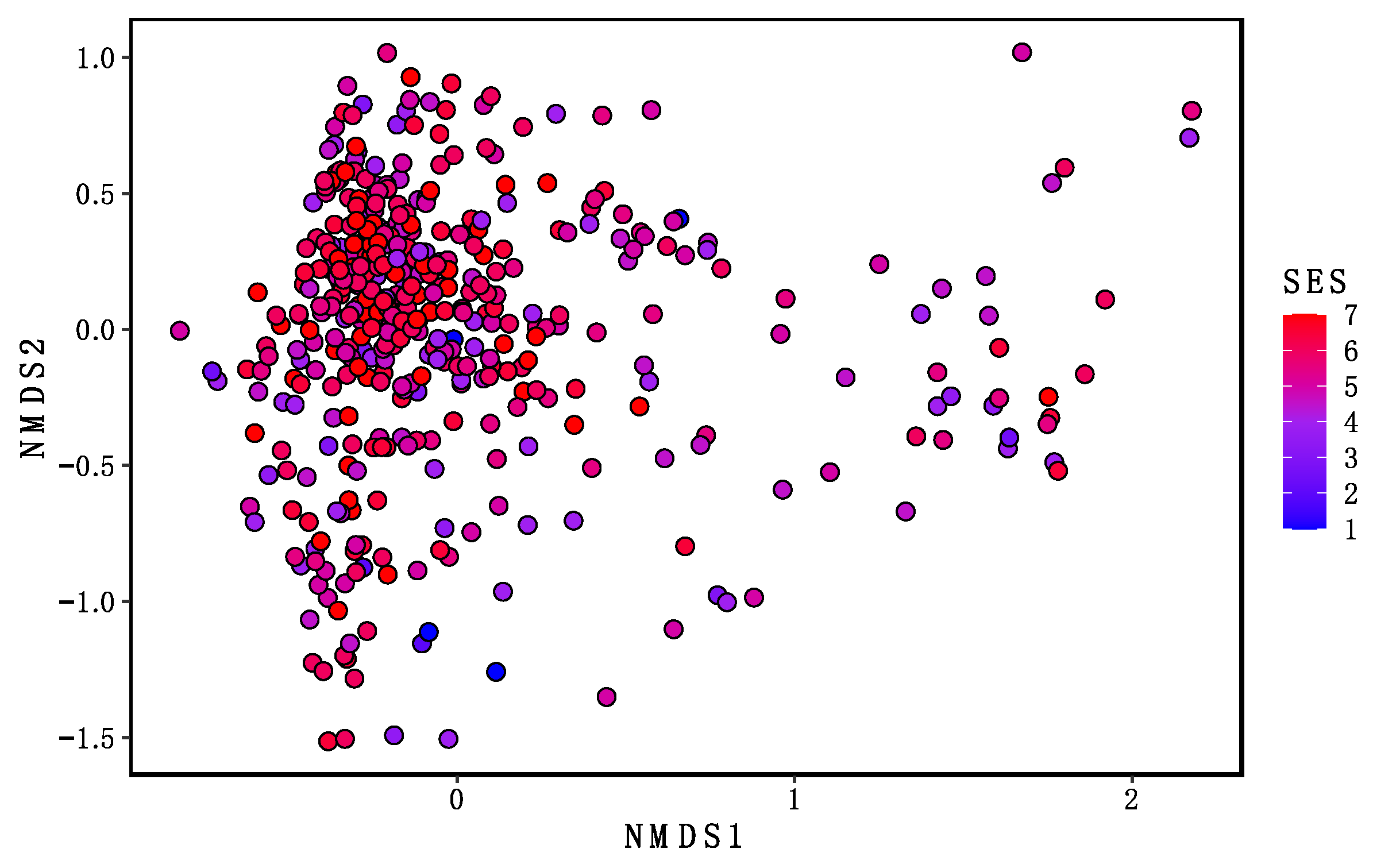
3.4. Beta Diversity
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cryan, J.F.; O’Riordan, K.J.; Sandhu, K.; Peterson, V.; Dinan, T.G. The gut microbiome in neurological disorders. Lancet Neurol. 2020, 19, 179–194. [Google Scholar] [CrossRef]
- Singhvi, N.; Gupta, V.; Gaur, M.; Sharma, V.; Puri, A.; Singh, Y.; Dubey, G.P.; Lal, R. Interplay of Human Gut Microbiome in Health and Wellness. Indian J. Microbiol. 2019, 60, 26–36. [Google Scholar] [CrossRef]
- Shreiner, A.B.; Kao, J.Y.; Young, V.B. The gut microbiome in health and in disease. Curr. Opin. Gastroenterol. 2015, 31, 69–75. [Google Scholar] [CrossRef]
- Gilbert, J.A.; Blaser, M.J.; Caporaso, J.G.; Jansson, J.; Lynch, S.V.; Knight, R. Current understanding of the human microbiome. Nat. Med. 2018, 24, 392–400. [Google Scholar] [CrossRef]
- Kim, H.; Sitarik, A.R.; Woodcroft, K.; Johnson, C.C.; Zoratti, E. Birth Mode, Breastfeeding, Pet Exposure, and Antibiotic Use: Associations with the Gut Microbiome and Sensitization in Children. Curr. Allergy Asthma Rep. 2019, 19, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sbihi, H.; Boutin, R.; Cutler, C.; Suen, M.; Finlay, B.B.; Turvey, S.E. Thinking bigger: How early-life environmental exposures shape the gut microbiome and influence the development of asthma and allergic disease. Allergy 2019, 74, 2103–2115. [Google Scholar] [CrossRef]
- Rothschild, D.; Weissbrod, O.; Barkan, E.; Kurilshikov, A.; Korem, T.; Zeevi, D.; Costea, P.I.; Godneva, A.; Kalka, I.N.; Bar, N.; et al. Environment dominates over host genetics in shaping human gut microbiota. Nat. Cell Biol. 2018, 555, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nat. Cell Biol. 2008, 457, 480–484. [Google Scholar] [CrossRef]
- Yatsunenko, T.; Rey, F.E.; Manary, M.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.; et al. Human gut microbiome viewed across age and geography. Nat. Cell Biol. 2012, 486, 222–227. [Google Scholar] [CrossRef]
- Goodrich, J.K.; Davenport, E.; Beaumont, M.; Jackson, M.; Knight, R.; Ober, C.; Spector, T.D.; Bell, J.; Clark, A.; Ley, R.E. Genetic Determinants of the Gut Microbiome in UK Twins. Cell Host Microbe 2016, 19, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, J.K.; Waters, J.L.; Poole, A.; Sutter, J.L.; Koren, O.; Blekhman, R.; Beaumont, M.; Van Treuren, W.; Knight, R.; Bell, J.; et al. Human Genetics Shape the Gut Microbiome. Cell 2014, 159, 789–799. [Google Scholar] [CrossRef]
- Benson, A.K.; Kelly, S.A.; Legge, R.; Ma, F.; Low, S.J.; Kim, J.; Zhang, M.; Oh, P.L.; Nehrenberg, D.; Hua, K.; et al. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc. Natl. Acad. Sci. USA 2010, 107, 18933–18938. [Google Scholar] [CrossRef]
- McKnite, A.; Perez-Munoz, M.E.; Lu, L.; Williams, E.; Brewer, S.; Andreux, P.A.; Bastiaansen, J.; Wang, X.; Kachman, S.; Auwerx, J.; et al. Murine Gut Microbiota Is Defined by Host Genetics and Modulates Variation of Metabolic Traits. PLoS ONE 2012, 7, e39191. [Google Scholar] [CrossRef]
