
Bacterial Extracellular DNA Promotes β-Amyloid Aggregation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sources and Procedures for DNA Extraction
2.2. DNA Fragment Size Characterization with an Agilent Bioanalyzer
2.3. DNA and RNA Extraction and Purification
2.4. Aβ Synthesis and Preparation
2.5. In Vitro Aggregation Assay and Half-Time Analysis
2.6. Statistical Analysis
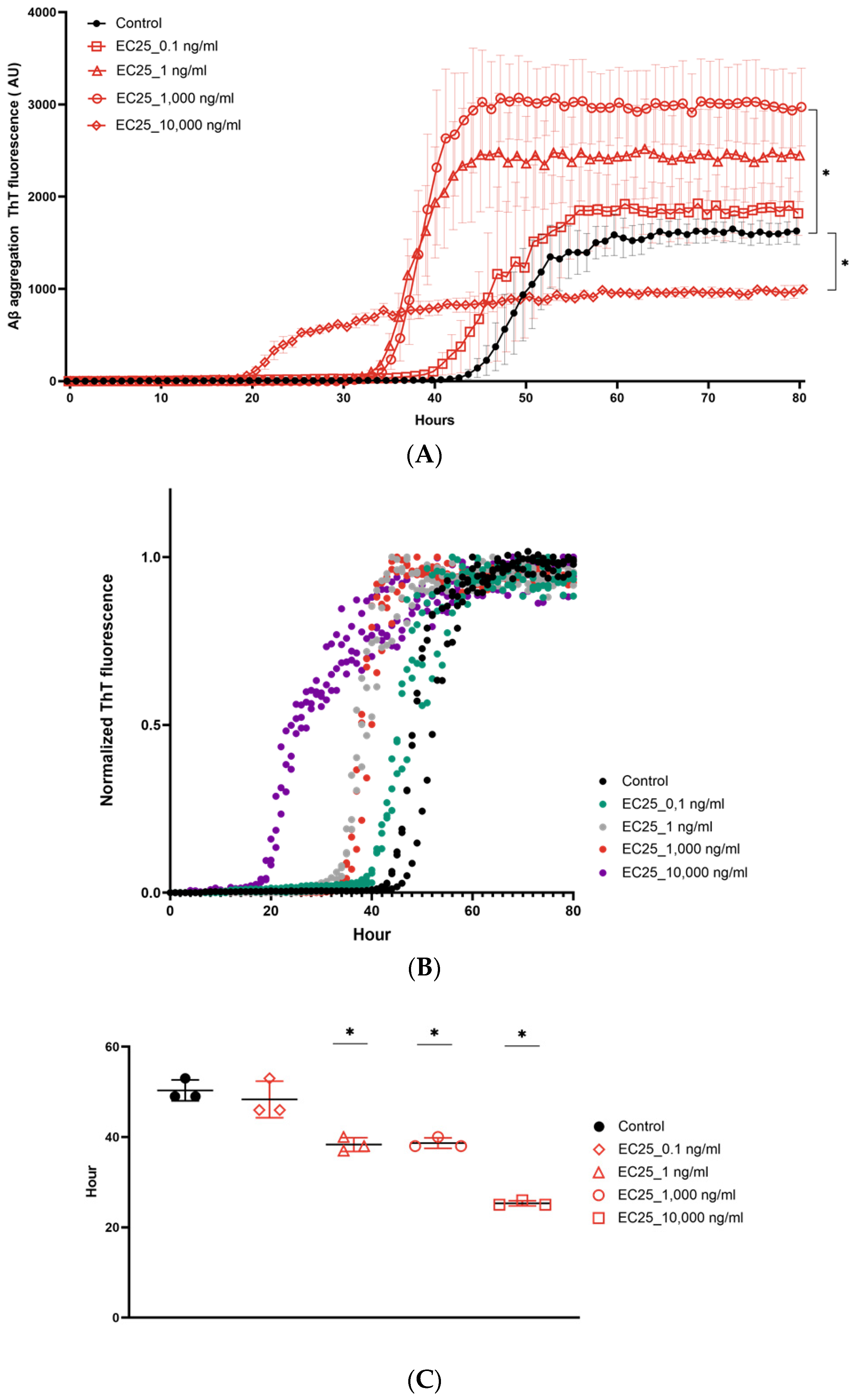
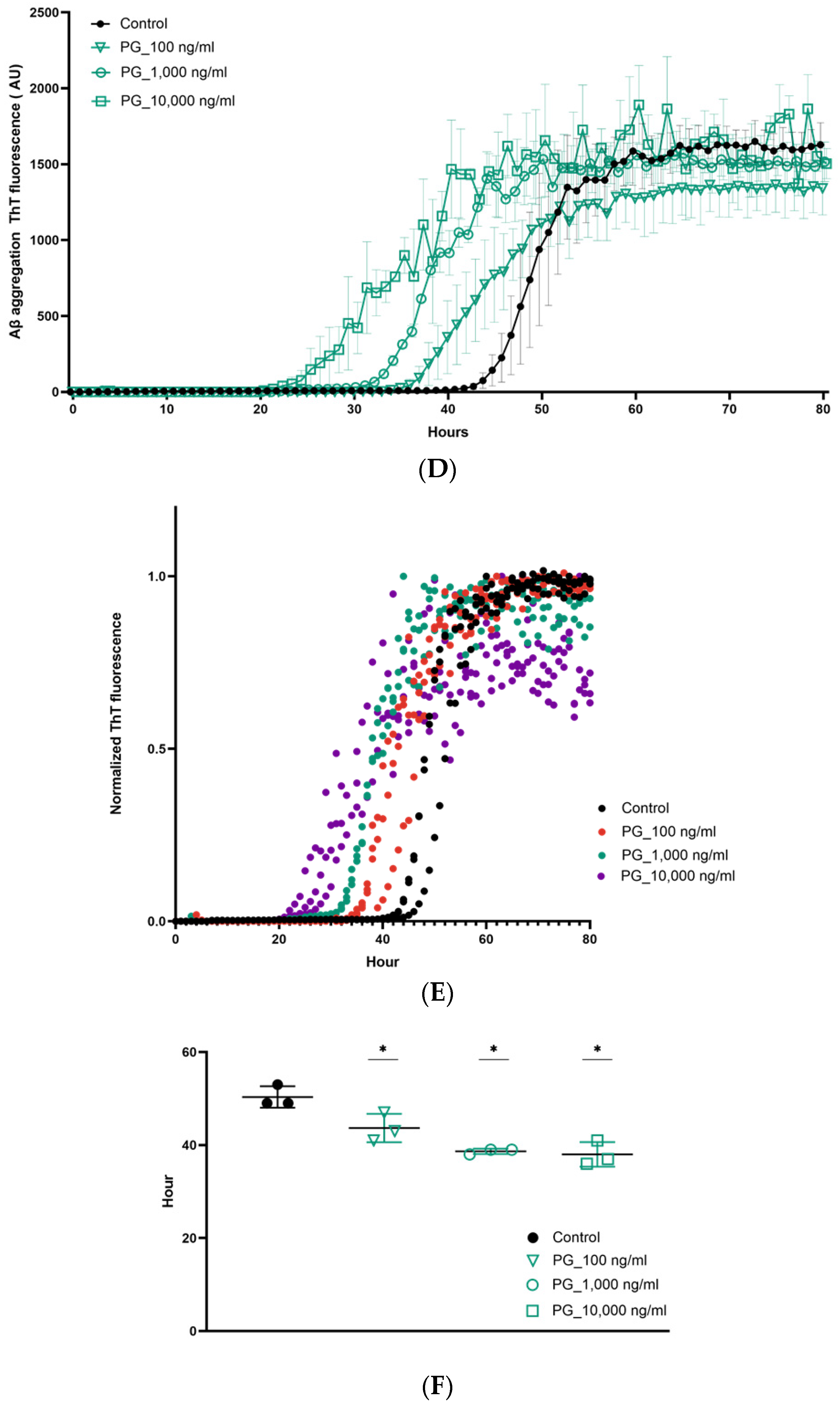
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018, 10, 1332–1340. [Google Scholar] [CrossRef]
- Aguzzi, A.; O’connor, T. Protein aggregation diseases: Pathogenicity and therapeutic perspectives. Nat. Rev. Drug Discov. 2010, 9, 237–248. [Google Scholar] [CrossRef]
- Kerman, A. Amyotrophic lateral sclerosis is a non-amyloid disease in which extensive misfolding of SOD1 is unique to the familial form. Acta Neuropathol. 2010, 1, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Soto, C. Prion-like protein aggregates and type 2 diabetes. Cold Spring Harb. Perspect. Med. 2017, 7, a024315. [Google Scholar] [CrossRef] [PubMed]
- McAlary, L.; Plotkin, S.S.; Yerbury, J.J.; Cashman, N.R. Prion-like propagation of protein misfolding and aggregation in amyotrophic lateral sclerosis. Front. Mol. Neurosci. 2019, 12, 262. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Wang, X.; Geng, M. Alzheimer’s disease hypothesis and related therapies. Trans. Neurodegen. 2018, 7, 1–7. [Google Scholar] [CrossRef]
- Gamblin, T.C. Caspase cleavage of tau: Linking amyloid and neurofibrillary tangles in Alzheimer’s disease. Proc. Nat. Acad. Sci. USA 2003, 100, 10032–10037. [Google Scholar] [CrossRef]
- Sakono, M.; Zako, T. Amyloid oligomers: Formation and toxicity of Aβ oligomers. FEBS J. 2010, 277, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, A. Amyloid β interacts with the amyloid precursor protein: A potential toxic mechanism in Alzheimer’s disease. Nat. Neurosci. 2000, 3, 460–464. [Google Scholar] [CrossRef]
- Du Yan, S. An intracellular protein that binds amyloid-β peptide and mediates neurotoxicity in Alzheimer’s disease. Nature 1997, 389, 689–695. [Google Scholar] [CrossRef]
- Walsh, D.M. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Soto, C. Transmissible proteins: Expanding the prion heresy. Cell 2012, 149, 968–977. [Google Scholar] [CrossRef] [PubMed]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Fu, Z.; Meng, L.; He, M.; Zhang, Z. The early events that initiate β-amyloid aggregation in Alzheimer’s disease. Front. Aging Neurosci. 2018, 10, 359. [Google Scholar] [CrossRef]
- Friesen, M.; Meyer-Luehmann, M. Aβ seeding as a tool to study cerebral amyloidosis and associated pathology. Front. Mol. Neurosci. 2019, 12, 233. [Google Scholar] [CrossRef]
