
Gut Dysbiosis and IL-21 Response in Patients with Severe COVID-19

 ,
,
Abstract
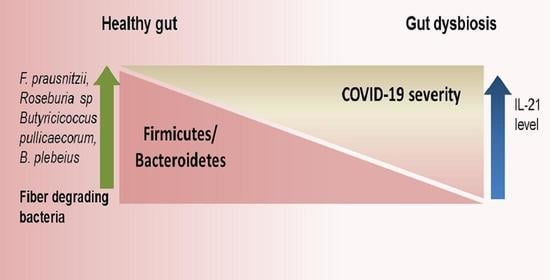
1. Background
2. Material and Methods
2.1. Study Design and Sample Collection
2.2. Study Groups
2.3. Exclusion and Inclusion Criteria
2.4. Gut Microbiome Analysis
2.4.1. DNA Extraction
2.4.2. The Next-Generation Sequencing of Metagenomic DNA
2.4.3. Sequence Data Processing and Statistical Analysis
2.5. Quantification of Plasma IL-21, TNF-α, and INF-γ
3. Results
3.1. COVID-19-Infected Patients
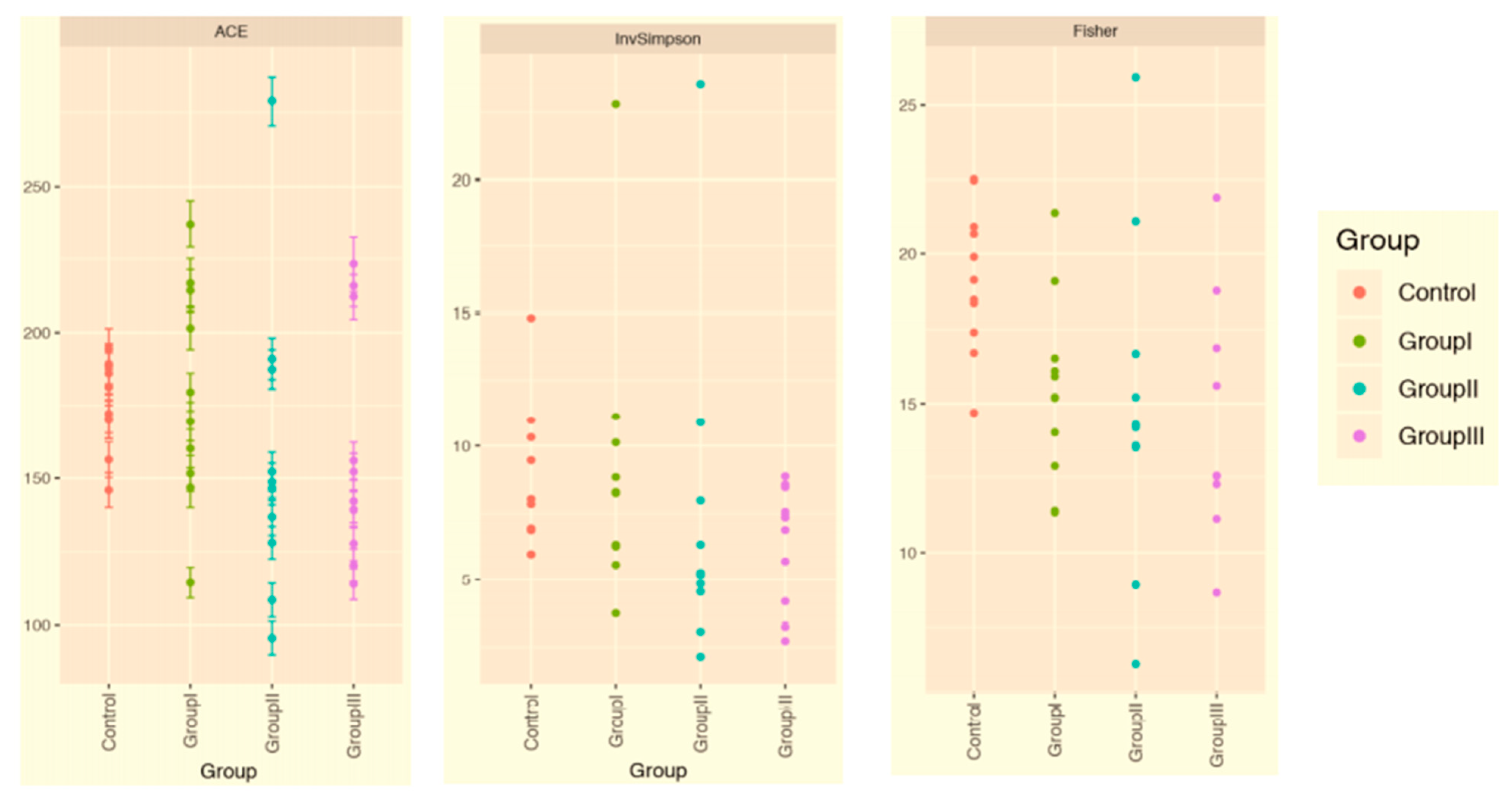
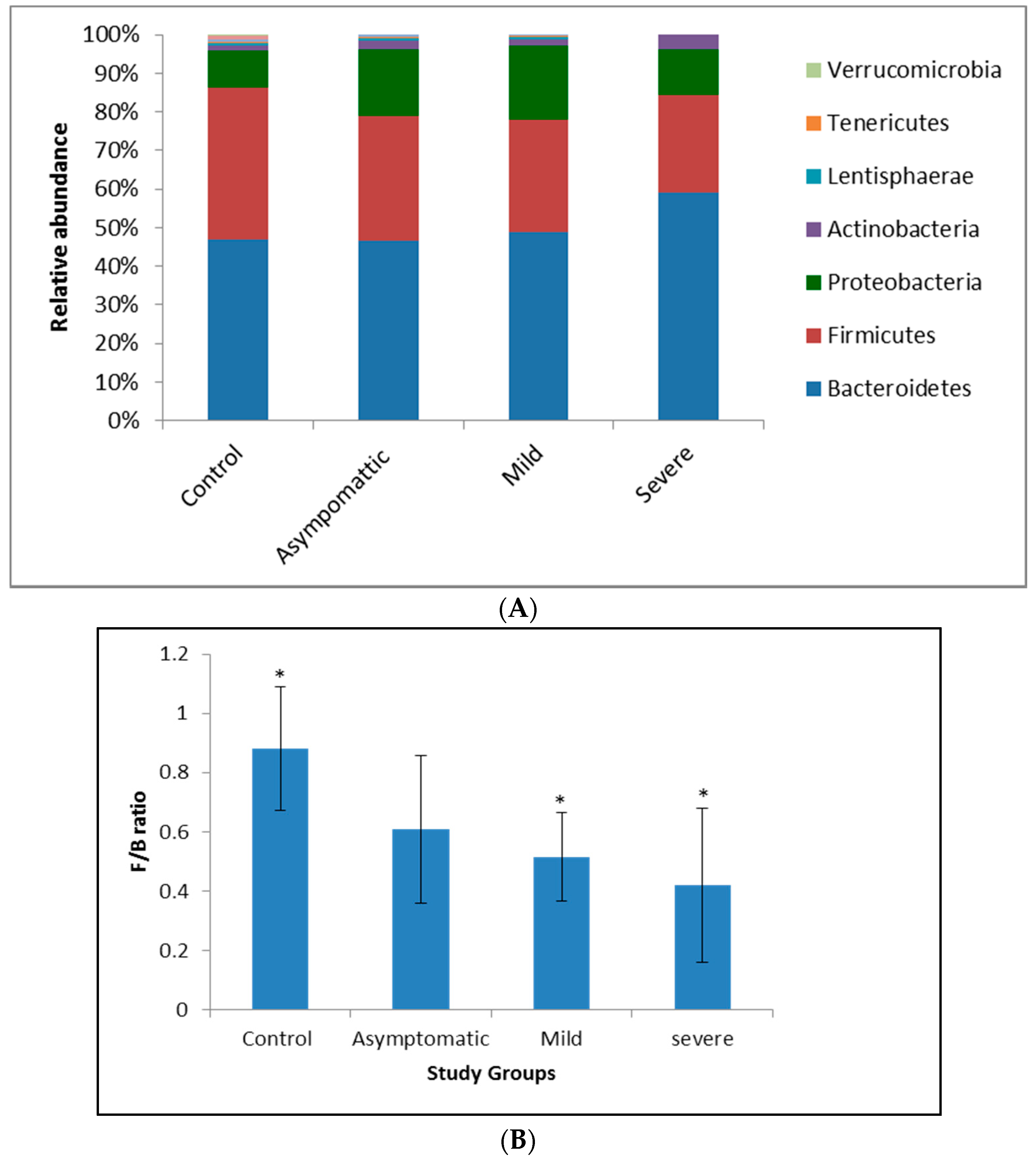
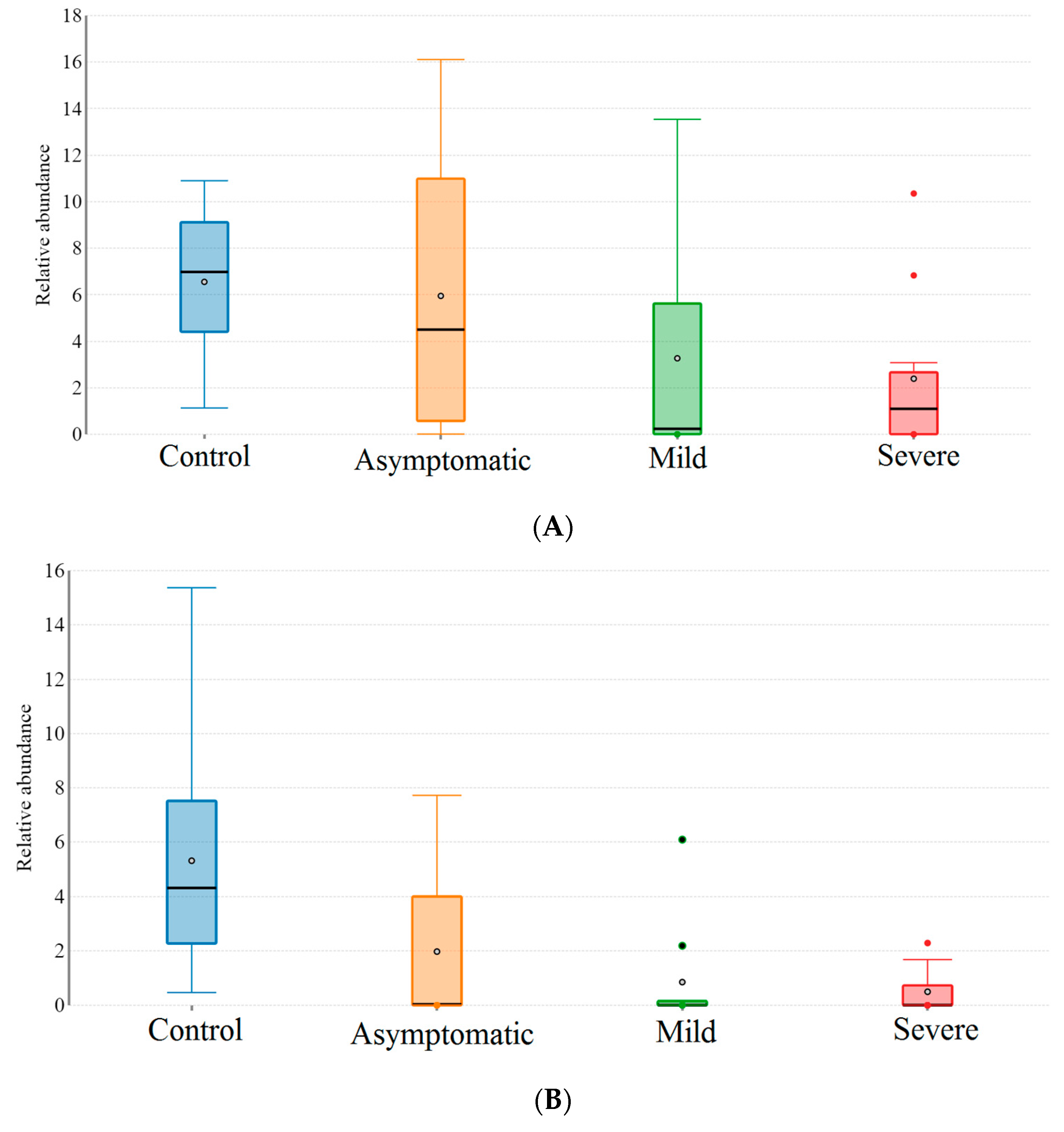
3.2. Patients with Different Severity Levels Had Altered Gut Microbiota That Was Less Diverse
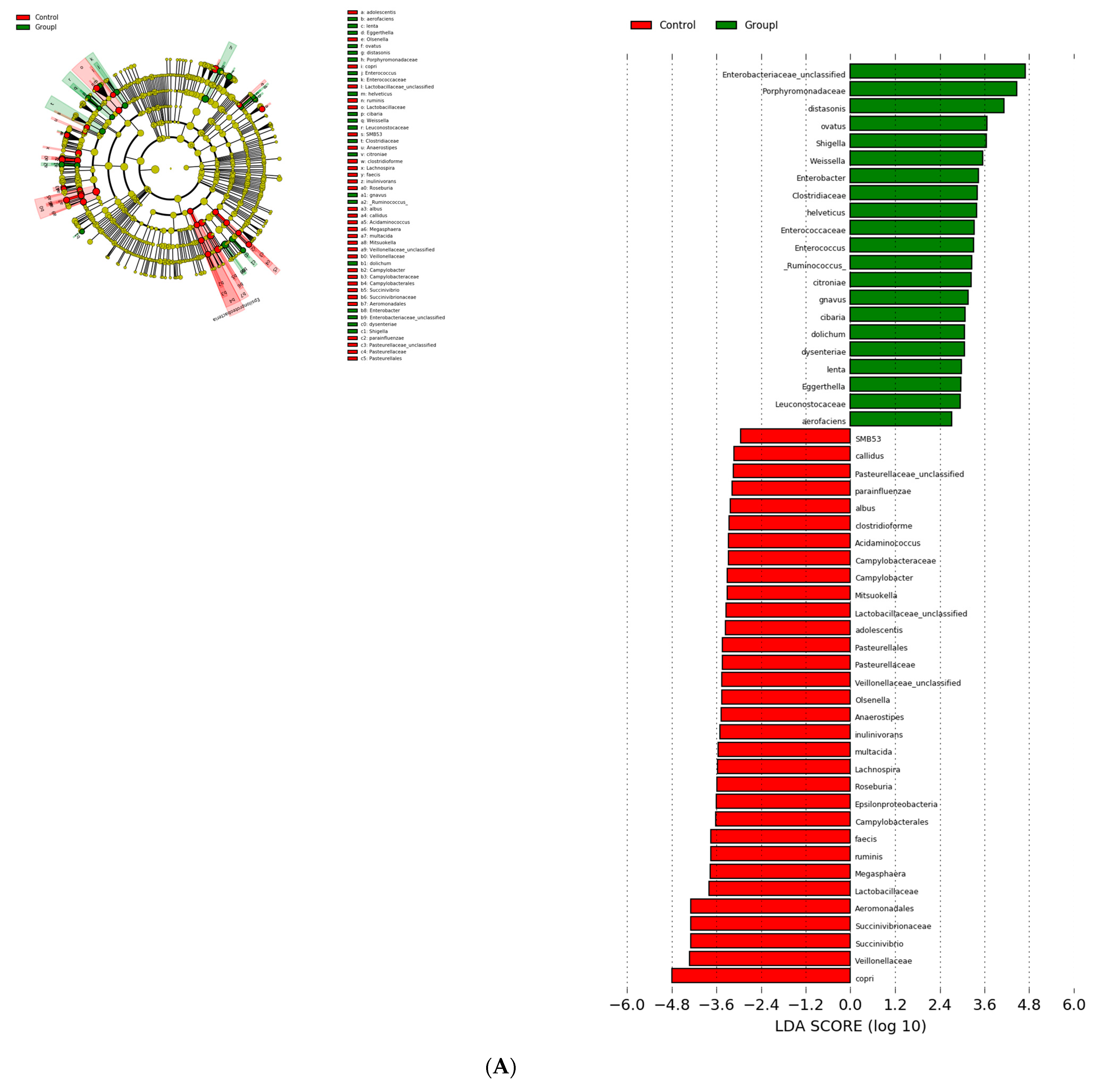
