1. Introduction
The prevalence of pasteurellosis in farmed lumpsuckers,
Cyclopterus lumpus, in Norway has increased in recent years since the first case was recorded in 2012 [
1]. Despite a decrease in the number of affected localities [
2],
Pasteurella atlantica [
3] remains a significant problem, and as the disease is non-notifiable, under-reporting of outbreaks cannot be excluded. Furthermore, while farmed lumpsuckers are vaccinated against vibriosis and atypical furunculosis, there are no commercially available vaccines against pasteurellosis.
The family
Pasteurellaceae is composed of commensals, opportunistic- and primary- pathogens and includes the genera
Pasteurella,
Actinobacillus, and
Hemophilus amongst others. The genus
Pasteurella has a broad host range, but little is known regarding the ecology of marine species.
P. atlantica isolated from diseased Norwegian lumpsuckers is related to but serologically distinct from
Pasteurella skyensis, which causes disease in Atlantic salmon (
Salmo salar L.) in Scotland, and a
P. atlantica isolate first detected in 2018, which causes disease in Norwegian farmed Atlantic salmon [
1,
3,
4]. Pasteurellosis affects all life stages of a lumpsucker, from fry to fish deployed in salmon cages [
4]. As
P. atlantica has been detected in lumpsucker eggs and milt, vertical transmission may also be possible [
5].
In a previous work [
6], we showed that whole cell-inactivated bacterin-based vaccines do not provide adequate protection against the disease, despite the high titers of specific antibodies raised. Such a situation highlights the need for a deeper characterization of the bacterium and the identification of immunogenic and protective antigens.
Most pathogenic bacteria have several tools in their genetic arsenal to avoid host defenses and to enhance their survival. Genetic determinants including pathogenicity islands, antibiotic resistance genes, toxins, and adhesins are often shared between bacterial populations via horizontal gene transfer and are commonly associated with plasmids, prophages, and other mobile genetic elements. As adhesins are involved in the early stages of colonization, they can be utilized as targets for vaccine development via reverse vaccinology (RV).
Using in silico bioinformatic analyses, immunogenic antigens for vaccine development can be predicted [
7,
8]. RV is a rapid process and can reduce vaccine development time by up to 2 years [
9]. It is also a more ethical approach due to a reduction in the number of experimental animals required for vaccine testing and results in effective vaccines. In human medicine, RV has been successful in the development of vaccines against several bacterial pathogens reviewed by Sharma et al. 2021 [
10]. Pathogens of significant concern such as
Neisseria meningitidis [
11,
12],
Mycobacterium tuberculosis [
13],
Chlamydia pneumoniae [
14],
Streptococcus pneumoniae [
15],
Helicobacter pylori [
16],
Porphyromonas gingivalis [
17], and
Bacillus anthracis [
18,
19] have been investigated using RV.
RV technologies have improved considerably since the principles were initially developed and applied. The main criteria required for reliable prediction of vaccine targets include subcellular localization, the probability of adhesion functionality, and the number of transmembrane domains [
20]. Additionally, using targets that are present only in virulent strains and having protein sequences that are dissimilar to host sequences results in more reliable vaccine targets that also induce strong immune responses [
9].
Adhesins are a class of surface-bound proteins involved in facilitating bacterial attachment to host tissues. They are classed as fimbrial or non-fimbrial based on the absence or presence, respectively, of an outer membrane anchor in the protein. Among the different types of adhesins, bacterial lectins are the most common [
15]. The mediation of attachment occurs through the recognition of specific carbohydrates, proteins, or lipids presented on the host cell surface.
In addition to attachment, adhesins also promote the delivery of toxins through the upregulation of virulence genes leading to invasion of the host. Furthermore, such interactions can trigger cytokine production or lectinophagocytosis by the host, compounding the severity of disease [
21]. For this reason, subunit vaccines that hinder microbial attachment could be of great advantage in combating pasteurellosis in lumpsuckers. Examples of successful adhesin vaccines include those against enterotoxigenic
E. coli (ETEC) strains in farm animals using the K88 fimbriae [
22] as well as uropathogenic
E. coli (UPEC) known to cause urinary tract infections (UTIs) using the FimCH complex [
23]. For the aquaculture industry, vaccines have been tested by targeting major bacterial adhesins from
Aeromonas hydrophila [
24,
25],
Vibrio harveyi [
26], and
Edwardsiella tarda [
27].