- Grant, W.B.; Campbell, A.; Itzhaki, R.F.; Savory, J. The significance of environmental factors in the etiology of Alzheimer’s disease. J. Alzheim. Dis. 2002, 4, 179–189. [Google Scholar] [CrossRef]
- Heneka, M.T. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Zhan, X.; Stamova, B.; Sharp, F. Lipopolysaccharide Associates with Amyloid Plaques, Neurons and Oligodendrocytes in Alzheimer’s Disease Brain: A Review. Front. Ag. Neurosci. 2018, 10, 42. [Google Scholar] [CrossRef]
- Friedland, R.P.; McMillan, J.D.; Kurlawala, Z. What are the molecular mechanisms by which functional bacterial amyloids influence amyloid beta deposition and neuroinflammation in neurodegenerative disorders? Int. J. Mol. Sci. 2020, 21, 1652. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Ritter, A.; Zhong, K. Alzheimer’s disease drug development pipeline: 2018. Alzheim. Dem. Trans. Res. Clin. Int. 2018, 4, 195–214. [Google Scholar] [CrossRef]
- Beffert, U.; Bertrand, P.; Champagne, D.; Gauthier, S.; Poirier, J. HSV-1 in brain and risk of Alzheimer’s disease. Lancet 1998, 351, 1330–1331. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Lukiw, W.J. Alzheimer’s disease and the microbiome. Front. Cellul. Neurosci. 2013, 7, 153. [Google Scholar] [CrossRef] [PubMed]
- Carter, C. Genetic, Transcriptome, Proteomic, and Epidemiological Evidence for Blood-Brain Barrier Disruption and Polymicrobial Brain Invasion as Determinant Factors in Alzheimer’s Disease. J. Alzheim. Dis. Rep. 2017, 1, 125–157. [Google Scholar] [CrossRef]
- Friedland, R.P.; Chapman, M.R. The role of microbial amyloid in neurodegeneration. PLoS Pathog. 2017, 13, e1006654. [Google Scholar] [CrossRef] [PubMed]
- Pisa, D.; Alonso, R.; Fernández-Fernández, A.; Rábano, A.; Carrasco, L. Polymicrobial Infections In Brain Tissue From Alzheimer’s Disease Patients. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Yeh, Y.H.; Gunasekharan, V.; Manuelidis, L. A prokaryotic viral sequence is expressed and conserved in mammalian brain. Proc. Nat. Acad. Sci. USA 2017, 114, 7118–7123. [Google Scholar] [CrossRef]
- Dominy, S.S. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 2019, 5, eaau3333. [Google Scholar] [CrossRef] [PubMed]
- Tetz, G.; Tetz, V. Bacteriophages as new human viral pathogens. Microorganisms 2018, 6, 54. [Google Scholar] [CrossRef]
- Cryan, J.F. The microbiota-gut-brain axis. Physiol. Rev. 2019, 99, 1877–2013. [Google Scholar] [CrossRef]
- Marizzoni, M. Short-chain fatty acids and lipopolysaccharide as mediators between gut dysbiosis and amyloid pathology in Alzheimer’s disease. J. Alzheimer’s Dis. Prepr. 2020, 78, 683–697. [Google Scholar] [CrossRef]