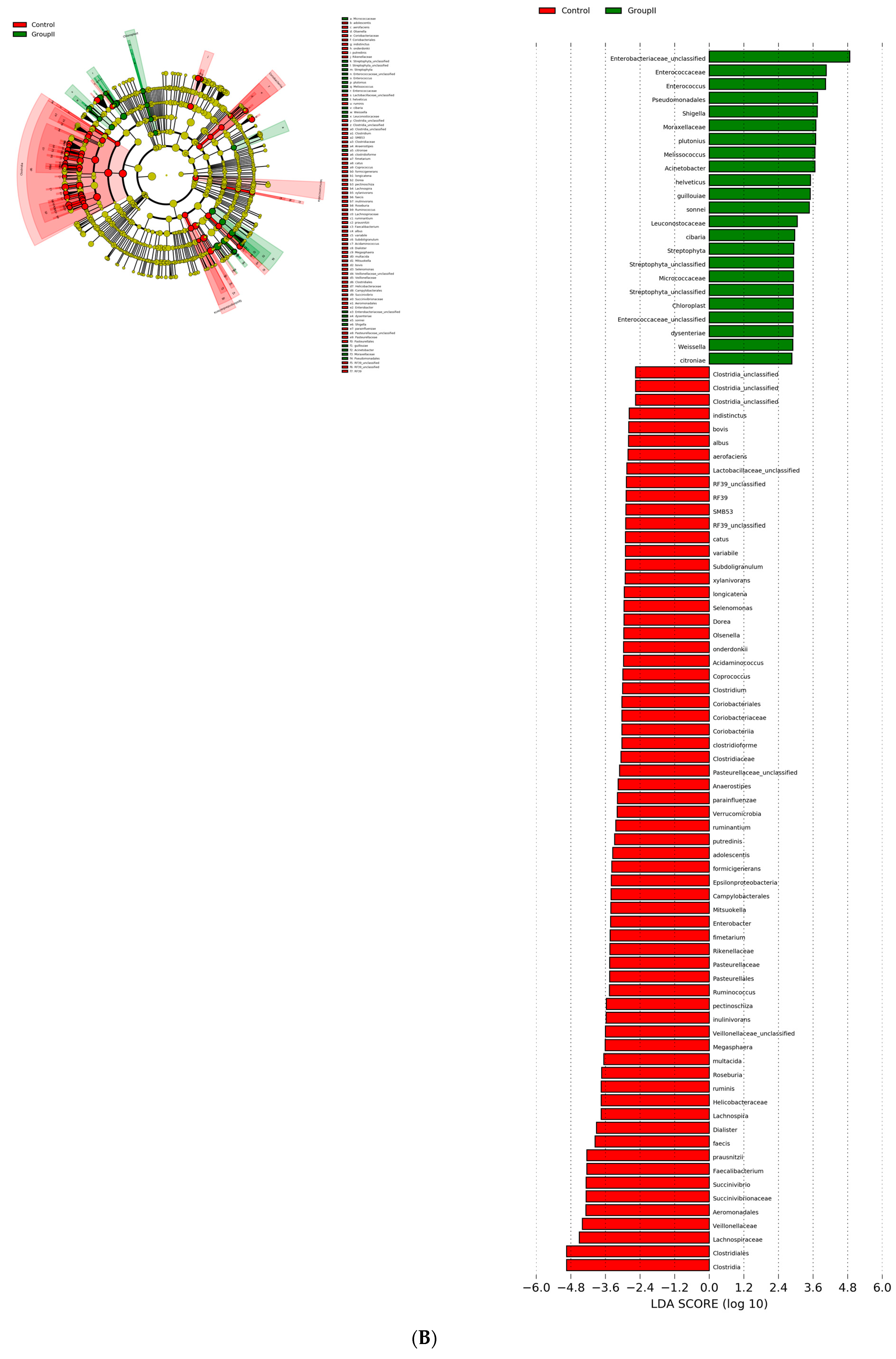
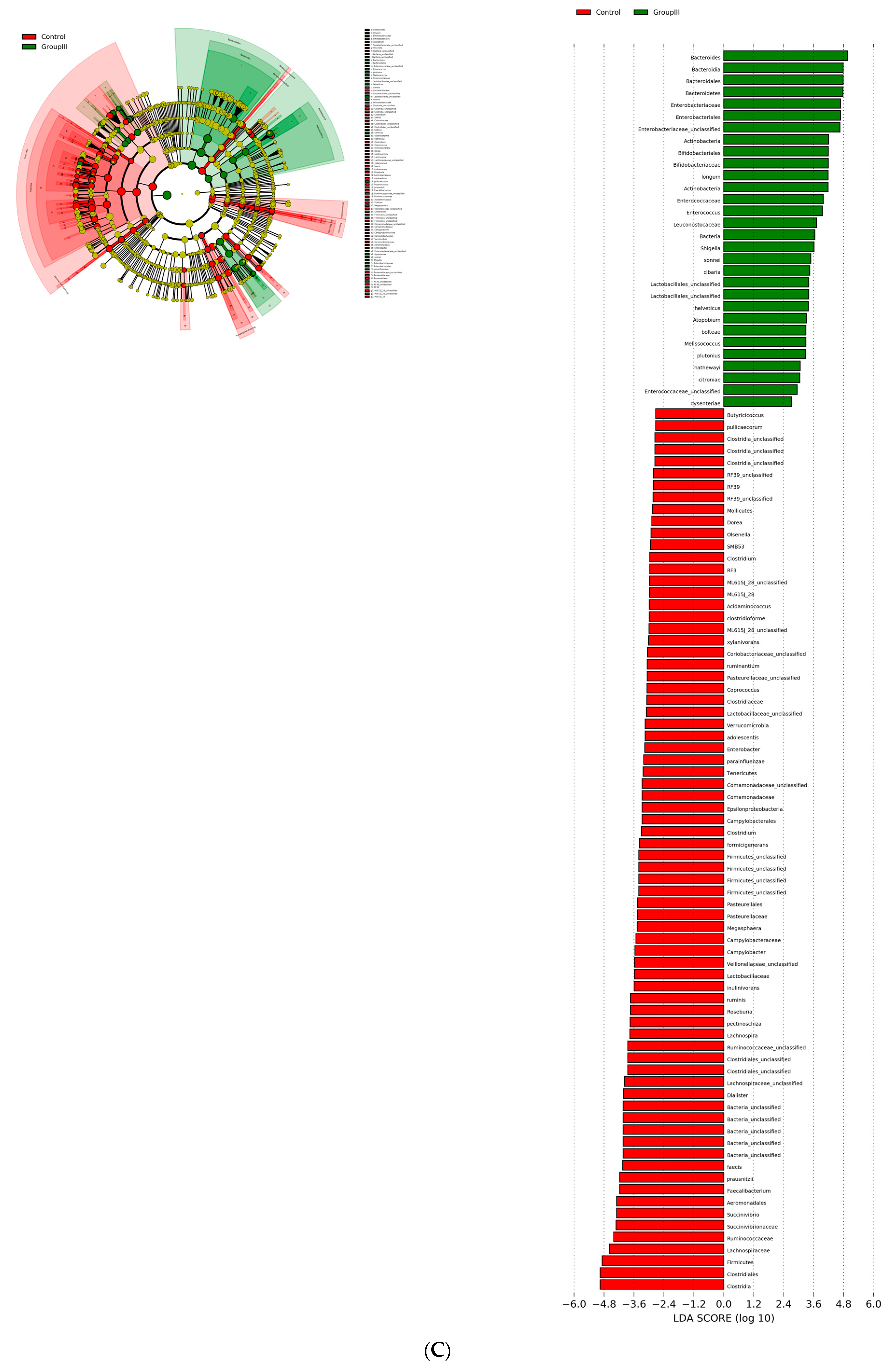
3.3. Bacterial Genera and Species Differences in the Guts of COVID-19 Patients
3.4. Severe COVID-19 Patients Have Elevated Circulating IL-21 Levels
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, Q.; Guan, X.; Wu, P.; Wang, X.; Zhou, L.; Tong, Y.; Ren, R.; Leung, K.S.M.; Lau, E.H.Y.; Wong, J.Y.; et al. Early Transmission Dynamics in Wuhan, China, of Novel Coronavirus-Infected Pneumonia. N. Engl. J. Med. 2020, 382, 1199–1207. [Google Scholar] [CrossRef]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor recognition by novel coronavírus from Wuhan: An analysis based on decade-long structural studies of SARS. J. Virol. 2020, 94, e00127-20. [Google Scholar] [CrossRef] [PubMed]