- Leamy, L.J.; Kelly, S.A.; Nietfeldt, J.; Legge, R.M.; Ma, F.; Hua, K.; Sinha, R.; Peterson, D.A.; Walter, J.; Benson, A.K.; et al. Host genetics and diet, but not immunoglobulin A expression, converge to shape compositional features of the gut microbiome in an advanced intercross population of mice. Genome Biol. 2014, 15, 552. [Google Scholar] [CrossRef]
- Blekhman, R.; Goodrich, J.K.; Huang, K.; Sun, Q.; Bukowski, R.; Bell, J.T.; Spector, T.D.; Keinan, A.; Ley, R.E.; Gevers, D.; et al. Host genetic variation impacts microbiome composition across human body sites. Genome Biol. 2015, 16, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Davenport, E.; Cusanovich, D.; Michelini, K.; Barreiro, L.; Ober, C.; Gilad, Y. Genome-Wide Association Studies of the Human Gut Microbiota. PLoS ONE 2015, 10, e0140301. [Google Scholar] [CrossRef] [PubMed]
- Bonder, M.J.; Kurilshikov, A.; Tigchelaar, E.F.; Mujagic, Z.; Imhann, F.; Vila, A.V.; Deelen, P.; Vatanen, T.; Schirmer, M.; Smeekens, S.P.; et al. The effect of host genetics on the gut microbiome. Nat. Genet. 2016, 48, 1407–1412. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Thingholm, L.B.; Skiecevičienė, J.; Rausch, P.; Kummen, M.; Hov, J.R.; Degenhardt, F.; Heinsen, F.-A.; Rühlemann, M.C.; Szymczak, S.; et al. Genome-wide association analysis identifies variation in vitamin D receptor and other host factors influencing the gut microbiota. Nat. Genet. 2016, 48, 1396–1406. [Google Scholar] [CrossRef] [PubMed]
- Turpin, W.; GEM Project Research Consortium; Espin-Garcia, O.; Xu, W.; Silverberg, M.S.; Kevans, D.; Smith, M.I.; Guttman, D.S.; Griffiths, A.; Panaccione, R.; et al. Association of host genome with intestinal microbial composition in a large healthy cohort. Nat. Genet. 2016, 48, 1413–1417. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.A.; Bacigalupe, R.; Wang, J.; Rühlemann, M.C.; Tito, R.Y.; Falony, G.; Joossens, M.; Vieira-Silva, S.; Henckaerts, L.; Rymenans, L.; et al. Genome-wide associations of human gut microbiome variation and implications for causal inference analyses. Nat. Microbiol. 2020, 5, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Fu, Y.; Sun, T.-Y.; Jiang, Z.; Miao, Z.; Shuai, M.; Gou, W.; Ling, C.-W.; Yang, J.; Wang, J.; et al. The interplay between host genetics and the gut microbiome reveals common and distinct microbiome features for complex human diseases. Microbiome 2020, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.A.; Rinaman, L.; Cryan, J.F. Stress & the gut-brain axis: Regulation by the microbiome. Neurobiol. Stress 2017, 7, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Moeller, A.H.; Foerster, S.; Wilson, M.L.; Pusey, A.E.; Hahn, B.H.; Ochman, H. Social behavior shapes the chimpanzee pan-microbiome. Sci. Adv. 2016, 2, e1500997. [Google Scholar] [CrossRef]