As research on
P. atlantica in lumpsuckers is still in its infancy, no commercial vaccines are available against pasteurellosis in lumpsuckers. Simple phylogenetic analyses using the
rpoB and 16S rRNA genes have shown that NVI-9100 is similar to other
Pasteurella sp. isolated from lumpsuckers [
4]. Whole genome analysis can, however, determine more accurate phylogenetic relationships. The purpose of this study is, therefore, to determine the taxonomic position of the species through whole genome sequencing and to predict potential vaccine antigens via in silico analysis. To further assess the predicted vaccine targets, their expression levels during exposure to lumpsucker leucocytes were analyzed in vitro.
2. Materials and Methods
2.1. Bacterial Culture, Genome Isolation, and Sequencing
Two
P. atlantica isolates described in previous studies (NVI 9100 and UiBP1-2013) [
4,
6,
28] collected from clinically sick lumpsuckers were whole-genome sequenced. Briefly, bacteria were grown in tryptic soy broth (TSB) (Becton Dickinson, Sparks, MD, USA) supplemented with 1.5% NaCl and 10% fetal bovine serum, Australian origin, (Gibco, Waltham, MA, USA) at 20 °C with shaking (200 rpm).
Total genomic DNA was isolated using the DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany) following manufacturer instructions. Briefly, 3 mL of an overnight culture of P. atlantica containing a maximum of 2 × 109 cells was centrifuged at 2500× g for 15 min and the pellet was resuspended in 180 µL of ATL buffer. Twenty microliters of proteinase K were then added, and the bacteria were incubated at 56 °C in a rotating heat block for 1 h. Two hundred microliters of AL buffer and two hundred microliters of 96% ethanol were added, vortexing well between additions. The mixture was then loaded onto DNeasy Mini Spin columns and washed with the appropriate buffers (AW1 and AW2) prior to elution using buffer AE. The eluted DNA was then purified and measured for concentration before storing at −20 °C until further processing.
Extracted DNA was sequenced using either an Illumina (UiBP1-2013) or PacBio (NVI 9100) platform. Illumina libraries were made using the Nextera DNA Flex Sample Prep kit (Illumina, San Diego, CA, USA) according to the manufacturer instructions and sequenced with Illumina MiSeq (Illumina, San Diego, CA, USA) using V3 chemistry. PacBio libraries were prepared with the Pacific Biosciences 20 kb library preparation protocol, and size was selected using BluePippin (Sage Science, Beverly, MA, USA), with subsequent sequencing performed on a Pacific Biosciences RS II instrument using P4-C2 chemistry and employing three SMRT cells (Pacific Biosciences, Menlo Park, CA, USA).
Raw Illumina sequences were adapter trimmed, quality filtered (Q > 20), de novo assembled using [
29,
30]. Contigs shorter than 1000 bp or with <5 times coverage were removed from each assembly prior to gene annotation. The genes of the Illumina sequenced genome were predicted by Glimmer 3, version 3.02b; Johns Hopkins University: USA, 1999 [
31].
PacBio sequences were de novo assembled using HGAP version 3 (Pacific Biosciences, SMRT Analysis Software version 2.2.0) and circularized with the Minimus2 software version 2 (Amos package). For post-circularization correction of bases, reads were subsequently mapped to the circularized sequences using RS_Resequencing.1 software (SMRT Analysis version version 2.3.0).
This whole genome project has been deposited at GenBank under the Accession Number PRJNA721926 (UiBP1-2013) and Accession Number CP074346 (NVI 9100).