- Penninger, J.M.; Grant, M.B.; Sung, J.J. The role of angiotensin converting enzyme 2 in modulating gut microbiota, intestinal inflammation, and coronavirus infection. Gastroenterology 2021, 160, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Viana, S.D.; Nunes, S.; Reis, F. ACE2 imbalance as a key player for the poor outcomes in COVID-19 patients with age-related comorbidities—Role of gut microbiota dysbiosis. Ageing Res. Rev. 2020, 62, 101123. [Google Scholar] [CrossRef]
- Camargo, S.M.R.; Vuille-Dit-Bille, R.N.; Meier, C.F.; Verrey, F. ACE2 and gut amino acid transport. Clin. Sci. 2020, 134, 2823–2833. [Google Scholar] [CrossRef] [PubMed]
- Byrne, C.S.; Chambers, E.S.; Alhabeeb, H.; Chhina, N.; Morrison, D.J.; Preston, T.; Tedford, C.; Fitzpatrick, J.; Irani, C.; Busza, A.; et al. Increased colonic propionate reduces anticipatory reward responses in the human striatum to high-energy foods. Am. J. Clin. Nutr. 2016, 104, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D. Human gut microbiome: Hopes, threats and promises. Gut 2018, 67, 1716–1725. [Google Scholar] [CrossRef]
- Pagliari, D.; Gambassi, G.; Piccirillo, C.A.; Cianci, R. The Intricate Link among Gut “Immunological Niche”, Microbiota, and Xenobiotics in Intestinal Pathology. Mediat. Inflamm. 2017, 2017, 8390595. [Google Scholar] [CrossRef]
- Geva-Zatorsky, N.; Sefik, E.; Kua, L.; Pasman, L.; Tan, T.G.; Ortiz-Lopez, A.; Yanortsang, T.B.; Yang, L.; Jupp, R.; Mathis, D.; et al. Mining the human gut microbiota for immunomodulatory organisms. Cell 2017, 168, 928–943.e11. [Google Scholar] [CrossRef]
- Zang, R.; Gomez Castro, M.F.; McCune, B.T.; Zeng, Q.; Rothlauf, P.W.; Sonnek, N.M.; Liu, Z.; Brulois, K.F.; Wang, X.; Greenberg, H.B.; et al. TMPRSS2 and TMPRSS4 promote SARS-CoV-2 infection of human small intestinal enterocytes. Sci. Immunol. 2020, 5, eabc3582. [Google Scholar] [CrossRef] [PubMed]
- Lamers, M.M.; Beumer, J.; Van Der Vaart, J.; Knoops, K.; Puschhof, J.; Breugem, T.I.; Ravelli, R.B.G.; Van Schayck, J.P.; Mykytyn, A.Z.; Duimel, H.Q.; et al. SARS-CoV-2 productively infects human gut enterocytes. Science 2020, 69, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Babraham Bioinformatics—Trim Galore. 2017. Available online: https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 12 October 2020).