- Amato, K.R.; Van Belle, S.; Di Fiore, A.; Estrada, A.; Stumpf, R.; White, B.; Nelson, K.E.; Knight, R.; Leigh, S. Patterns in Gut Microbiota Similarity Associated with Degree of Sociality among Sex Classes of a Neotropical Primate. Microb. Ecol. 2017, 74, 250–258. [Google Scholar] [CrossRef]
- Amaral, W.Z.; Lubach, G.R.; Proctor, A.; Lyte, M.; Phillips, G.J.; Coe, C.L. Social Influences on Prevotella and the Gut Microbiome of Young Monkeys. Psychosom. Med. 2017, 79, 888–897. [Google Scholar] [CrossRef]
- Tung, J.; Barreiro, L.; Burns, M.; Grenier, J.-C.; Lynch, J.; Grieneisen, L.E.; Altmann, J.; Alberts, S.C.; Blekhman, R.; Archie, E.A. Social networks predict gut microbiome composition in wild baboons. eLife 2015, 4, e05224. [Google Scholar] [CrossRef]
- Bailey, M.T. Psychological Stress, Immunity, and the Effects on Indigenous Microflora. Microb. Endocrinol. Interkingdom Signal. Infect. Dis. Health 2016, 874, 225–246. [Google Scholar] [CrossRef]
- Grzywacz, J.; Almeida, D.M.; Neupert, S.D.; Ettner, S.L. Socioeconomic Status and Health: A Micro-level Analysis o Exposure and Vulnerability to Daily Stressors. J. Health Soc. Behav. 2004, 45, 1–16. [Google Scholar] [CrossRef]
- Tannock, G.W.; Savage, D.C. Influences of dietary and environmental stress on microbial populations in the murine gastrointestinal tract. Infect. Immun. 1974, 9, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M.T.; Dowd, S.; Galley, J.D.; Hufnagle, A.R.; Allen, R.G.; Lyte, M. Exposure to a social stressor alters the structure of the intestinal microbiota: Implications for stressor-induced immunomodulation. Brain, Behav. Immun. 2011, 25, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Sudo, N.; Chida, Y.; Aiba, Y.; Sonoda, J.; Oyama, N.; Yu, X.-N.; Kubo, C.; Koga, Y. Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice. J. Physiol. 2004, 558, 263–275. [Google Scholar] [CrossRef]
- O’Mahony, S.M.; Marchesi, J.; Scully, P.; Codling, C.; Ceolho, A.-M.; Quigley, E.M.; Cryan, J.; Dinan, T. Early Life Stress Alters Behavior, Immunity, and Microbiota in Rats: Implications for Irritable Bowel Syndrome and Psychiatric Illnesses. Biol. Psychiatry 2009, 65, 263–267. [Google Scholar] [CrossRef]
- Jašarević, E.; Howerton, C.L.; Howard, C.D.; Bale, T.L. Alterations in the Vaginal Microbiome by Maternal Stress Are Associated With Metabolic Reprogramming of the Offspring Gut and Brain. Endocrinol. 2015, 156, 3265–3276. [Google Scholar] [CrossRef]
- Bailey, M.T.; Coe, C.L. Maternal separation disrupts the integrity of the intestinal microflora in infant rhesus monkeys. Dev. Psychobiol. 1999. [Google Scholar] [CrossRef]
- Dowd, J.B.; Renson, A. “Under the Skin” and into the Gut: Social Epidemiology of the Microbiome. Curr. Epidemiology Rep. 2018, 5, 432–441. [Google Scholar] [CrossRef]
- Herd, P.; Palloni, A.; Rey, F.; Dowd, J. Social and population health science approaches to understand the human microbiome. Nat. Hum. Behav. 2018, 2, 808–815. [Google Scholar] [CrossRef]
