
Pan-Genome Analysis Reveals Host-Specific Functional Divergences in Burkholderia gladioli


Abstract
1. Introduction
2. Materials and Methods
2.1. Public Genomic Resources
2.2. Bacterial Strain, Culture Conditions, and Genomic DNA Extraction
2.3. Genome Sequencing and Assembly
2.4. Phylogenomic Analysis
2.5. Pan-Genomic Analysis
2.6. Functional Enrichment Analyses
2.7. Identification of Secondary Metabolite Biosynthetic Gene Clusters and CRISPR/Cas
3. Results and Discussion
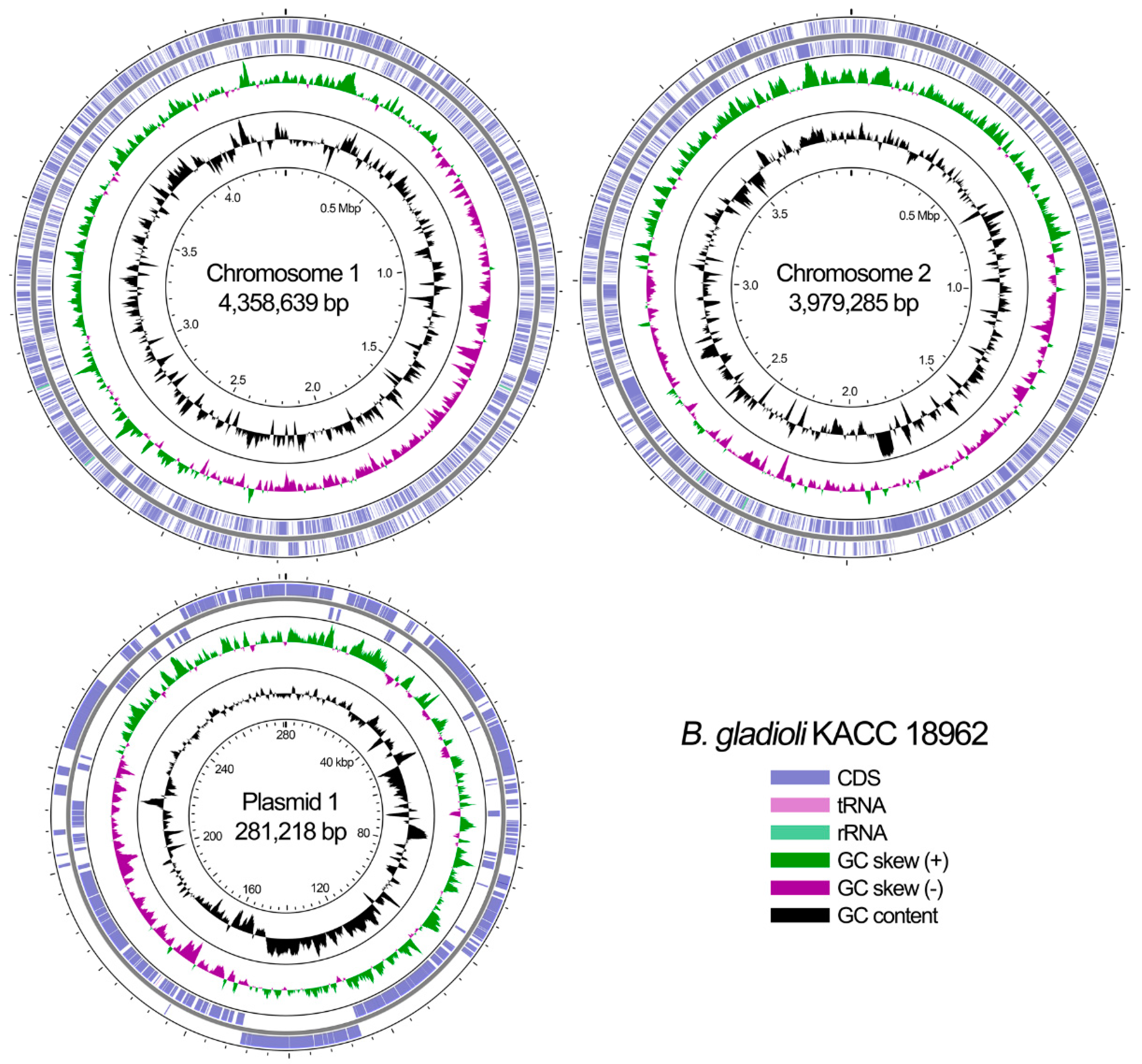
3.1. Available Genomic Information for B. gladioli
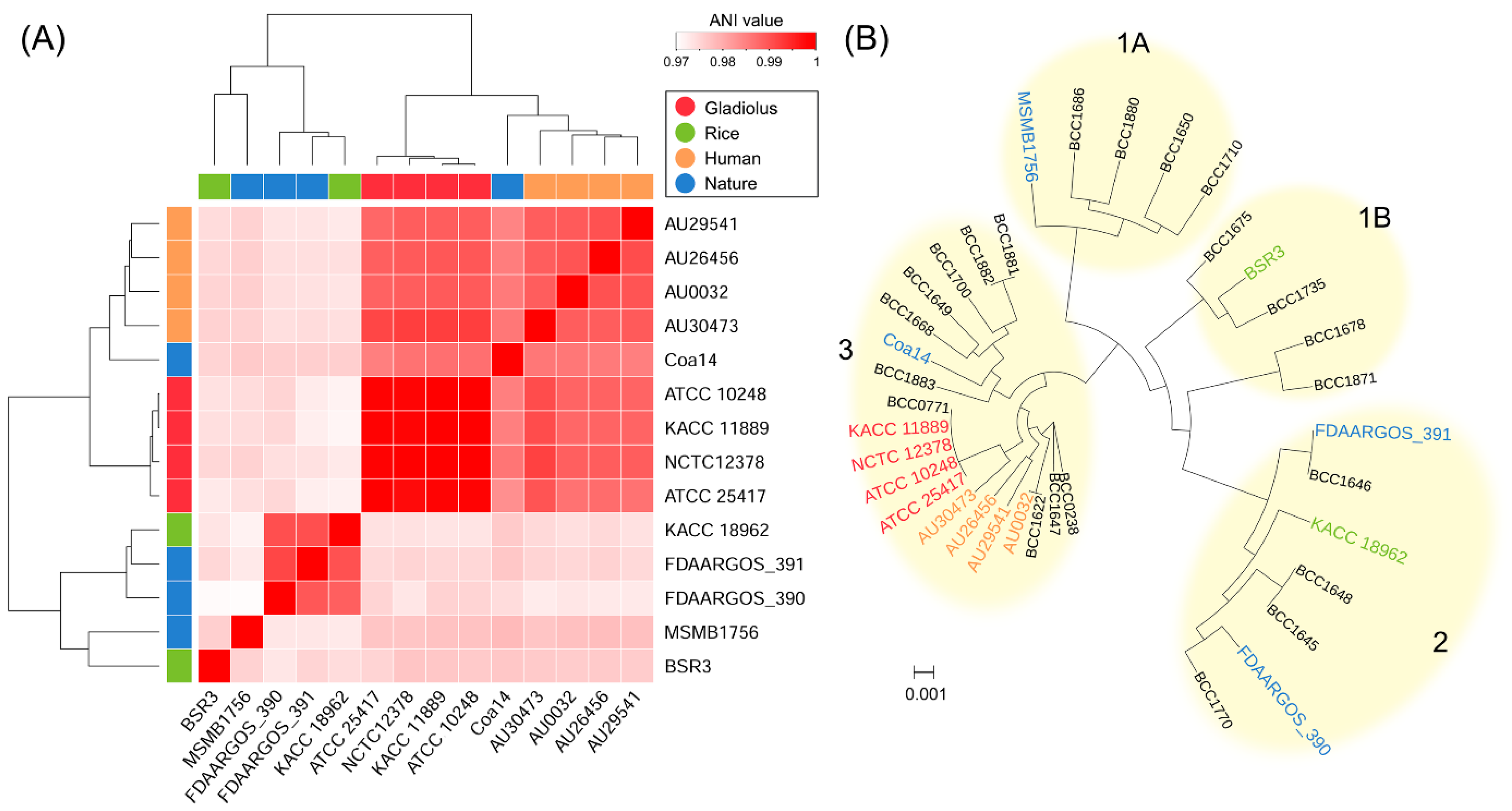
3.2. Phylogenomic Analysis
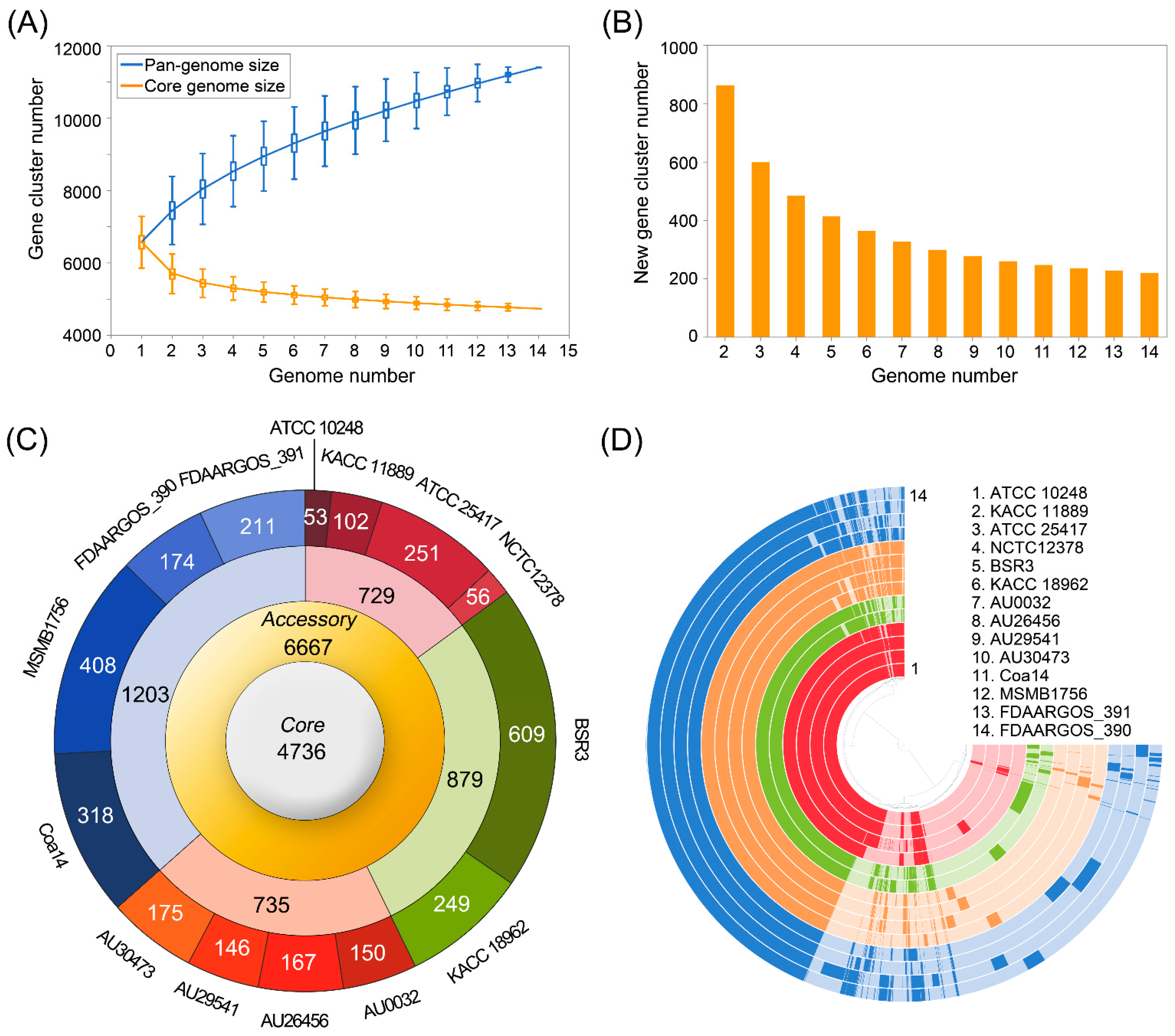
3.3. Pan-Genome Analysis
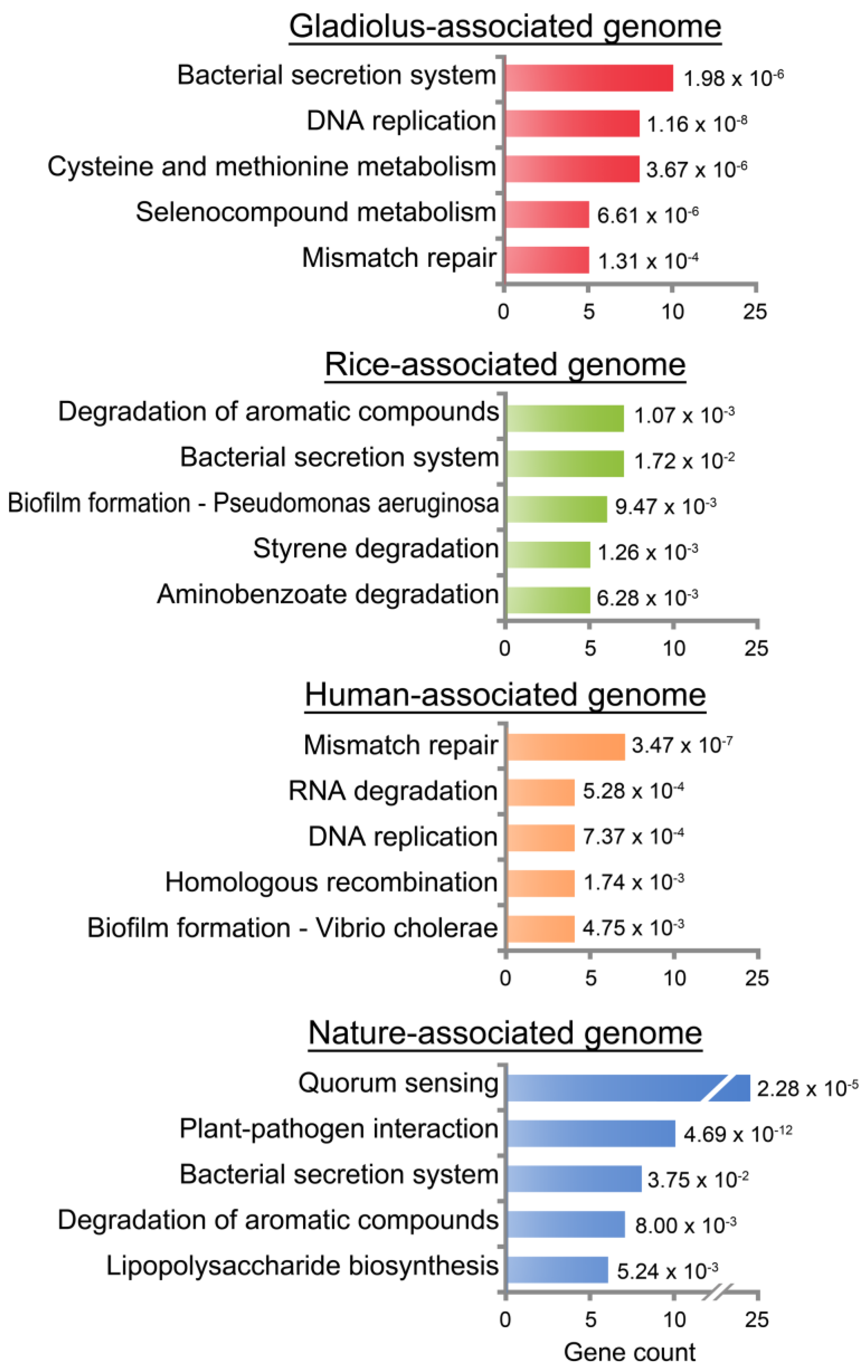
3.4. Functional Analysis for Core and Niche-Associated Genome
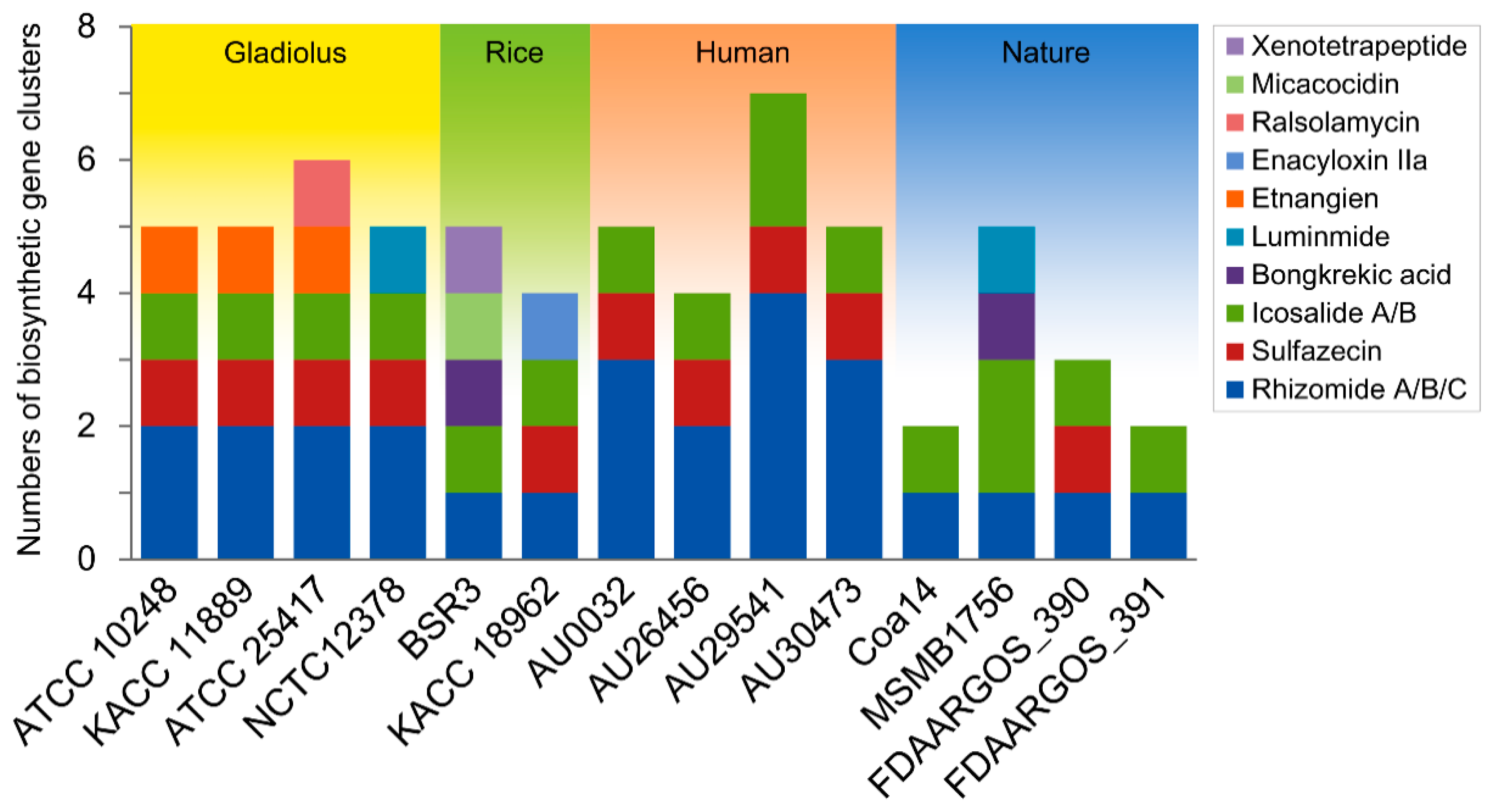
3.5. In Silico Analysis of Nonribosomal Peptide Synthetases
3.6. Prediction of CRISPR/Cas System
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dutta, C.; Paul, S. Microbial lifestyle and genome signatures. Curr. Genom. 2012, 13, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Di Cenzo, G.C.; Finan, T.M. The divided bacterial genome: Structure, function, and evolution. Microbiol. Mol. Biol. Rev. 2017, 81. [Google Scholar] [CrossRef]
- Toft, C.; Andersson, S.G.E. Evolutionary microbial genomics: Insights into bacterial host adaptation. Nat. Rev. Genet. 2010, 11, 465–475. [Google Scholar] [CrossRef]
- Cases, I.; De Lorenzo, V.; Ouzounis, C.A. Transcription regulation and environmental adaptation in bacteria. Trends Microbiol. 2003, 11, 248–253. [Google Scholar] [CrossRef]
- Retchless, A.C.; Lawrence, J.G. Ecological adaptation in bacteria: Speciation driven by codon selection. Mol. Biol. Evol. 2012, 29, 3669–3683. [Google Scholar] [CrossRef]
- Fraser-Liggett, C.M. Insights on biology and evolution from microbial genome sequencing. Genome Res. 2005, 15, 1603–1610. [Google Scholar] [CrossRef]
- Zhang, J.; Chiodini, R.; Badr, A.; Zhang, G. The impact of next-generation sequencing on genomics. J. Genet. Genom. 2011, 38, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.; Makova, K.D.; Nekrutenko, A.; Hardison, R.C. Comparative genomics. Annu. Rev. Genom. Hum. Genet. 2004, 5, 15–56. [Google Scholar] [CrossRef] [PubMed]
- Tettelin, H.; Riley, D.; Cattuto, C.; Medini, D. Comparative genomics: The bacterial pan-genome. Curr. Opin. Microbiol. 2008, 11, 472–477. [Google Scholar] [CrossRef]
- Tettelin, H.; Masignani, V.; Cieslewicz, M.J.; Donati, C.; Medini, D.; Ward, N.L.; Angiuoli, S.V.; Crabtree, J.; Jones, A.L.; Durkin, A.S.; et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. USA 2005, 102, 13950–13955. [Google Scholar] [CrossRef]
- Marschall, T.; Marz, M.; Abeel, T.; Dijkstra, L.; Dutilh, B.E.; Ghaffaari, A.; Kersey, P.; Kloosterman, W.P.; Mäkinen, V.; Novak, A.M.; et al. Computational pan-genomics: Status, promises and challenges. Brief. Bioinform. 2018, 19, 118–135. [Google Scholar] [CrossRef]
- Mira, A.; Martín-Cuadrado, A.B.; D’Auria, G.; Rodríguez-Valera, F. The bacterial pan-genome: A new paradigm in microbiology. Int. Microbiol. 2010, 13, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Nunvar, J.; Capek, V.; Fiser, K.; Fila, L.; Drevinek, P. What matters in chronic Burkholderia cenocepacia infection in cystic fibrosis: Insights from comparative genomics. PLoS Pathog. 2017, 13. [Google Scholar] [CrossRef]
- Vandamme, P.; Peeters, C.; De Smet, B.; Price, E.P.; Sarovich, D.S.; Henry, D.A.; Hird, T.J.; Zlosnik, J.E.A.; Mayo, M.; Warner, J.; et al. Comparative genomics of Burkholderia singularis sp. nov., a low G+C content, free-living bacterium that defies taxonomic dissection of the genus Burkholderia. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef]
- Gislason, A.S.; Turner, K.; Domaratzki, M.; Cardona, S.T. Comparative analysis of the Burkholderia cenocepacia K56-2 essential genome reveals cell envelope functions that are uniquely required for survival in species of the genus Burkholderia. Microb. Genom. 2017, 3. [Google Scholar] [CrossRef]
- Seo, Y.-S.; Lim, J.Y.; Park, J.; Kim, S.; Lee, H.-H.; Cheong, H.; Kim, S.-M.; Moon, J.S.; Hwang, I. Comparative genome analysis of rice-pathogenic Burkholderia provides insight into capacity to adapt to different environments and hosts. BMC Genom. 2015, 16, 349. [Google Scholar] [CrossRef]