- Parker, A.; Fonseca, S.; Carding, S.R. Gut microbes and metabolites as modulators of blood-brain barrier integrity and brain health. Gut Microb. 2020, 11, 135–157. [Google Scholar] [CrossRef] [PubMed]
- Tetz, V.; Tetz, G. Bacterial DNA induces the formation of heat-resistant disease-associated proteins in human plasma. Sci. Rep. 2019, 9, 1–10. [Google Scholar]
- Tetz, G.; Pinho, M.; Pritzkow, S.; Mendez, N.; Soto, C.; Tetz, V. Bacterial DNA promotes Tau aggregation. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tetz, G.; Brown, S.; Hao, Y.; Tetz, V. Parkinson’s disease and bacteriophages as its overlooked contributors. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Tetz, G.; Brown, S.M.; Hao, Y.; Tetz, V. Type 1 diabetes: An association between autoimmunity, the dynamics of gut amyloid-producing E. coli and their phages. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Tetz, V.; Tetz, G. Effect of deoxyribonuclease I treatment for dementia in end-stage Alzheimer’s disease: A case report. J. Med. Case Rep. 2016, 10, 1–3. [Google Scholar] [CrossRef]
- Coureuil, M.; Lécuyer, H.; Bourdoulous, S.; Nassif, X. A journey into the brain: Insight into how bacterial pathogens cross blood–brain barriers. Nat. Rev. Microb. 2017, 15, 149–159. [Google Scholar] [CrossRef]
- Dehhaghi, M.; Kazemi Shariat Panahi, H.; Guillemin, G.J. Microorganisms’ footprint in neurodegenerative diseases. Front. Cellul. Neurosci. 2018, 12, 466. [Google Scholar] [CrossRef]
- Tkáčová, Z. Identification of the proteins of Borrelia garinii interacting with human brain microvascular endothelial cells. Ticks Tick Borne Dis. 2020, 11, 101451. [Google Scholar] [CrossRef]
- Van De Haar, H.J.; Burgmans, S.; Jansen, J.F.; Van Osch, M.J.; Van Buchem, M.A.; Muller, M.; Hofman, P.A.; Verhey, F.R.; Backes, W.H. Blood-brain barrier leakage in patients with early Alzheimer disease. Radiology 2016, 281, 527–535. [Google Scholar] [CrossRef]
- Li, F.; Hearn, M.; Bennett, L.E. The role of microbial infection in the pathogenesis of Alzheimer’s disease and the opportunity for protection by anti-microbial peptides. Crit. Rev. Microbiol. 2021, 5, 1–4. [Google Scholar]
- Olsen, I.; Singhrao, S. Can oral infection be a risk factor for Alzheimer’s disease? J. Oral Microbiol. 2015, 7, 29143. [Google Scholar] [CrossRef]
- Kamer, A.R.; Craig, R.G.; Dasanayake, A.P.; Brys, M.; Glodzik-Sobanska, L.; de Leon, M.J. Inflammation and Alzheimer’s disease: Possible role of periodontal diseases. Alzheim. Dem. 2008, 4, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Gade Malmos, K. ThT 101: A primer on the use of thioflavin T to investigate amyloid formation. Amyloid 2017, 24, 1–6. [Google Scholar] [CrossRef]