- Miller, G.E.; Engen, P.A.; Gillevet, P.M.; Shaikh, M.; Sikaroodi, M.; Forsyth, C.B.; Mutlu, E.; Keshavarzian, A. Lower Neighborhood Socioeconomic Status Associated with Reduced Diversity of the Colonic Microbiota in Healthy Adults. PLoS ONE 2016, 11, e0148952. [Google Scholar] [CrossRef]
- Bowyer, R.C.E.; Jackson, M.A.; Le Roy, C.I.; Ni Lochlainn, M.; Spector, T.D.; Dowd, J.B.; Steves, C.J. Socioeconomic Status and the Gut Microbiome: A TwinsUK Cohort Study. Microorg. 2019, 7, 17. [Google Scholar] [CrossRef]
- Jankovic, J.; Stojisavljević, D.; Erić, M.; Marinković, J. Association of socioeconomic status measured by education, and cardiovascular health: A population-based cross-sectional study. BMJ Open 2014, 4, e005222. [Google Scholar] [CrossRef]
- Taliun, D.; NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium; Harris, D.N.; Kessler, M.D.; Carlson, J.; Szpiech, Z.A.; Torres, R.; Taliun, S.A.G.; Corvelo, A.; Gogarten, S.M.; et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nat. Cell Biol. 2021, 590, 290–299. [Google Scholar] [CrossRef]
- Marees, A.T.; De Kluiver, H.; Stringer, S.; Vorspan, F.; Curis, E.; Marie-Claire, C.; Derks, E. A tutorial on conducting genome-wide association studies: Quality control and statistical analysis. Int. J. Methods Psychiatr. Res. 2018, 27, e1608. [Google Scholar] [CrossRef] [PubMed]
- Comeau, A.M.; Douglas, G.M.; Langille, M.G.I. Microbiome Helper: A Custom and Streamlined Workflow for Microbiome Research. mSystems 2017, 2, e00127-16. [Google Scholar] [CrossRef] [PubMed]
- McIver, L.J.; Abu-Ali, G.; Franzosa, E.A.; Schwager, R.; Morgan, X.C.; Waldron, L.; Segata, N.; Huttenhower, C. bioBakery: A meta’omic analysis environment. Bioinform. 2017, 34, 1235–1237. [Google Scholar] [CrossRef] [PubMed]
- Beghini, F.; McIver, L.J.; Blanco-Míguez, A.; Dubois, L.; Asnicar, F.; Maharjan, S.; Mailyan, A.; Manghi, P.; Scholz, M.; Thomas, A.M.; et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. eLife 2021, 10. [Google Scholar] [CrossRef]
- Rosseel, Y. lavaan: AnRPackage for Structural Equation Modeling. J. Stat. Softw. 2012, 48, 1–36. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2020. [Google Scholar]
- Weston, R.; Gore, P.A. A Brief Guide to Structural Equation Modeling. Couns. Psychol. 2006, 34, 719–751. [Google Scholar] [CrossRef]
- Lovén, J.; Orlando, D.A.; Sigova, A.A.; Lin, C.Y.; Rahl, P.B.; Burge, C.B.; Levens, D.L.; Lee, T.I.; Young, R.A. Revisiting Global Gene Expression Analysis. Cell 2012, 151, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Gloor, G.B.; Macklaim, J.M.; Pawlowsky-Glahn, V.; Egozcue, J.J. Microbiome Datasets Are Compositional: And This Is Not Optional. Front. Microbiol. 2017, 8, 2224. [Google Scholar] [CrossRef]
- Wu, X.; Berkow, K.; Frank, D.N.; Li, E.; Gulati, A.S.; Zhu, W. Comparative analysis of microbiome measurement platforms using latent variable structural equation modeling. BMC Bioinform. 2013, 14, 79. [Google Scholar] [CrossRef][Green Version]