- O’Sullivan, L.A.; Mahenthiralingam, E. Biotechnological potential within the genus Burkholderia. Lett. Appl. Microbiol. 2005, 41, 8–11. [Google Scholar] [CrossRef] [PubMed]
- Compant, S.; Nowak, J.; Coenye, T.; Clément, C.; Ait Barka, E. Diversity and occurrence of Burkholderia spp. in the natural environment. FEMS Microbiol. Rev. 2008, 32, 607–626. [Google Scholar] [CrossRef]
- Mahenthiralingam, E.; Urban, T.A.; Goldberg, J.B. The multifarious, multireplicon Burkholderia cepacia complex. Nat. Rev. Microbiol. 2005, 3, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Eberl, L.; Vandamme, P. Members of the genus Burkholderia: Good and bad guys. F1000Research 2016, 5, 1007. [Google Scholar] [CrossRef]
- Seo, Y.S.; Lim, J.; Choi, B.S.; Kim, H.; Goo, E.; Lee, B.; Lim, J.S.; Choi, I.Y.; Moon, J.S.; Kim, J.; et al. Complete genome sequence of Burkholderia gladioli BSR3. J. Bacteriol. 2011, 193, 3149. [Google Scholar] [CrossRef]
- McCulloch, L. A bacterial disease of gladiolus. Science 1921, 54, 115–116. [Google Scholar] [CrossRef]
- Nandakumar, R.; Rush, M.C.; Correa, F. Association of Burkholderia glumae and B. gladioli with panicle blight symptoms on rice in Panama. Plant Dis. 2007, 91, 767. [Google Scholar] [CrossRef]
- Nandakumar, R.; Shahjahan, A.K.M.; Yuan, X.L.; Dickstein, E.R.; Groth, D.E.; Clark, C.A.; Cartwright, R.D.; Rush, M.C. Burkholderia glumae and B. gladioli cause bacterial panicle blight in rice in the Southern United States. Plant Dis. 2009, 93, 896–905. [Google Scholar] [CrossRef]
- Ura, H.; Furuya, N.; Iiyama, K.; Hidaka, M.; Tsuchiya, K.; Matsuyama, N. Burkholderia gladioli associated with symptoms of bacterial grain rot and leaf-sheath browning of rice plants. J. Gen. Plant Pathol. 2006, 72, 98–103. [Google Scholar] [CrossRef]
- Hotta, H.; Yano, I.; Yabuuchi, E.; Kosako, Y.; Oyaizu, H.; Ezaki, T.; Hashimoto, Y.; Arakawa, M. Proposal of Burkholderia gen. nov. and transfer of seven species of the genus Pseudomonas homology group II to the new genus, with the type species Burkholderia cepacia (Palleroni and Holmes 1981) comb. nov. Microbiol. Immunol. 1992, 36, 1251–1275. [Google Scholar] [CrossRef]
- Chowdhury, P.R.; Heinemann, J.A. The general secretory pathway of Burkholderia gladioli pv. agaricicola BG164R is necessary for cavity disease in white button mushrooms. Appl. Environ. Microbiol. 2006, 72, 3558–3565. [Google Scholar] [CrossRef]
- Lopes, E.F.; Da Costa, J.G.; Wolf, I.R.; de Lima, J.P.A.; Astolfi-Filho, S. Draft genome sequence of Burkholderia gladioli Coa14, a bacterium with petroleum bioremediation potential isolated from Coari Lake, Amazonas, Brazil. Genome Announc. 2018, 6. [Google Scholar] [CrossRef]
- Collymore, C.; Giuliano, F.; Banks, E.K. Head tilt in immunodeficient mice due to contamination of drinking water by Burkholderia gladioli. J. Am. Assoc. Lab. Anim. Sci. 2019, 58, 246–250. [Google Scholar] [CrossRef]
- Brizendine, K.D.; Baddley, J.W.; Pappas, P.G.; Leon, K.J.; Rodriguez, J.M. Fatal Burkholderia gladioli infection misidentified as empedobacter brevis in a lung transplant recipient with cystic fibrosis. Transpl. Infect. Dis. 2012, 14. [Google Scholar] [CrossRef] [PubMed]
- Quon, B.S.; Reid, J.D.; Wong, P.; Wilcox, P.G.; Javer, A.; Wilson, J.M.; Levy, R.D. Burkholderia gladioli—A predictor of poor outcome in cystic fibrosis patients who receive lung transplants? A case of locally invasive rhinosinusitis and persistent bacteremia in a 36-year-old lung transplant recipient with cystic fibrosis. Can. Respir. J. 2011, 18. [Google Scholar] [CrossRef]
- Jones, C.; Webster, G.; Mullins, A.J.; Jenner, M.; Bull, M.J.; Dashti, Y.; Spilker, T.; Parkhill, J.; Connor, T.R.; Lipuma, J.J.; et al. Kill and cure: Genomic phylogeny and bioactivity of Burkholderia gladioli bacteria capable of pathogenic and beneficial lifestyles. Microb. Genom. 2021, 7, 1–13. [Google Scholar] [CrossRef]
- Chin, C.S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E.; et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI reference sequences (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007, 35. [Google Scholar] [CrossRef]
- Pritchard, L.; Glover, R.H.; Humphris, S.; Elphinstone, J.G.; Toth, I.K. Genomics and taxonomy in diagnostics for food security: Soft-rotting enterobacterial plant pathogens. Anal. Methods 2016, 8, 12–24. [Google Scholar] [CrossRef]