- Salvadores, N.; Shahnawaz, M.; Scarpini, E.; Tagliavini, F.; Soto, C. Detection of Misfolded Aβ Oligomers for Sensitive Biochemical Diagnosis of Alzheimer’s Disease. Cell Rep. 2014, 7, 261–268. [Google Scholar] [CrossRef]
- Kumar, S.; Walter, J. Phosphorylation of amyloid beta (Aβ) peptides–A trigger for formation of toxic aggregates in Alzheimer’s disease. Aging 2013, 3, 803. [Google Scholar] [CrossRef] [PubMed]
- Friedland, R.P. Mechanisms of molecular mimicry involving the microbiota in neurodegeneration. J. Alzheim. Dise. 2015, 45, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Vogt, N.M. Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Alonso, R.; Pisa, D.; Fernández-Fernández, A.M.; Carrasco, L. Infection of fungi and bacteria in brain tissue from elderly persons and patients with Alzheimer’s disease. Front. Ag. Neurosci. 2018, 10, 159. [Google Scholar] [CrossRef]
- Tetz, V.V.; Tetz, G.V. A new biological definition of life. Biomol. Concepts 2019, 11, 1–6. [Google Scholar] [CrossRef]
- Shoemark, D.K.; Allen, S.J. The microbiome and disease: Reviewing the links between the oral microbiome, aging, and Alzheimer’s disease. J. Alzheim. Dis. 2013, 43, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Weiss, N.; Miller, F.; Cazaubon, S.; Couraud, P.O. The blood-brain barrier in brain homeostasis and neurological diseases. Biochim. Biophys. Acta (BBA) Biomem. 2009, 1788, 842–857. [Google Scholar] [CrossRef] [PubMed]
- Hashioka, S. The possible causal link of periodontitis to neuropsychiatric disorders: More than psychosocial mechanisms. Int. J. Molecul. Sci. 2019, 20, 3723. [Google Scholar] [CrossRef] [PubMed]
- Chatani, E.; Yamamoto, N. Recent progress on understanding the mechanisms of amyloid nucleation. Biophyss. Rev. 2008, 10, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K. presence of intrinsically disordered proteins can inhibit the nucleation phase of amyloid fibril formation of Aβ (1–42) in amino acid sequence independent manner. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Kurihara, K.; Tamura, M.; Shohda, K.I.; Toyota, T.; Suzuki, K.; Sugawara, T. Self-reproduction of supramolecular giant vesicles combined with the amplification of encapsulated DNA. Nat. Chem. 2011, 3, 775. [Google Scholar] [CrossRef]
- Vilasi, S.; Sarcina, R.; Maritato, R.; De Simone, A.; Irace, G.; Sirangelo, I. Heparin induces harmless fibril formation in amyloidogenic W7FW14F apomyoglobin and amyloid aggregation in wild-type protein in vitro. PLoS ONE 2011, 13, e22076. [Google Scholar] [CrossRef] [PubMed]
- Iannuzzi, C.; Irace, G.; Sirangelo, I. The effect of glycosaminoglycans (GAGs) on amyloid aggregation and toxicity. Molecules 2015, 20, 2510–2528. [Google Scholar] [CrossRef]