- Vu, K.; Lou, W.; Tun, H.M.; Konya, T.B.; Morales-Lizcano, N.; Chari, R.S.; Field, C.J.; Guttman, D.S.; Mandal, R.; Wishart, D.S.; et al. From Birth to Overweight and Atopic Disease: Multiple and Common Pathways of the Infant Gut Microbiome. Gastroenterol. 2021, 160, 128–144.e10. [Google Scholar] [CrossRef]
- Enders, C.; Bandalos, D. The Relative Performance of Full Information Maximum Likelihood Estimation for Missing Data in Structural Equation Models. Struct. Equ. Model. A Multidiscip. J. 2001, 8, 430–457. [Google Scholar] [CrossRef]
- McDonald, R.P.; Ho, M.-H.R. Principles and practice in reporting structural equation analyses. Psychol. Methods 2002, 7, 64–82. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Bentler, P.M. Cutoff criteria for fit indexes in covariance structure analysis: Conventional criteria versus new alternatives. Struct. Equ. Model. A Multidiscip. J. 1999, 6, 1–55. [Google Scholar] [CrossRef]
- Bäckhed, F.; Roswall, J.; Peng, Y.; Feng, Q.; Jia, H.; Kovatcheva-Datchary, P.; Li, Y.; Xia, Y.; Xie, H.; Zhong, H.; et al. Dynamics and Stabilization of the Human Gut Microbiome during the First Year of Life. Cell Host Microbe 2015, 17, 690–703. [Google Scholar] [CrossRef]
- Fernández, L.; Langa, S.; Martin, V.; Maldonado, A.; Jiménez, E.; Martín, R.; Rodríguez, J.M. The human milk microbiota: Origin and potential roles in health and disease. Pharmacol. Res. 2013, 69, 1–10. [Google Scholar] [CrossRef]
- Vangay, P.; Ward, T.; Gerber, J.S.; Knights, D. Antibiotics, Pediatric Dysbiosis, and Disease. Cell Host Microbe 2015, 17, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.; Miquel, S.; Benevides, L.; Bridonneau, C.; Robert, V.; Hudault, S.; Chain, F.; Berteau, O.; Azevedo, V.; Chatel, J.M.; et al. Functional Characterization of Novel Faecalibacterium prausnitzii Strains Isolated from Healthy Volunteers: A Step Forward in the Use of F. prausnitzii as a Next-Generation Probiotic. Front. Microbiol. 2017, 8, 1226. [Google Scholar] [CrossRef]
- Fitzgerald, C.B.; Shkoporov, A.N.; Sutton, T.D.S.; Chaplin, A.V.; Velayudhan, V.; Ross, R.P.; Hill, C. Comparative analysis of Faecalibacterium prausnitzii genomes shows a high level of genome plasticity and warrants separation into new species-level taxa. BMC Genom. 2018, 19, 1–20. [Google Scholar] [CrossRef]
- Miquel, S.; Martin, R.; Rossi, O.; Humaran, L.G.B.; Chatel, J.; Sokol, H.; Thomas, M.; Wells, J.; Langella, P. Faecalibacterium prausnitzii and human intestinal health. Curr. Opin. Microbiol. 2013, 16, 255–261. [Google Scholar] [CrossRef]
- Gschwendtner, S.; Kang, H.; Thiering, E.; Kublik, S.; Foesel, B.; Schulz, H.; Krauss-Etschmann, S.; Heinrich, J.; Schöler, A.; Schloter, M.; et al. Early life determinants induce sustainable changes in the gut microbiome of six-year-old children. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef]
- Hagerty, S.L.; Hutchison, K.E.; Lowry, C.A.; Bryan, A.D. An empirically derived method for measuring human gut microbiome alpha diversity: Demonstrated utility in predicting health-related outcomes among a human clinical sample. PLoS ONE 2020, 15, e0229204. [Google Scholar] [CrossRef]