2.2. Phylogenetic Analysis
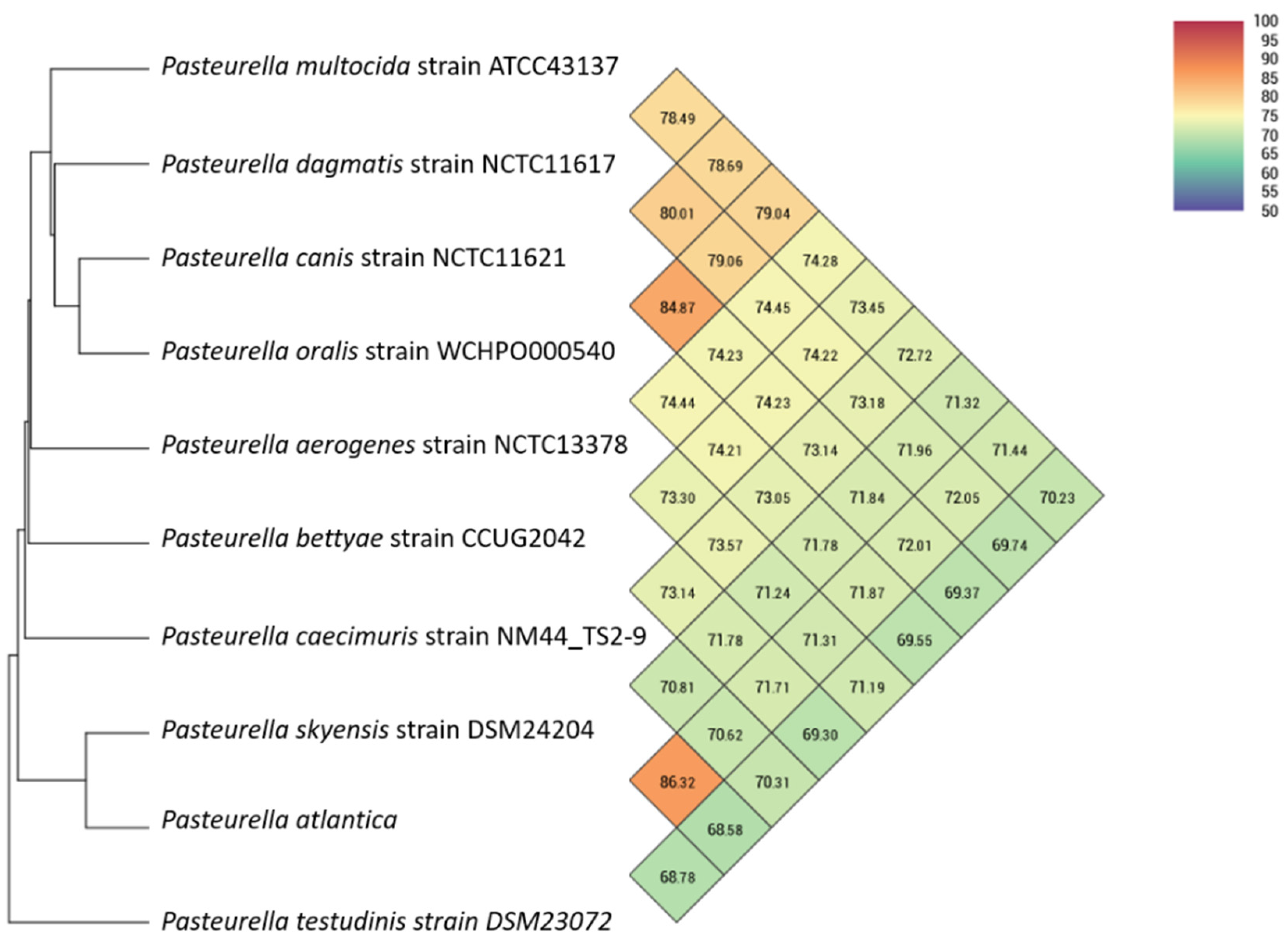
Twenty-three
Pasteurellaceae genomes (
Table 1) were included for phylogenetic analyses.
In order to evaluate similarity among whole genomes, Orthologous Average Nucleotide Identity Tool using OrthoANI [
43] was used to provide reliable and fast assessment of average nucleotide identity (ANI) for taxonomic classification purposes. A Similar Genome Finder with high similarity parameters (max hits: 50;
p-value threshold: 1; distance: 0.5) utilizing Mash, a fast genome distance estimation tool [