- Bromfield, S.M.; Smith, D.K. Heparin versus DNA: Chiral preferences in polyanion binding to self-assembled multivalent (SAMul) nanostructures. J. Am. Chem. Soc. 2015, 137, 10056–10059. [Google Scholar] [CrossRef] [PubMed]
- Glebova, K.; Konorova, I.; Poleshchuk, V.; Baidakova, G.; Veiko, N. Properties of Extracellular DNA from the Cerebrospinal Fluid and Blood Plasma during Parkinson’s Disease. Bullet. Exp. Biol. Med. 2014, 156, 826–828. [Google Scholar] [CrossRef] [PubMed]
- Connolly, I. A pilot study on the use of cerebrospinal fluid cell-free DNA in intramedullary spinal ependymoma. J. Neuro-Oncol. 2017, 135, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.; Keeney, P.; Brohawn, D. RNA Sequencing Reveals Small and Variable Contributions of Infectious Agents to Transcriptomes of Postmortem Nervous Tissues From Amyotrophic Lateral Sclerosis, Alzheimer’s Disease and Parkinson’s Disease Subjects, and Increased Expression of Genes From Disease-Activated Microglia. Front. Neurosci. 2019, 13, 235. [Google Scholar] [PubMed]
- Crespo, R.; Villar-Alvarez, E.; Taboada, P.; Rocha, F.A.; Damas, A.M.; Martins, P.M. What can the kinetics of amyloid fibril formation tell about off-pathway aggregation? J. Biol. Chem. 2016, 291, 2018–2032. [Google Scholar] [CrossRef]
- Walsh, D.M. Amyloid β-protein fibrillogenesis: Structure and biological activity of protofibrillar intermediates. J. Biol. Chem. 1999, 274, 25945–25952. [Google Scholar] [CrossRef] [PubMed]
- Habicht, G. Directed selection of a conformational antibody domain that prevents mature amyloid fibril formation by stabilizing Aβ protofibrils. Proc. Nat. Acad. Sci. USA 2007, 104, 19232–19237. [Google Scholar] [CrossRef]
- Peng, C. Cellular milieu imparts distinct pathological α-synuclein strains in α-synucleinopathies. Nature 2018, 557, 558–563. [Google Scholar] [CrossRef]
- Ferreira, N. Multiple system atrophy-associated oligodendroglial protein p25α stimulates formation of novel α-synuclein strain with enhanced neurodegenerative potential. Acta Neuropathol. 2021, 12, 1–29. [Google Scholar]
- Cohen, M.; Appleby, B.; Safar, J.G. Distinct prion-like strains of amyloid beta implicated in phenotypic diversity of Alzheimer’s disease. Prion 2016, 10, 9–17. [Google Scholar] [CrossRef]
- Pietronigro, E.C.; Della Bianca, V.; Zenaro, E.; Constantin, G. NETosis in Alzheimer’s disease. Front. Immunol. 2017, 8, 211. [Google Scholar] [CrossRef]
- Cerovic, M.; Forloni, G.; Balducci, C. Neuroinflammation and the gut microbiota: Possible alternative therapeutic targets to counteract Alzheimer’s disease? Front. Aging Neurosci. 2019, 11, 284. [Google Scholar] [CrossRef]