- Vogt, N.M.; Kerby, R.L.; Dill-McFarland, K.A.; Harding, S.J.; Merluzzi, A.P.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Zetterberg, H.; Blennow, K.; et al. Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Prehn-Kristensen, A.; Zimmermann, A.; Tittmann, L.; Lieb, W.; Schreiber, S.; Baving, L.; Fischer, A. Reduced microbiome alpha diversity in young patients with ADHD. PLoS ONE 2018, 13, e0200728. [Google Scholar] [CrossRef]
- Jiang, H.; Ling, Z.; Zhang, Y.; Mao, H.; Ma, Z.; Yin, Y.; Wang, W.; Tang, W.; Tan, Z.; Shi, J.; et al. Altered fecal microbiota composition in patients with major depressive disorder. Brain, Behav. Immun. 2015, 48, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Laursen, M.F.; Andersen, L.B.B.; Michaelsen, K.F.; Mølgaard, C.; Trolle, E.; Bahl, M.I.; Licht, T.R. Infant Gut Microbiota Development Is Driven by Transition to Family Foods Independent of Maternal Obesity. mSphere 2016, 1, e00069-15. [Google Scholar] [CrossRef]
- Odamaki, T.; Kato, K.; Sugahara, H.; Hashikura, N.; Takahashi, S.; Xiao, J.-Z.; Abe, F.; Osawa, R. Age-related changes in gut microbiota composition from newborn to centenarian: A cross-sectional study. BMC Microbiol. 2016, 16, 1–12. [Google Scholar] [CrossRef]
- Hoffmann, T.J.; Ehret, G.B.; Nandakumar, P.; Ranatunga, D.; Schaefer, C.; Kwok, P.-Y.; Iribarren, C.; Chakravarti, G.B.E.P.N.A.; Risch, D.R.C.S.C.I.N. Genome-wide association analyses using electronic health records identify new loci influencing blood pressure variation. Nat. Genet. 2016, 49, 54–64. [Google Scholar] [CrossRef]
- Ehret, G.B.; CHARGE-EchoGen Consortium; Ferreira, T.; Chasman, D.I.; Jackson, A.U.; Schmidt, E.; Johnson, T.; Thorleifsson, G.; Luan, J.; Donnelly, L.A.; et al. The genetics of blood pressure regulation and its target organs from association studies in 342,415 individuals. Nat. Genet. 2016, 48, 1171–1184. [Google Scholar] [CrossRef]
- Wood, A.R.; The Electronic Medical Records and Genomics (eMERGE) Consortium; Esko, T.; Yang, J.; Vedantam, S.; Pers, T.; Gustafsson, S.; Chu, A.Y.; Estrada, K.; Luan, J.; et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat. Genet. 2014, 46, 1173–1186. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.C.; Clayton, D.G.; Concannon, P.; Akolkar, B.; Cooper, J.D.; Erlich, H.A.; Julier, C.; Morahan, G.; Nerup, J.; Nierras, C.; et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat. Genet. 2009, 41, 703–707. [Google Scholar] [CrossRef]
- Torkamani, A.; Topol, E. Polygenic Risk Scores Expand to Obesity. Cell 2019, 177, 518–520. [Google Scholar] [CrossRef]
- Ronald, A.; de Bode, N.; Polderman, T.J. Systematic Review: How the Attention-Deficit/Hyperactivity Disorder Polygenic Risk Score Adds to Our Understanding of ADHD and Associated Traits. J. Am. Acad. Child Adolesc. Psychiatry 2021. [Google Scholar] [CrossRef] [PubMed]