- Lin, Y.T.; Lee, C.C.; Leu, W.M.; Wu, J.J.; Huang, Y.C.; Meng, M. Fungicidal activity of volatile organic compounds emitted by Burkholderia gladioli strain BBB-01. Molecules 2021, 26, 745. [Google Scholar] [CrossRef]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wu, J.; Yang, J.; Sun, S.; Xiao, J.; Yu, J. PGAP: Pan-genomes analysis pipeline. Bioinformatics 2012, 28, 416–418. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Enright, A.J.; Van Dongen, S.; Ouzounis, C.A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30, 1575–1584. [Google Scholar] [CrossRef]
- Zhao, Y.; Jia, X.; Yang, J.; Ling, Y.; Zhang, Z.; Yu, J.; Wu, J.; Xiao, J. PanGP: A tool for quickly analyzing bacterial pan-genome profile. Bioinformatics 2014, 30, 1297–1299. [Google Scholar] [CrossRef] [PubMed]
- Eren, A.M.; Esen, O.C.; Quince, C.; Vineis, J.H.; Morrison, H.G.; Sogin, M.L.; Delmont, T.O. Anvi’o: An advanced analysis and visualization platformfor ’omics data. PeerJ 2015, 2015. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Smirnov, S.; Nikolskaya, A.N.; et al. The COG database: An updated vesion includes eukaryotes. BMC Bioinform. 2003, 4. [Google Scholar] [CrossRef] [PubMed]
- Carbon, S.; Douglass, E.; Dunn, N.; Good, B.; Harris, N.L.; Lewis, S.E.; Mungall, C.J.; Basu, S.; Chisholm, R.L.; Dodson, R.J.; et al. The gene ontology resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, R.; Sacan, A. Weighted set enrichment of gene expression data. BMC Syst. Biol. 2013, 7, S10. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. AntiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef] [PubMed]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Néron, B.; Rocha, E.P.C.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef]
- Biswas, A.; Gagnon, J.N.; Brouns, S.J.J.; Fineran, P.C.; Brown, C.M. CRISPRTarget: Bioinformatic prediction and analysis of crRNA targets. RNA Biol. 2013, 10, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.L.; Bishop-Lilly, K.A.; Ladner, J.T.; Daligault, H.E.; Davenport, K.W.; Jaissle, J.; Frey, K.G.; Koroleva, G.I.; Bruce, D.C.; Coyne, S.R.; et al. Complete genome sequences for 59 Burkholderia isolates, both pathogenic and near neighbor. Genome Announc. 2016, 3, e00159-15. [Google Scholar] [CrossRef]
- Chen, H.; Sun, T.; Bai, X.; Yang, J.; Yan, F.; Yu, L.; Tu, Q.; Li, A.; Tang, Y.; Zhang, Y.; et al. Genomics-driven activation of silent biosynthetic gene clusters in Burkholderia gladioli by screening recombineering system. Molecules 2021, 26, 700. [Google Scholar] [CrossRef]
- Clode, F.E.; Kaufmann, M.E.; Malnick, H.; Pitt, T.L. Evaluation of three oligonucleotide primer sets in PCR for the identification of Burkholderia cepacia and their differentiation from Burkholderia gladioli. J. Clin. Pathol. 1999, 52, 173–176. [Google Scholar] [CrossRef]
- Lee, J.; Park, J.; Kim, S.; Park, I.; Seo, Y.S. Differential regulation of toxoflavin production and its role in the enhanced virulence of Burkholderia gladioli. Mol. Plant Pathol. 2016, 17, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Daligault, H.; Davenport, K.; Minogue, T.; Bishop-Lilly, K.; Broomall, S.; Bruce, D.; Chain, P.; Coyne, S.; Frey, K.; Gibbons, H.; et al. Whole-genome assemblies of 56 Burkholderia species. Genome Announc. 2014, 2, e01106-14. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Oliver, R.A.; Townsend, C.A. Identification and characterization of the sulfazecin monobactam biosynthetic gene cluster. Cell Chem. Biol. 2017, 24, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Fory, P.A.; Triplett, L.; Ballen, C.; Abello, J.F.; Duitama, J.; Aricapa, M.G.; Prado, G.A.; Correa, F.; Hamilton, J.; Leach, J.E.; et al. Comparative analysis of two emerging rice seed bacterial pathogens. Phytopathology 2014, 104, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Naughton, L.M.; An, S.Q.; Hwang, I.; Chou, S.H.; He, Y.Q.; Tang, J.L.; Ryan, R.P.; Dow, J.M. Functional and genomic insights into the pathogenesis of Burkholderia species to rice. Environ. Microbiol. 2016, 18, 780–790. [Google Scholar] [CrossRef]
- Kim, S.; Park, J.; Choi, O.; Kim, J.; Seo, Y.S. Investigation of quorum sensing-dependent gene expression in Burkholderia gladioli BSR3 through RNA-seq analyses. J. Microbiol. Biotechnol. 2014, 24, 1609–1621. [Google Scholar] [CrossRef]