44], mounted on the Pathosystems Resource Integration Center (PATRIC) platform (
https://patricbrc.org/ (accessed on 3 June 2021)) [
45,
46] was also used to assess the genomic similarity of
P. atlantica to the bacterial database.
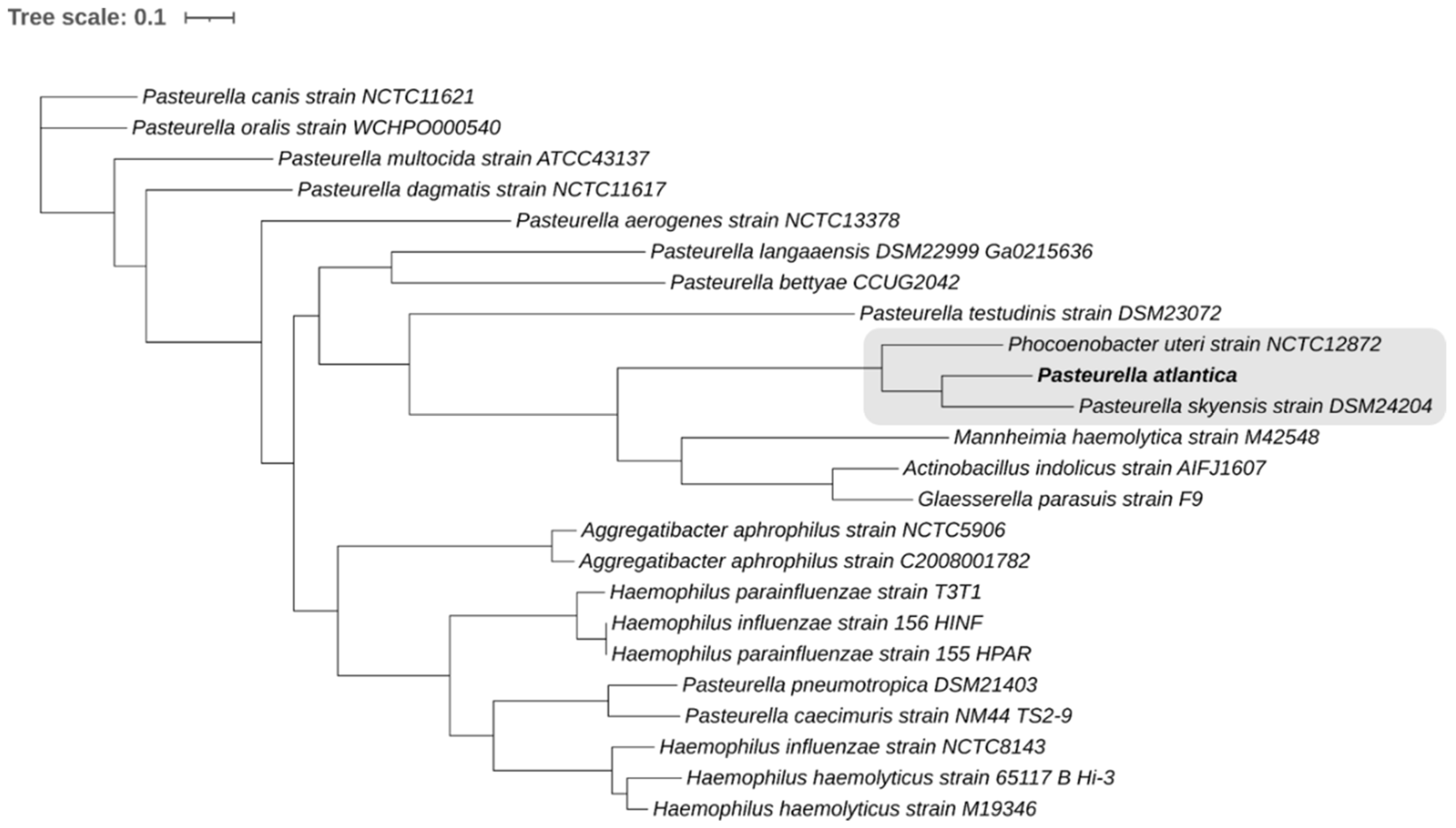
PATRIC [
45,
46] was used to construct a whole-genome codon-based tree built on 500 single-copy genes [
47] present in all genomes studied (
Table 1). Both amino acid and nucleotide sequences were aligned using MUSCLE [
48] and the codon align function of Biopython [
49], respectively. The concatenated alignments of protein and nucleotide sequences were used in order to generate a RAxML method-based phylogenetic tree with branch support values determined by 100 rounds of rapid bootstrapping [
50,
51]. The tree was visualized using iTOL online platform [
52].
2.3. Analysis of Genomic Regions and Reverse Vaccinology Approach
The novel genomes were screened against major virulence factor databases. Specifically, the proteome of
P. atlantica was blasted against VFDB [
53] and VICTORS [
54] using an e-value of 1 × 10
−10. A moderate e-value was applied to avoid false positive hits while identifying as many presumptive virulence factors as possible. The prediction was complemented by screening against PATRIC_VF [
55], the integrated virulence factor database inside the PATRIC platform (
https://patricbrc.org/ (accessed on 3 June 2021)) [
45,
46]. Subcellular localization for all presumptive virulence factors was examined by PSORTb 3.0 [
56] to evaluate putative interaction with the extracellular environment (localization score threshold >7.5). Proteins with both extracellular and outer membrane subcellular localization were imported to SignalP server [
57] in order to further assess any known types of signal peptides as well as the pathways in which they are involved during their secretion or anchoring process (threshold >90%). The detection of possible adhesion-related functionality was tested in SPAAN [
58]. A Protein Fold Recognition Server, Phyre2 [
59], was applied to obtain insights on structure and folding properties of presumptive virulence factors using confidence level of above 97%.