- Andrews, S. FastQC A Quality Control Tool for High Throughput Sequence Data. 2017. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 12 October 2020).
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef]
- Lucas, C.; Wong, P.; Klein, J.; Castro, T.; Silva, J.; Sundaram, M.; Ellingson, M.K.; Mao, T.; Oh, J.E.; Israelow, B.; et al. Longitu-dinal analyses reveal immunological misfiring in severe COVID-19. Nature 2020, 584, 463–469. [Google Scholar] [CrossRef]
- Schirmer, M.; Smeekens, S.P.; Vlamakis, H.; Jaeger, M.; Oosting, M.; Franzosa, E.A.; Ter Horst, R.; Jansen, T.; Jacobs, L.; Bonder, M.J.; et al. Linking the human gut microbiome to inflammatory cytokine production capacity. Cell 2016, 167, 1125–1136.e8. [Google Scholar] [CrossRef]
- Calder, P.C. Nutrition, immunity and COVID-19. BMJ Nutr. Prevand Health 2020, 3, 74–92. [Google Scholar] [CrossRef]
- Cai, Q.; Chen, F.; Wang, T.; Luo, F.; Liu, X.; Wu, Q.; He, Q.; Wang, Z.; Liu, Y.; Liu, L.; et al. Obesity and COVID-19 Severity in a Designated Hospital in Shenzhen, China. Diabetes Care 2020, 43, 1392–1398. [Google Scholar] [CrossRef]
- Hill, M.A.; Mantzoros, C.; Sowers, J.R. Commentary: COVID-19 in patients with diabetes. Metabolism 2020, 107, 154217. [Google Scholar] [CrossRef] [PubMed]
- Mazza, S.; Sorce, A.; Peyvandi, F.; Vecchi, M.; Caprioli, F. A fatal case of COVID-19 pneumonia occurring in a patient with severe acute ulcerative colitis. Gut 2020, 69, 1148–1149. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Zmora, N.; Adolph, T.E.; and Elinav, E. The intestinal microbiota fuelling metabolic inflammation. Nat. Rev. Immunol. 2020, 20, 40–54. [Google Scholar] [CrossRef]
- Mariat, D.; Firmesse, O.; Levenez, F.; Guimarăes, V.; Sokol, H.; Doré, J.; Corthier, G.; Furet, J.P. The Firmicutes/Bacteroidetes ratio of the human microbiota changes with age. BMC Microbial. 2009, 9, 123. [Google Scholar] [CrossRef] [PubMed]
- Claesson, M.J.; Cusack, S.; O’Sullivan, O.; Greene-Diniz, R.; de Weerd, H.; Flannery, E.; Marchesi, J.R.; Falush, D.; Dinan, T.; Fitzgerald, G.; et al. Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4586–4591. [Google Scholar] [CrossRef]
- Lin, C.H.; Chen, C.C.; Chiang, H.L.; Liou, J.M.; Chang, C.M.; Lu, T.P.; Chuang, E.Y.; Tai, Y.C.; Cheng, C.; Lin, H.Y.; et al. Altered gut microbiota and inflammatory cytokine responses in patients with Parkinson’s disease. J. Neuroinflamm. 2019, 16, 129. [Google Scholar] [CrossRef] [PubMed]
- Hevia, A.; Milani, C.; López, P.; Cuervo, A.; Arboleya, S.; Duranti, S.; Turroni, F.; González, S.; Suárez, A.; Gueimonde, M.; et al. Intestinal dysbiosis associated with systemic lupus erythematosus. mBio 2014, 5, e01548-14. [Google Scholar] [CrossRef]
- Johnson, B.M.; Gaudreau, M.C.; Al-Gadban, M.M.; Gudi, R.; Vasu, C. Impact of dietary deviation on disease progression and gut microbiome composition in lupus-prone SNF1 mice. Clin. Exp. Immunol. 2015, 181, 323–337. [Google Scholar] [CrossRef]
- Zuo, T.; Zhang, F.; Lui, G.C.; Yeoh, Y.K.; Li, A.Y.; Zhan, H.; Wan, Y.; Chung, A.C.; Cheung, C.P.; Chen, N.; et al. Alterations in gut microbiota of patients with COVID-19 during time of hospitalization. Gastroenterology 2020, 159, 944–955. [Google Scholar] [CrossRef]
- Hashimoto, T.; Perlot, T.; Rehman, A.; Trichereau, J.; Ishiguro, H.; Paolino, M.; Sigl, V.; Hanada, T.; Hanada, R.; Lipinski, S.; et al. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature 2012, 487, 477–481. [Google Scholar] [CrossRef]
- Perlot, T.; Penninger, J.M. ACE2—from the renin-angiotensin system to gut microbiota and malnutrition. Microbes Infect. 2013, 15, 866–873. [Google Scholar] [CrossRef]