- Zeiser, E.T.; Becka, S.A.; Wilson, B.M.; Barnes, M.D.; LiPuma, J.J.; Papp-Wallace, K.M. “Switching partners”: Piperacillin-Avibactam is a highly potent combination against multidrug-resistant Burkholderia cepacia complex and Burkholderia gladioli cystic fibrosis isolates. J. Clin. Microbiol. 2019, 57, e00181-19. [Google Scholar] [CrossRef]
- Zeiser, E.T.; Becka, S.A.; Barnes, M.D.; Taracila, M.A.; LiPuma, J.J.; Papp-Wallace, K.M. Resurrecting old -lactams: Potent inhibitory activity of temocillin against multidrug-resistant Burkholderia species isolates from the United States. Antimicrob. Agents Chemother. 2019, 63, e02315-18. [Google Scholar] [CrossRef]
- Stothard, P.; Wishart, D.S. Circular genome visualization and exploration using CGView. Bioinformatics 2005, 21, 537–539. [Google Scholar] [CrossRef]
- Kim, M.; Oh, H.S.; Park, S.C.; Chun, J. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 2014, 64, 346–351. [Google Scholar] [CrossRef]
- Lipuma, J.J. The changing microbial epidemiology in cystic fibrosis. Clin. Microbiol. Rev. 2010, 23, 299–323. [Google Scholar] [CrossRef]
- Diene, S.M.; Merhej, V.; Henry, M.; El Filali, A.; Roux, V.; Robert, C.; Azza, S.; Gavory, F.; Barbe, V.; La Scola, B.; et al. The rhizome of the multidrug-resistant enterobacter aerogenes genome reveals how new “Killer Bugs” are created because of a sympatric lifestyle. Mol. Biol. Evol. 2013, 30, 369–383. [Google Scholar] [CrossRef]
- Tuanyok, A.; Leadem, B.R.; Auerbach, R.K.; Beckstrom-Sternberg, S.M.; Beckstrom-Sternberg, J.S.; Mayo, M.; Wuthiekanun, V.; Brettin, T.S.; Nierman, W.C.; Peacock, S.J.; et al. Genomic islands from five strains of Burkholderia pseudomallei. BMC Genom. 2008, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Ren, H.; Hu, M.; Zhou, J.; Li, B.; Kong, N.; Zhang, Q.; Jin, Y.; Liang, L.; Yue, J. Characterization of Burkholderia cepacia complex core genome and the underlying recombination and positive selection. Front. Genet. 2020, 11. [Google Scholar] [CrossRef]
- Georgiades, K.; Raoult, D. Defining pathogenic bacterial species in the genomic era. Front. Microbiol. 2011, 1. [Google Scholar] [CrossRef]
- Siew, H.S.; Yu, Y.; Chi, H.L.; Karuturi, R.K.M.; Wuthiekanun, V.; Tuanyok, A.; Hui, H.C.; Ong, C.; Paramalingam, S.S.; Tan, G.; et al. The core and accessory genomes of Burkholderia pseudomallei: Implications for human melioidosis. PLoS Pathog. 2008, 4. [Google Scholar] [CrossRef]
- Konstantinidis, K.T.; Tiedje, J.M. Trends between gene content and genome size in prokaryotic species with larger genomes. Proc. Natl. Acad. Sci. USA 2004, 101, 3160–3165. [Google Scholar] [CrossRef]
- Romero, H.; Zhang, Y.; Gladyshev, V.N.; Salinas, G. Evolution of selenium utilization traits. Genome Biol. 2005, 6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Turanov, A.A.; Hatfield, D.L.; Gladyshev, V.N. In silico identification of genes involved in selenium metabolism: Evidence for a third selenium utilization trait. BMC Genom. 2008, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Romero, H.; Salinas, G.; Gladyshev, V.N. Dynamic evolution of selenocysteine utilization in bacteria: A balance between selenoprotein loss and evolution of selenocysteine from redox active cysteine residues. Genome Biol. 2006, 7. [Google Scholar] [CrossRef]
- Whanger, P.D. Selenocompounds in plants and animals and their biological significance. J. Am. Coll. Nutr. 2002, 21, 223–232. [Google Scholar] [CrossRef]
- Feng, R.; Wei, C.; Tu, S. The roles of selenium in protecting plants against abiotic stresses. Environ. Exp. Bot. 2013, 87, 58–68. [Google Scholar] [CrossRef]
- Pilon-Smits, E.A.; Quinn, C.F.; Tapken, W.; Malagoli, M.; Schiavon, M. Physiological functions of beneficial elements. Curr. Opin. Plant Biol. 2009, 12, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Kaur, N.; Sharma, S.; Kaur, S.; Nayyar, H. Selenium in agriculture: A nutrient or contaminant for crops? Arch. Agron. Soil Sci. 2014, 60, 1593–1624. [Google Scholar] [CrossRef]