- Poole, S.; Singhrao, S.; Kesavalu, L.; Curtis, M.; Crean, S. Determining the Presence of Periodontopathic Virulence Factors in Short-Term Postmortem Alzheimer’s Disease Brain Tissue. J. Alzheim. Disease 2013, 36, 665–677. [Google Scholar] [CrossRef]
- Tohidpour, A. Neuroinflammation and infection: Molecular mechanisms associated with dysfunction of neurovascular unit. Front. Cellul. Infect. Microbiol. 2017, 7, 276. [Google Scholar] [CrossRef]
- Guzman-Martinez, L.; Maccioni, R.B.; Andrade, V.; Navarrete, L.P.; Pastor, M.G.; Ramos-Escobar, N. Neuroinflammation as a common feature of neurodegenerative disorders. Front. Pharmacol. 2019, 10, 1008. [Google Scholar] [CrossRef] [PubMed]
- Ismail, R. The relationships between neuroinflammation, beta-amyloid and tau deposition in Alzheimer’s disease: A longitudinal PET study. J. Neuroinf. 2020, 17, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.G. Exposure to the functional bacterial amyloid protein curli enhances alpha-synuclein aggregation in aged Fischer 344 rats and Caenorhabditis elegans. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Van Den Berge, N. Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of alpha-synuclein in rats. Acta Neuropathol. 2019, 138, 535–550. [Google Scholar] [CrossRef]
- Kim, S. Transneuronal propagation of pathologic α-synuclein from the gut to the brain models Parkinson’s disease. Neuron 2019, 103, 627–641. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Sommerville, N.R.; Liu, J.Y.; Ngan, M.P.; Poon, D.; Ponomarev, E.D.; Lu, Z.; Kung, J.S.; Rudd, J.A. Intra-gastrointestinal amyloid-β1–42 oligomers perturb enteric function and induce Alzheimer’s disease pathology. J. Physiol. 2020, 598, 4209–4223. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, N. Trans-synaptic spreading of alpha-synuclein pathology through sensory afferents leads to sensory nerve degeneration and neuropathic pain. Acta Neuropathol. Commun. 2021, 9, 1–17. [Google Scholar] [CrossRef]
- Braak, H.; Rub, U.; Gai, W.; Del Tredici, K. Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural Transm. 2003, 110, 517–536. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tetz, G.; Tetz, V. Bacterial Extracellular DNA Promotes β-Amyloid Aggregation. Microorganisms 2021, 9, 1301. https://doi.org/10.3390/microorganisms9061301
Tetz G, Tetz V. Bacterial Extracellular DNA Promotes β-Amyloid Aggregation. Microorganisms. 2021; 9(6):1301. https://doi.org/10.3390/microorganisms9061301
Chicago/Turabian StyleTetz, George, and Victor Tetz. 2021. "Bacterial Extracellular DNA Promotes β-Amyloid Aggregation" Microorganisms 9, no. 6: 1301. https://doi.org/10.3390/microorganisms9061301
APA StyleTetz, G., & Tetz, V. (2021). Bacterial Extracellular DNA Promotes β-Amyloid Aggregation. Microorganisms, 9(6), 1301. https://doi.org/10.3390/microorganisms9061301