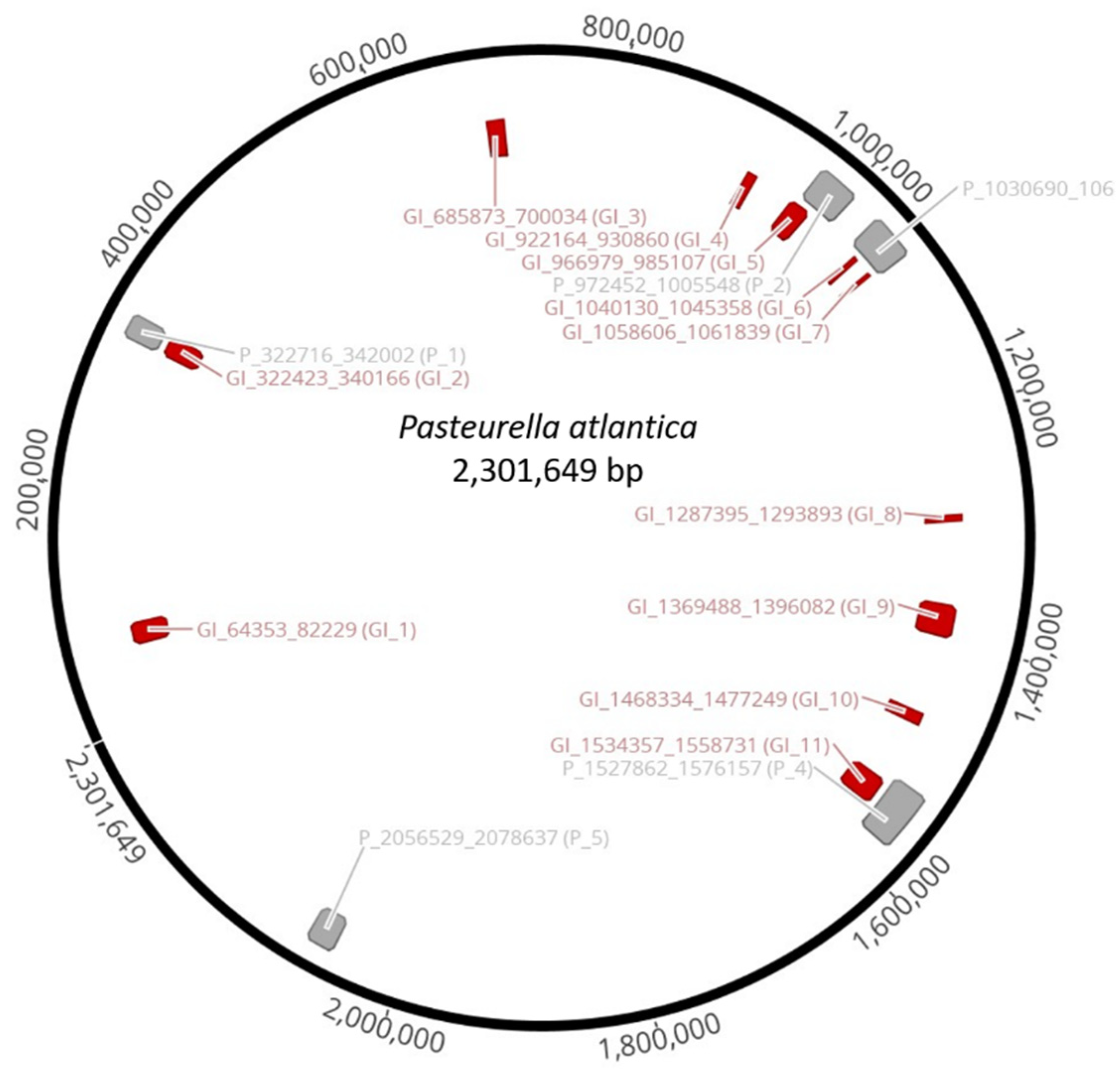
Clusters of genes of probable horizontal origin, known as genomic islands (GIs), were identified by IslandViewer 4, version 4; Simon Fraser University: Canada, 2017 [
60]. Inducible prophages were detected using the prophage finder tool, PHASTER [
61], while VIRFAM [
62] was used to classify intact prophages.
In line with reverse vaccinology principles, the identification of the strongest virulence candidates was deduced through Dynamic Vaxign Analysis, Vaxign [
63], a software platform that has been developed and dedicated to vaccine design. The pipeline of Vaxign has integrated PSORTb [
56] and SPAAN [
58] so it can also be used to validate the previously identified virulence factors. Vaxign-ML [
20] was also included in the reverse vaccinology analysis as a machine learning model to improve the prediction of bacterial protective antigens. VaxiJen [
64] complemented the analysis to corroborate that the finally selected candidates for vaccine development also can function as protective antigens, which is a prerequisite for vaccine development.