- Chauhan, R.; Awasthi, S.; Tripathi, P.; Mishra, S.; Dwivedi, S.; Niranjan, A.; Mallick, S.; Tripathi, P.; Tripathi, R.D.; Chauhan, R.; et al. Selenite modulates the level of phenolics and nutrient element to alleviate the toxicity of arsenite in rice (Oryza sativa L.). Ecotoxicol. Environ. Saf. 2017, 138, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Balk, J.; Pilon, M. Ancient and essential: The assembly of iron-sulfur clusters in plants. Trends Plant Sci. 2011, 16, 218–226. [Google Scholar] [CrossRef]
- Rahmanto, A.S.; Davies, M.J. Selenium-containing amino acids as direct and indirect antioxidants. IUBMB Life 2012, 64, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Sumner, S.E.; Markley, R.L.; Kirimanjeswara, G.S. Role of selenoproteins in bacterial pathogenesis. Biol. Trace Elem. Res. 2019, 192, 69–82. [Google Scholar] [CrossRef]
- Brink, D.P.; Ravi, K.; Lidén, G.; Gorwa-Grauslund, M.F. Mapping the diversity of microbial lignin catabolism: Experiences from the eLignin database. Appl. Microbiol. Biotechnol. 2019, 103, 3979–4002. [Google Scholar] [CrossRef]
- Bugg, T.D.H.; Winfield, C.J. Enzymatic cleavage of aromatic rings: Mechanistic aspects of the catechol dioxygenases and later enzymes of bacterial oxidative cleavage pathways. Nat. Prod. Rep. 1998, 15, 513–530. [Google Scholar] [CrossRef]
- Romero-Silva, M.J.; Méndez, V.; Agulló, L.; Seeger, M. Genomic and functional analyses of the gentisate and protocatechuate ring-cleavage pathways and related 3-hydroxybenzoate and 4-hydroxybenzoate peripheral pathways in Burkholderia xenovorans LB400. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Pérez-Pantoja, D.; Donoso, R.; Agulló, L.; Córdova, M.; Seeger, M.; Pieper, D.H.; González, B. Genomic analysis of the potential for aromatic compounds biodegradation in Burkholderiales. Environ. Microbiol. 2012, 14, 1091–1117. [Google Scholar] [CrossRef]
- Cottyn, B.; Regalado, E.; Lanoot, B.; De Cleene, M.; Mew, T.W.; Swings, J. Bacterial populations associated with rice seed in the tropical environment. Phytopathology 2001, 91, 282–292. [Google Scholar] [CrossRef]
- Nandakumar, R.; Bollich, P.A.; Shahjahan, A.K.M.; Groth, D.E.; Rush, M.C. Evidence for the soilborne nature of the rice sheath rot and panicle blight pathogen, Burkholderia gladioli. Can. J. Plant Pathol. 2008, 30, 148–154. [Google Scholar] [CrossRef]
- Ma’Ruf, A.; Pramudono, B.; Aryanti, N. Lignin isolation process from rice husk by alkaline hydrogen peroxide: Lignin and silica extracted. In Proceedings of the AIP Conference Proceedings; American Institute of Physics Inc.: Yogyakarta, Indonesia, 2017; Volume 1823, p. 020013. [Google Scholar]
- Seo, J.S.; Keum, Y.S.; Li, Q.X. Bacterial degradation of aromatic compounds. Int. J. Environ. Res. Public Health 2009, 6, 278–309. [Google Scholar] [CrossRef]
- Ali, N.; Dashti, N.; Khanafer, M.; Al-Awadhi, H.; Radwan, S. Bioremediation of soils saturated with spilled crude oil. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef]
- Andreoni, V.; Gianfreda, L. Bioremediation and monitoring of aromatic-polluted habitats. Appl. Microbiol. Biotechnol. 2007, 76, 287–308. [Google Scholar] [CrossRef] [PubMed]
- Amoroso, A.; Crespan, E.; Wimmer, U.; Hubscher, U.; Maga, G. DNA polymerases and oxidative damage: Friends or foes? Curr. Mol. Pharmacol. 2010, 1, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Dorer, M.S.; Sessler, T.H.; Salama, N.R. Recombination and DNA repair in Helicobacter pylori. Annu. Rev. Microbiol. 2011, 65, 329–348. [Google Scholar] [CrossRef] [PubMed]
- Sahan, A.Z.; Hazra, T.K.; Das, S. The pivotal role of DNA repair in infection mediated-inflammation and cancer. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef]
- Guillemet, E.; Leréec, A.; Tran, S.L.; Royer, C.; Barbosa, I.; Sansonetti, P.; Lereclus, D.; Ramarao, N. The bacterial DNA repair protein Mfd confers resistance to the host nitrogen immune response. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Stich, M.; Manrubia, S.C.; Lázaro, E. Variable mutation rates as an adaptive strategy in replicator populations. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Wielgoss, S.; Schneider, D.; Barrick, J.E.; Tenaillon, O.; Cruveiller, S.; Chane-Woon-Ming, B.; Médigue, C.; Lenski, R.E. Mutation rate inferred from synonymous substitutions in a long-term evolution experiment with Escherichia coli. G3 Genes Genomes Genet 2011, 1, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Suerbaum, S.; Maynard Smith, J.; Bapumia, K.; Morelli, G.; Smith, N.H.; Kunstmann, E.; Dyrek, I.; Achtman, M. Free recombination within Helicobacter pylori. Proc. Natl. Acad. Sci. USA 1998, 95, 12619–12624. [Google Scholar] [CrossRef] [PubMed]