- Yun, E.J.; Lee, S.; Kim, J.H.; Kim, B.B.; Kim, H.T.; Lee, S.H.; Pelton, J.G.; Kang, N.J.; Choi, I.G.; Kim, K.H. Enzymatic production of 3,6-anhydro-L-galactose from agarose and its purification and in vitro skin whitening and anti-inflammatory activities. Appl. Microbiol. Biotechnol. 2013, 97, 2961–2970. [Google Scholar] [CrossRef]
- Zou, Y.; Fu, X.; Liu, N.; Duan, D.; Wang, X.; Xu, J.; Gao, X. The synergistic anti-inflammatory activities of agaro-oligosaccharides with different degrees of polymerization. J. Appl. Phycol. 2019, 31, 2547–2558. [Google Scholar] [CrossRef]
- Park, N.J.; Yu, S.; Kim, D.H.; Yun, E.J.; Kim, K.H. Characterization of Bp GH16A of Bacteroides plebeius, a key enzyme initiating the depolymerization of agarose in the human gut. Appl. Microbiol. Biotechnol. 2021, 105, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Shang, Q.; Li, G.; Wang, X.; Yu, G. Degradation of Marine Algae-Derived Carbohydrates by Bacteroidetes Isolated from Human Gut Microbiota. Mar. Drugs 2017, 15, 92. [Google Scholar] [CrossRef]
- Wang, W.; Liu, P.; Hao, C.; Wu, L.; Wan, W.; Mao, X. Neoagaro-oligosaccharide monomers inhibit inflammation in LPS-stimulated macrophages through suppression of MAPK and NF-κB pathways. Sci. Rep. 2017, 7, 44252. [Google Scholar] [CrossRef]
- Shin, N.-R.; Whon, T.W.; Bae, J.-W. Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Lavelle, A.; Lennon, G.; O’Sullivan, O.; Docherty, N.; Balfe, A.; Maguire, A.; Mulcahy, H.E.; Doherty, G.; O’Donoghue, D.; Hyland, J.; et al. Spatial variation of the colonic microbiota in patients with ulcerative colitis and control volunteers. Gut 2015, 64, 1553–1561. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Long, W.; Hao, B.; Ding, D.; Ma, X.; Zhao, L.; Pang, X. A human stool-derived Bilophila wadsworthia strain caused systemic inflammation in specific-pathogen-free mice. Gut Pathog. 2017, 9, 59. [Google Scholar] [CrossRef]
- Yuan, J.; Chen, C.; Cui, J.; Lu, J.; Yan, C.; Wei, X.; Zhao, X.; Li, N.; Li, S.; Xue, G.; et al. Fatty Liver Disease Caused by High-Alcohol-Producing Klebsiella pneumoniae. Cell Metab. 2019, 30, 675–688.e7. [Google Scholar] [CrossRef]
- Litvak, Y.; Byndloss, M.X.; and Bäumler, A.J. Colonocyte metabolism shapes the gut microbiota. Science 2018, 362, eaat9076. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.; Pigneur, B.; Watterlot, L.; Lakhdari, O.; Bermúdez-Humarán, L.G.; Gratadoux, J.J.; Blugeon, S.; Bridonneau, C.; Furet, J.P.; Corthier, G.; et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. USA 2008, 105, 16731–16736. [Google Scholar] [CrossRef]
- Quévrain, E.; Maubert, M.A.; Michon, C.; Chain, F.; Marquant, R.; Tailhades, J.; Miquel, S.; Carlier, L.; Bermúdez-Humarán, L.G.; Pigneur, B.; et al. Identification of an anti-inflammatory protein from Faecalibacterium prausnitzii a commensal bacterium deficient in Crohn’s disease. Gut 2016, 65, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Sun, L.; Liu, Y.; Ren, H.; Shen, Y.; Bi, F.; Zhang, T.; Wang, X. Alter between gut bacteria and blood metabolites and the anti-tumor effects of Faecalibacterium prausnitzii in breast cancer. BMC Microbiol. 2020, 20, 82. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, J.; Kubota, T.; Takada, E.; Takai, K.; Fujiwara, N.; Arimitsu, N.; Ueda, Y.; Wakisaka, S.; Suzuki, T.; Suzuki, N. Bifidobacteria abundance-featured gut microbiota compositional change in patients with Behcet’s disease. PLoS ONE 2016, 11, e0153746. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters/Patient Group | Asymptomatic (Mean ± SD) | Mild (Mean ± SD) | Severe (Mean ± SD) | p-Value * |