2.4. Functional Studies
2.4.1. Processing Bacteria for qPCR Analysis
Cultures of
P. atlantica (UiBP1-2013) used for the in vitro exposure experiment (
Section 2.4.5) were cultivated as described in
Section 2.1, harvested in the late exponential growth phase (18 h post inoculation) and centrifuged at 2500×
g (Beckman Coulter Allegra X-15R) for 15 min at 4 °C. For the gene expression analysis during bacterial cell proliferation in growth medium, 1 mL samples were harvested 14, 16, 18, and 20 h after inoculation. Bacterial cell counts were measured at harvest using a cell counter (CASY Model TT (Innovatis) and CASY worX version 1.26) followed by centrifugation at 4000×
g (Beckman Coulter Allegra X-15R) for 10 min at 4 °C. For both methods, the growth medium was then discarded, and the samples were stored on ice.
Total RNA was immediately extracted using the Bacterial RNA Kit (E.Z.N.A., Norcross, GA, USA) according to manufacturer instructions. The RNA was then DNase-treated (Sigma-Aldrich, Saint-Louis, MO, USA), converted to cDNA using qScript cDNA Synthesis Kit (Quantabio, Beverly, MA, USA), and stored at −20 °C.
2.4.2. qPCR
Each qPCR reaction contained a volume of 25 μL and consisted of 12.5 μL 2× SYBR Green JumpStart Taq Ready Mix (Thermo-Fisher Scientific, Waltham, MA, USA), 1 μL each of the forward and reverse primers (10 μM final working concentration,
Table 2), 0.5 μL of RNase and DNase free water (Sigma-Aldrich), and 10 μL of cDNA (concentration depended on the specific analysis). A C1000 Touch thermal cycler with CFX96 Real-Time System (Bio-Rad, Oslo, Norway) was used for qPCR, with the following cycle conditions: 94 °C for 5 min followed by 40 cycles of 94 °C for 15 s and 60 °C for 1 min. Melting curve analyses were performed after each run (60 to 92 °C at a rate of 1 °C/5 s) to ensure that the specificity of the primers and the qPCR products were visualized on a 2% agarose gel. Three parallel reactions were performed for each gene, and negative controls excluding cDNA (NTC) and cDNA reactions without reverse transcriptase (NRT) were included for all master mixes. The gene expression levels were calculated by the ΔΔCt method [
65].
2.4.3. Primer Design and Validation
Genes considered for this work were based on the results from an analysis of genomic regions and potential vaccine targets. The genes selected as reference genes were
rpoD and
gyrA. The target gene selected was the putative uncharacterized protein (<Hia>) (