- Doroghazi, J.R.; Buckley, D.H. Widespread homologous recombination within and between Streptomyces species. ISME J. 2010, 4, 1136–1143. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.C.; Tap, J.; Aron-Wisnewsky, J.; Pelloux, V.; Basdevant, A.; Bouillot, J.L.; Zucker, J.D.; Doré, J.; Clément, K. Gut microbiota after gastric bypass in human obesity: Increased richness and associations of bacterial genera with adipose tissue genes. Am. J. Clin. Nutr. 2013, 98, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Cooper, T.F. Recombination speeds adaptation by reducing competition between beneficial mutations in populations of Escherichia coli. PLoS Biol. 2007, 5, 1899–1905. [Google Scholar] [CrossRef] [PubMed]
- Van Acker, H.; Sass, A.; Bazzini, S.; De Roy, K.; Udine, C.; Messiaen, T.; Riccardi, G.; Boon, N.; Nelis, H.J.; Mahenthiralingam, E.; et al. Biofilm-grown Burkholderia cepacia complex cells survive antibiotic treatment by avoiding production of reactive oxygen species. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Lawrence, J.G.; Hendrickson, H. Lateral gene transfer: When will adolescence end? Mol. Microbiol. 2003, 50, 739–749. [Google Scholar] [CrossRef]
- Beiko, R.G.; Harlow, T.J.; Ragan, M.A. Highways of gene sharing in prokaryotes. Proc. Natl. Acad. Sci. USA 2005, 102, 14332–14337. [Google Scholar] [CrossRef]
- Wallden, K.; Rivera-Calzada, A.; Waksman, G. Type IV secretion systems: Versatility and diversity in function. Cell. Microbiol. 2010, 12, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Bertani, B.; Ruiz, N. Function and biogenesis of lipopolysaccharides. EcoSal Plus 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Guo, G.; Ma, Q.; Zhang, F.; Ma, F.; Liu, J.; Xiao, D.; Yang, X.; Sun, M. Diversity in S-layers. Prog. Biophys. Mol. Biol. 2017, 123, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Sleytr, U.B.; Schuster, B.; Egelseer, E.M.; Pum, D. S-layers: Principles and applications. FEMS Microbiol. Rev. 2014, 38, 823–864. [Google Scholar] [CrossRef]
- Sleytr, U.B.I. Basic and applied S-layer research: An overview. FEMS Microbiol. Rev. 1997, 20, 5–12. [Google Scholar] [CrossRef]
- Hibbing, M.E.; Fuqua, C.; Parsek, M.R.; Peterson, S.B. Bacterial competition: Surviving and thriving in the microbial jungle. Nat. Rev. Microbiol. 2010, 8, 15–25. [Google Scholar] [CrossRef]
- Marahiel, M.A. Working outside the protein-synthesis rules: Insights into non-ribosomal peptide synthesis. J. Pept. Sci. 2009, 15, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.X.; Zhong, Z.; Zhang, W.P.; Qian, P.Y. Discovery of cationic nonribosomal peptides as gram-negative antibiotics through global genome mining. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Esmaeel, Q.; Pupin, M.; Jacques, P.; Leclère, V. Nonribosomal peptides and polyketides of Burkholderia: New compounds potentially implicated in biocontrol and pharmaceuticals. Environ. Sci. Pollut. Res. 2018, 25, 29794–29807. [Google Scholar] [CrossRef]
- Lin, Z.; Falkinham, J.O.; Tawfik, K.A.; Jeffs, P.; Bray, B.; Dubay, G.; Cox, J.E.; Schmidt, E.W. Burkholdines from Burkholderia ambifaria: Antifungal agents and possible virulence factors. J. Nat. Prod. 2012, 75, 1518–1523. [Google Scholar] [CrossRef]
- Franke, J.; Ishida, K.; Ishida-Ito, M.; Hertweck, C. Nitro versus hydroxamate in siderophores of pathogenic bacteria: Effect of missing hydroxylamine protection in malleobactin biosynthesis. Angew. Chem. Int. Ed. 2013, 52, 8271–8275. [Google Scholar] [CrossRef]
- Carr, G.; Seyedsayamdost, M.R.; Chandler, J.R.; Greenberg, E.P.; Clardy, J. Sources of diversity in bactobolin biosynthesis by Burkholderia thailandensis E264. Org. Lett. 2011, 13, 3048–3051. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhou, H.; Chen, H.; Jing, X.; Zheng, W.; Li, R.; Sun, T.; Liu, J.; Fu, J.; Huo, L.; et al. Discovery of recombinases enables genome mining of cryptic biosynthetic gene clusters in Burkholderiales species. Proc. Natl. Acad. Sci. USA 2018, 115, E4255–E4263. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Yu, Y.; Zhang, Y.; Zhao, G.; Ding, X. Complete genome sequence of the glidobactin producing strain [Polyangium] brachysporum DSM 7029. J. Biotechnol. 2015, 210, 83–84. [Google Scholar] [CrossRef] [PubMed]