|---|---|---|---|---|
| Age (years) | 57.9 ± 17.99 | 49.4 ± 13.12 | 45.9 ± 8.82 | 0.34 |
| Total Leukocyte Count (×109/µL) | 6927.78 ± 4342.4 | 10,481.11 ± 5772.13 | 6878 ± 3566.17 | 0.17 |
| Neutrophils | 58.56 ± 22.20 | 77.22 ±17.06 | 76 ± 12.42 | 0.27 |
| Lymphocytes | 32.11 ± 19.34 | 15.55 ± 13.28 | 18.67 ± 11.98 | 0.23 |
| C reactive protein (mg/L) | 11.41 ± 10.54 | 44.66 ± 31.76 | 99.18 ± 41.15 | 0.02 |
| Eosinophil | 1.56 ± 2.65 | 0.5 ± 1.06 | 0.4 ± 0.89 | 0.65 |
| Aspartate aminotransferase (U/L) | 41.08 ± 24.34 | 54.62 ± 28.63 | 54.47 ± 31.59 | 0.78 |
| Alanine aminotransferase (U/L) | 53.74 ± 78.56 | 66.17 ± 58.76 | 53.97 ± 41.72 | 0.76 |
| S.No | Comparison | OTU (abs LDA Score > 2.0) |
|---|---|---|
| 1 | Healthy vs. asymptomatic | 53 |
| 2 | Healthy vs. mildly infected | 89 |
| 3 | Healthy vs. severely infected | 104 |
| Phylum | Species | Percent Reduction in Mean Relative Abundance | ||
|---|---|---|---|---|
| COVID-19 Infected | ||||
| Asymptomatic | Mild | Severe | ||
| Bacteroidetes | * Bacteroides plebeius | 29.3 | 62.2 | 96.6 |
| Firmicutes | Faecalibacterium prausnitzii | 3.32 | 27.9 | 50.59 |
| Firmicutes | Roseburia faecis | 54.70 | 77.24 | 85.87 |
| Firmicutes | Roseburia inulinivorans | 85.2 | 41.52 | 96.17 |
| Firmicutes | Dorea formicigenerans | 30.12 | 69.87 | 61.44 |
| Firmicutes | Lachnospira pectinoschiza | 61.29 | 77.71 | 96.77 |
| Firmicutes | Pseudobutyrivibrio xylanivorans | 50.0 | Absent(100) | Absent(100) |
| Firmicutes | Clostridium ruminantium | 59.09 | Absent(100) | Absent(100) |
| Firmicutes | Butyricicoccus pullicaecorum | 40 | 90.09 | Absent(100) |
| Percent increase in Mean Relative abundance | ||||
| Actinobacteria | Bifidobacterium sp | 87.61 | 126.43 | 347.24 |
| Phylum | Species # | Mean Relative Abundance (%) | p-Value | |
|---|---|---|---|---|
| Control | COVID-19 | |||
| Bacteroidetes | Bacteroides caccae | 1.35 | 6.35 | 0.003 |
| Bacteroidetes | Bacteroides ovatus | 0.22 | 1.85 | 0.0284 |
| Bacteroidetes | Parabacteroides distasonis | 0.736 | 7.887 | 0.376 |
| Bacteroidetes | Bacteroides fragilis | 0.015 | 3.36 | 0.002 |
| Firmicutes | Ruminococcus gnavus | 0.021 | 1.96 | 0.135 |
| Firmicutes | Clostridium bolteae | 2.29 | 3.29 | 0.036 |
| Firmicutes | Clostridium citroniae | 0.02 | 0.888 | 0.013 |
| Firmicutes | Clostridium hathewayi | 0.001 | 0.971 | 0.03 |
| Proteobacteria | Shigella sonnei | 1.95 | 3.48 | 0.0052 |
| Proteobacteria | Shigella dysenteriae | 0.001 | 0.33 | 0.022 |
| Actinobacteria | Atopobium rimae | 4.18 | 4.63 | 0.0303 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, M.; Mathew, B.J.; Gupta, P.; Garg, G.; Khadanga, S.; Vyas, A.K.; Singh, A.K. Gut Dysbiosis and IL-21 Response in Patients with Severe COVID-19. Microorganisms 2021, 9, 1292. https://doi.org/10.3390/microorganisms9061292
Khan M, Mathew BJ, Gupta P, Garg G, Khadanga S, Vyas AK, Singh AK. Gut Dysbiosis and IL-21 Response in Patients with Severe COVID-19. Microorganisms. 2021; 9(6):1292. https://doi.org/10.3390/microorganisms9061292
Chicago/Turabian StyleKhan, Mahejibin, Bijina J. Mathew, Priyal Gupta, Garima Garg, Sagar Khadanga, Ashish Kumar Vyas, and Anirudh K. Singh. 2021. "Gut Dysbiosis and IL-21 Response in Patients with Severe COVID-19" Microorganisms 9, no. 6: 1292. https://doi.org/10.3390/microorganisms9061292
APA StyleKhan, M., Mathew, B. J., Gupta, P., Garg, G., Khadanga, S., Vyas, A. K., & Singh, A. K. (2021). Gut Dysbiosis and IL-21 Response in Patients with Severe COVID-19. Microorganisms, 9(6), 1292. https://doi.org/10.3390/microorganisms9061292