Table 2).
qPCR assays were designed using the software Primer Premier version 6.24 (Premier Biosoft, San Francisco, CA, USA). The five highest rated assays for each target sequence were then chosen for testing. The length of the amplicons was kept between 100 and 250 bp for optimal amplification efficiency. The specificity of the primers was confirmed by qPCR (20 ng of cDNA used in each qPCR reaction), and product size was observed by electrophoresis on 2% agarose gels. All of the qPCR assays produced single amplification products. The best assay for each target gene based on Cq value, non-template controls, melting curves, and the results of electrophoresis were then chosen for further work. The resulting primers used for qPCR are listed in
Table 2.
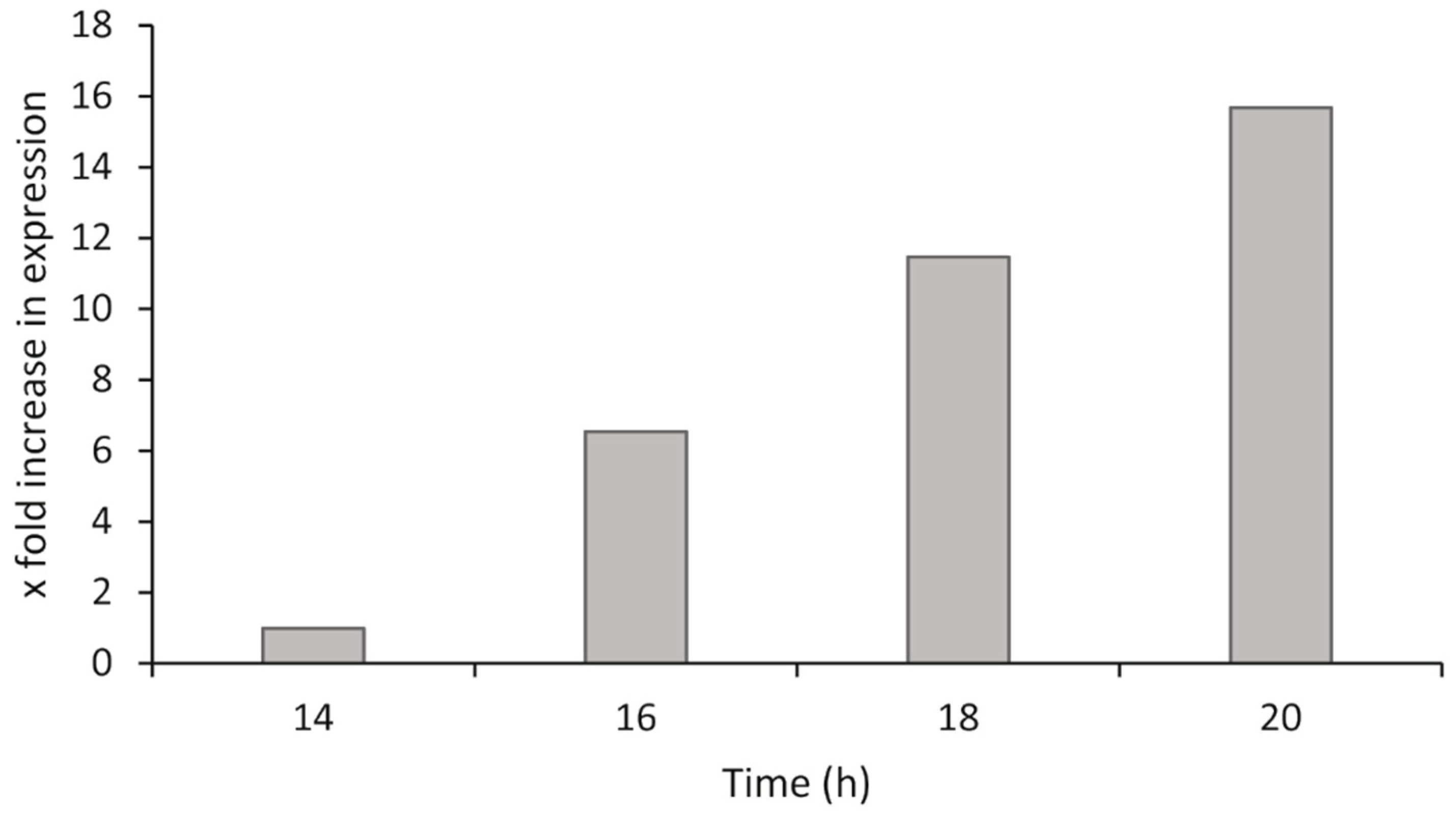
2.4.4. Bacterial Exponential Growth Phase Analysis
P. atlantica was grown as described in
Section 2.1. At different time points after inoculation, the bacteria were harvested, the RNA was isolated, and cDNA synthesis was performed as described in
Section 2.4.1. The synthesized cDNA was diluted across a twofold dilution series to give a range from 10 ng/µL to 0.625 ng/µL of cDNA for both the target and reference genes in qPCR (100 ng to 6.25 ng in each qPCR reaction). The relative gene expression was calculated by the ΔΔCq method using the most suitable dilution from the range tested, and comparisons of gene expression were made to the lowest stable time point of the analysis.
2.4.5. Head Kidney Leucocyte (HKL) Exposure Analysis
Four lumpsuckers were quickly netted and killed by a sharp blow to the head. Leucocytes were isolated from the head kidney on discontinuous Percoll gradients as described previously [
66] with the following modifications. The supplemented L-15 medium did not contain gentamicin sulphate since the cells were to be exposed to viable
P. atlantica. Additionally, resuspension of the isolated leucocytes was performed in L-15 supplemented with 5% fetal calf serum (L-15/FCS). The leucocytes were counted in a CASY-TT Cell Counter TM (Innovatis AG), and the viability (95%) and aggregation factor (1.2) of the cells were determined. The concentration of the isolated leucocytes was then adjusted to 3.3 × 10
6 cells mL
−1 in L-15/FCS, and 250 μL volumes were added to each well of the 24-well Nunc plates (approximately 8 × 10
5 cells per well) (Thermo-Fisher Scientific) and incubated overnight at 15 °C prior to exposure to