- Lackner, G.; Moebius, N.; Partida-Martinez, L.; Hertweck, C. Complete genome sequence of Burkholderia rhizoxinica, an endosymbiont of Rhizopus microsporus. J. Bacteriol. 2011, 193, 783–784. [Google Scholar] [CrossRef] [PubMed]
- Irschik, H.; Schummer, D.; Höfle, G.; Reichenbach, H.; Steinmetz, H.; Jansen, R. Etnangien, a macrolide-polyene antibiotic from Sorangium cellulosum that inhibits nucleic acid polymerases. J. Nat. Prod. 2007, 70, 1060–1063. [Google Scholar] [CrossRef]
- Jansen, R.; Van Embden, J.D.A.; Gaastra, W.; Schouls, L.M. Identification of genes that are associated with DNA repeats in prokaryotes. Mol. Microbiol. 2002, 43, 1565–1575. [Google Scholar] [CrossRef]
- Mojica, F.J.; Díez-Villaseñor, C.; Soria, E.; Juez, G. Biological significance of a family of regularly spaced repeats in the genomes of archaea, bacteria and mitochondria. Mol. Microbiol. 2000, 36, 244–246. [Google Scholar] [CrossRef]
- Louwen, R.; Staals, R.; Endtz, H.; van Baarlen, P.; van der Oost, J. The role of CRISPR-Cas systems in virulence of pathogenic bacteria. Microbiol. Mol. Biol. Rev. 2014, 78, 74–88. [Google Scholar] [CrossRef]
- Gao, R.; Krysciak, D.; Petersen, K.; Utpatel, C.; Knapp, A.; Schmeisser, C.; Daniel, R.; Voget, S.; Jaeger, K.E.; Streit, W.R. Genome-wide RNA sequencing analysis of quorum sensing-controlled regulons in the plant-associated Burkholderia glumae PG1 strain. Appl. Environ. Microbiol. 2015, 81, 7993–8007. [Google Scholar] [CrossRef]
- Høyland-Kroghsbo, N.M.; Paczkowski, J.; Mukherjee, S.; Broniewski, J.; Westra, E.; Bondy-Denomy, J.; Bassler, B.L. Quorum sensing controls the Pseudomonas aeruginosa CRISPR-Cas adaptive immune system. Proc. Natl. Acad. Sci. USA 2017, 114, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, M.; Catrice, O.; Batut, J.; Masson-Boivin, C. Cupriavidus taiwanensis bacteroids in Mimosa pudica indeterminate nodules are not terminally differentiated. Appl. Environ. Microbiol. 2011, 77, 2161–2164. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.M.; Laevens, S.; Lee, T.M.; Coenye, T.; De Vos, P.; Mergeay, M.; Vandamme, P. Ralstonia taiwanensis sp. nov., isolated from root nodules of Mimosa species and sputum of a cystic fibrosis patient. Int. J. Syst. Evol. Microbiol. 2001, 51, 1729–1735. [Google Scholar] [CrossRef] [PubMed]
- Estrada-de los Santos, P.; Vacaseydel-Aceves, N.B.; Martínez-Aguilar, L.; Cruz-Hernández, M.A.; Mendoza-Herrera, A.; Caballero-Mellado, J. Cupriavidus and Burkholderia species associated with agricultural plants that grow in alkaline soils. J. Microbiol. 2011, 49, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Klonowska, A.; Melkonian, R.; Miché, L.; Tisseyre, P.; Moulin, L. Transcriptomic profiling of Burkholderia phymatum STM815, Cupriavidus taiwanensis LMG19424 and Rhizobium mesoamericanum STM3625 in response to Mimosa pudica root exudates illuminates the molecular basis of their nodulation competitiveness and symbiotic ev. BMC Genom. 2018, 19, 1–22. [Google Scholar] [CrossRef]
- Barrett, C.F.; Parker, M.A. Coexistence of Burkholderia, Cupriavidus, and Rhizobium sp. nodule bacteria on two Mimosa spp. in Costa Rica. Appl. Environ. Microbiol. 2006, 72, 1198–1206. [Google Scholar] [CrossRef]
- Kim, J.; Yoon, S.; Park, Y.; Kim, S.; Ryu, C. Crossing the kingdom border: Human diseases caused by plant pathogens. Environ. Microbiol. 2020, 22, 2485–2495. [Google Scholar] [CrossRef] [PubMed]
- Kirzinger, M.W.B.; Nadarasah, G.; Stavrinides, J. Insights into cross-kingdom plant pathogenic bacteria. Genes 2011, 2, 980–997. [Google Scholar] [CrossRef] [PubMed]
- Gong, B.; Shin, M.; Sun, J.; Jung, C.H.; Bolt, E.L.; Van Der Oost, J.; Kim, J.S. Molecular insights into DNA interference by CRISPR-associated nuclease-helicase Cas3. Proc. Natl. Acad. Sci. USA 2014, 111, 16359–16364. [Google Scholar] [CrossRef]
- Rollins, M.C.F.; Chowdhury, S.; Carter, J.; Golden, S.M.; Wilkinson, R.A.; Bondy-Denomy, J.; Lander, G.C.; Wiedenheft, B. Cas1 and the Csy complex are opposing regulators of Cas2/3 nuclease activity. Proc. Natl. Acad. Sci. USA 2017, 114, E5113–E5121. [Google Scholar] [CrossRef]
- Buyukyoruk, M.; Wiedenheft, B. Type I-F CRISPR–Cas provides protection from DNA, but not RNA phages. Cell Discov. 2019, 5. [Google Scholar] [CrossRef] [PubMed]