- Wray, N.R.; Ripke, S.; Mattheisen, M.; Trzaskowski, M.; Byrne, E.M.; Abdellaoui, A.; Adams, M.J.; Agerbo, E.; Air, T.M.; Andlauer, T.; et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 2018, 50, 668–681. [Google Scholar] [CrossRef]
- Adolfsson, R. Biological insights from 108 schizophrenia-associated genetic loci. Nat. Cell Biol. 2014, 511, 421–427. [Google Scholar] [CrossRef]
- Wells, P.; Adebayo, A.; Bowyer, R.C.E.; Freidin, M.B.; Finckh, A.; Strowig, T.; Lesker, T.R.; Alpízar-Rodríguez, D.; Gilbert, B.; Kirkham, B.; et al. Associations between gut microbiota and genetic risk for rheumatoid arthritis in the absence of disease: A cross-sectional study. Lancet Rheumatol. 2020, 2, e418–e427. [Google Scholar] [CrossRef]
- Tobin, C.S.T.; Erving, C.L.; Barve, A. Race and SES Differences in Psychosocial Resources: Implications for Social Stress Theory. Soc. Psychol. Q. 2020, 84, 1–25. [Google Scholar] [CrossRef]
- Flies, E.J.; Skelly, C.; Negi, S.S.; Prabhakaran, P.; Liu, Q.; Liu, K.; Goldizen, F.C.; Lease, C.; Weinstein, P. Biodiverse green spaces: A prescription for global urban health. Front. Ecol. Environ. 2017, 15, 510–516. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | N | Mean (SD) or % | Range |
|---|---|---|---|
| Metagenomics | 588 | - | - |
| Age (years) | 315 | 4.5 (3.63) | 1 m–15 y |
| Sex (Female) | 547 | 45% | - |
| Socioeconomic status (SES) | 434 | 4.2 (1.87) | 1–7 |
| PGS | 358 | 0.36 (1.42) | −2.72–2.79 |
| Birth type | 370 | 69% (Vaginal) | - |
| Race | 406 | 60.6% White; 26.8% Mixed; 7.6% African American; 1.2% Asian; 1.2% Native American; 2.5% Declined | - |
| --Alpha-Diversity (Shannon) | 588 | 2.05 (0.56) | 0.09–3.02 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lewis, C.R.; Bonham, K.S.; McCann, S.H.; Volpe, A.R.; D’Sa, V.; Naymik, M.; De Both, M.D.; Huentelman, M.J.; Lemery-Chalfant, K.; Highlander, S.K.; et al. Family SES Is Associated with the Gut Microbiome in Infants and Children. Microorganisms 2021, 9, 1608. https://doi.org/10.3390/microorganisms9081608
Lewis CR, Bonham KS, McCann SH, Volpe AR, D’Sa V, Naymik M, De Both MD, Huentelman MJ, Lemery-Chalfant K, Highlander SK, et al. Family SES Is Associated with the Gut Microbiome in Infants and Children. Microorganisms. 2021; 9(8):1608. https://doi.org/10.3390/microorganisms9081608
Chicago/Turabian StyleLewis, Candace R., Kevin S. Bonham, Shelley Hoeft McCann, Alexandra R. Volpe, Viren D’Sa, Marcus Naymik, Matt D. De Both, Matthew J. Huentelman, Kathryn Lemery-Chalfant, Sarah K. Highlander, and et al. 2021. "Family SES Is Associated with the Gut Microbiome in Infants and Children" Microorganisms 9, no. 8: 1608. https://doi.org/10.3390/microorganisms9081608
APA StyleLewis, C. R., Bonham, K. S., McCann, S. H., Volpe, A. R., D’Sa, V., Naymik, M., De Both, M. D., Huentelman, M. J., Lemery-Chalfant, K., Highlander, S. K., Deoni, S. C. L., & Klepac-Ceraj, V. (2021). Family SES Is Associated with the Gut Microbiome in Infants and Children. Microorganisms, 9(8), 1608. https://doi.org/10.3390/microorganisms9081608