P. atlantica.
A late exponential phase (18 h) culture of
P. atlantica was prepared as described in
Section 2.4.1, re-suspended in L-15/FCS, and adjusted to 1.5 × 10
8 bacteria mL
−1. Volumes of 250 μL were then added to each well (approximately 4 × 10
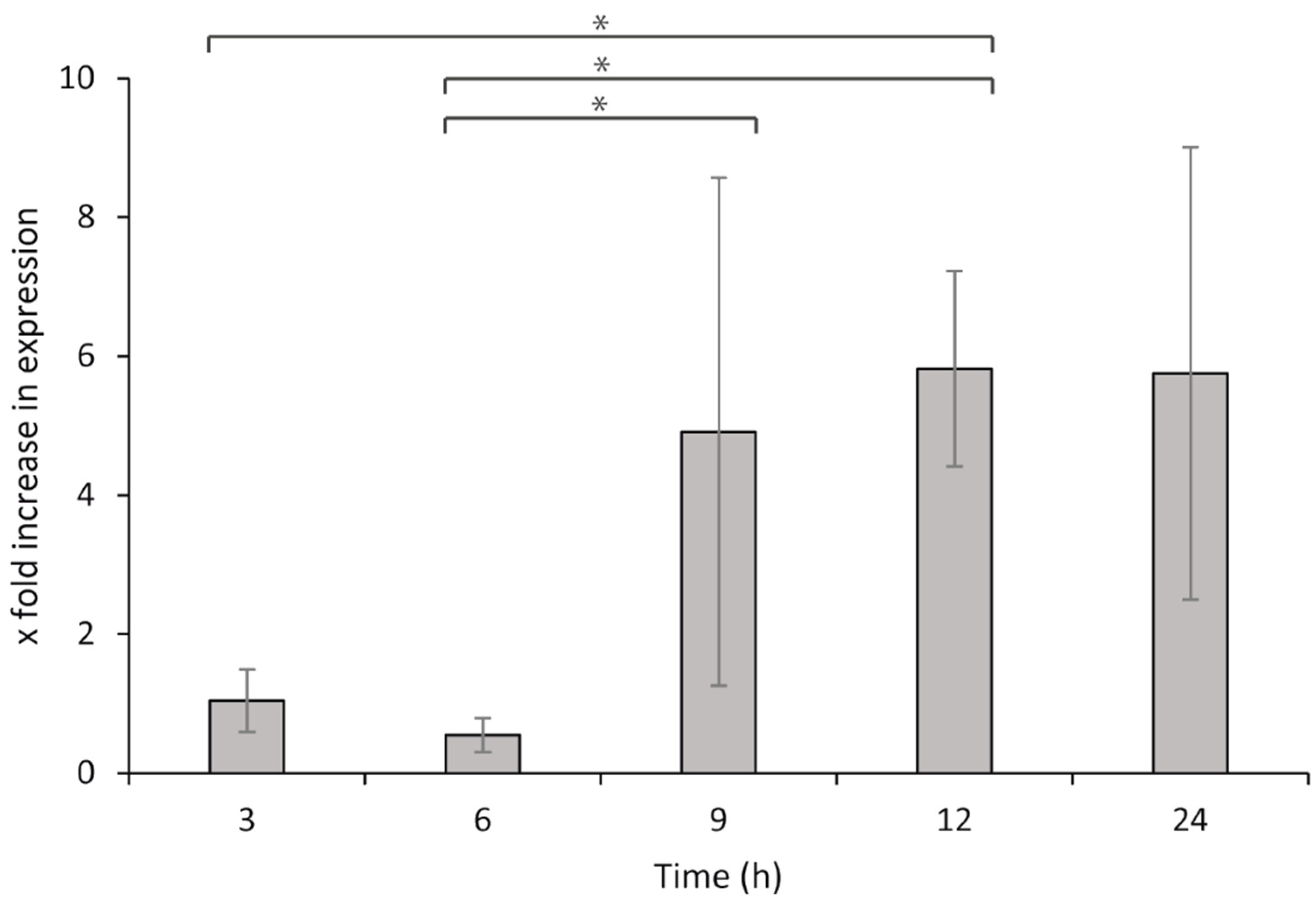
7 bacteria per well). Sterile L-15/FCS medium was supplied to the leucocytes and used for the non-challenged controls. The cells were then incubated at 15 °C, and samples were collected at 3, 6, 9, 12, and 24 h after exposure. Samples were collected from the wells and centrifuged to remove all media prior to storage at −80 °C, until the total RNA was isolated, DNase treated, and converted to cDNA as described above. For qPCR, 80 ng per reaction was used as the template for both the target and reference genes.
2.4.6. Statistical Analysis
The relative gene expression during the HKL exposure was calculated by the ΔΔCq method [
65] and comparisons were made to the negative controls (bacteria without leucocytes). The results were analyzed using two-way ANOVA and Fisher’s LSD for post hoc tests, and differences were considered significant when
p < 0.05. All statistical analyses were carried out using SigmaPlot version 12 (Systat Software, San Jose, CA, USA).


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}