- Watve, M.G.; Tickoo, R.; Jog, M.M.; Bhole, B.D. How many antibiotics are produced by the genus Streptomyces? Arch. Microbiol. 2001, 176, 386–390. [Google Scholar] [CrossRef]
- Petković, H.; Cullum, J.; Hranueli, D.; Hunter, I.S.; Perić-Concha, N.; Pigac, J.; Thamchaipenet, A.; Vujaklija, D.; Long, P.F. Genetics of Streptomyces rimosus, the oxytetracycline producer. Microbiol. Mol. Biol. Rev. 2006, 70, 704–728. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.S.; Al-Dhabi, N.A.; Duraipandiyan, V.; Balachandran, C.; Kumar, P.P.; Ignacimuthu, S. In vitro antimicrobial, antioxidant and cytotoxic properties of Streptomyces lavendulae strain SCA5. BMC Microbiol. 2014, 14. [Google Scholar] [CrossRef]
- Schlatter, D.C.; Kinkel, L.L. Global biogeography of Streptomyces antibiotic inhibition, resistance, and resource use. FEMS Microbiol. Ecol. 2014, 88, 386–397. [Google Scholar] [CrossRef]
- Genin, S.; Denny, T.P. Pathogenomics of the Ralstonia solanacearum species complex. Annu. Rev. Phytopathol. 2012, 50, 67–89. [Google Scholar] [CrossRef] [PubMed]
- Schell, M.A. Control of virulence and pathogenicity genes of Ralstonia solanacearum by an elaborate sensory network. Annu. Rev. Phytopathol. 2000, 38, 263–292. [Google Scholar] [CrossRef] [PubMed]
- Palmer, K.L.; Gilmore, M.S. Multidrug-resistant enterococci lack CRISPR-cas. mBio 2010, 1. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | Chromosome 1 | Chromosome 2 | Plasmid 1 |
|---|---|---|---|
| Genome sequencing level | Complete | ||
| Total number of reads | 10,350,496 | ||
| Total yield (bp) | 1,562,924,896 | ||
| Bases with a phred value > 20 (%) | 92.02 | ||
| Bases with a phred value > 30 (%) | 82.95 | ||
| Sequencing depth | 190 | 187 | 190 |
| Genome size (bp) | 4,358,639 | 3,979,285 | 281,218 |
| Genome G + C content (%) | 67.56 | 68.62 | 61.1 |
| No. of genes | 3982 | 3250 | 243 |
| No. of coding genes | 3820 | 3169 | 227 |
| No. of pseudogenes | 93 | 65 | 15 |
| No. of RNA genes (16S/5S/23S) | 3/3/3 | 2/2/2 | 0 |
| No. of tRNA genes | 56 | 10 | 1 |
| Other RNA | 4 | 0 | 0 |
| B. gladioli Strains | Source | Assembly | Size (Mb) | GC (%) | Level | Proteins |
|---|---|---|---|---|---|---|
| ATCC 10248 | Gladiolus | GCA_000959725.1 | 8.9 | 67.6 | Complete | 7514 |
| KACC 11889 | Gladiolus | GCA_002208175.1 | 8.9 | 67.7 | Complete | 7221 |
| ATCC 25417 | Gladiolus | GCA_000756855.1 | 9.3 | 67.3 | Scaffold | 7941 |
| NCTC 12378 | Gladiolus | GCA_900446225.1 | 8.4 | 68.0 | Contig | 7098 |
| BSR3 | Rice | GCA_000194745.1 | 9.1 | 67.4 | Complete | 7639 |
| KACC 18962 | Rice | This study | 8.6 | 67.8 | Complete | 7216 |
| AU0032 | Sputum a | GCA_002980975.1 | 8.0 | 68.2 | Contig | 6769 |
| AU26456 | Sputum | GCA_002981405.1 | 8.1 | 68.2 | Contig | 6874 |
| AU29541 | Sputum | GCA_002981475.1 | 8.4 | 68.2 | Contig | 7112 |
| AU30473 | Sputum | GCA_002981875.1 | 8.1 | 68.3 | Contig | 6928 |
| Coa14 | Water | GCA_002917905.1 | 8.5 | 68.0 | Contig | 7189 |
| MSMB1756 | Soil | GCA_001527485.1 | 8.2 | 68.1 | Contig | 6946 |
| FDAARGOS_390 | Nature b | GCA_002554225.1 | 8.8 | 67.6 | Contig | 7470 |
| FDAARGOS_391 | Nature | GCA_002554395.1 | 8.4 | 68.0 | Contig | 7058 |
| Core | COG | No. Cluster | p-Value |
|---|---|---|---|
| Class | |||
| J | Translation, ribosomal structure and biogenesis | 172 | 1.52 × 10−4 |
| F | Nucleotide transport and metabolism | 91 | 4.79 × 10−4 |
| Dispensable | |||
| Class | COG | No. cluster | P-value |
| L | Replication, recombination and repair | 234 | 0 |
| Q | Secondary metabolites biosynthesis, transport and catabolism | 297 | 0 |
| - | Unclassified | 3929 | 0 |
| W | Extracellular structures | 29 | 5.52 × 10−12 |
| U | Intracellular trafficking, secretion, and vesicular transport | 147 | 6.91 × 10−7 |
| V | Defense mechanisms | 53 | 6.36 × 10−4 |
| R | General function prediction only | 541 | 3.76 × 10−3 |
| I | Lipid transport and metabolism | 176 | 1.27 × 10−2 |
| Strain | CRISPR Count | Type | Cas Gene Count | Cas Gene | No. Spacers | Spacer Match |
|---|---|---|---|---|---|---|
| ATCC 10248 | 4 | - | - | - | 1,1,1,1 | ND a |
| KACC 11889 | 4 | - | - | - | 1,1,1,1 | ND |
| ATCC 25417 | 2 | - | - | - | 1,1 | ND |
| NCTC 12378 | 4 | - | - | - | 1,1,1,1 | ND |
| BSR3 | 2 | - | - | - | 1,2 | Cupriavidus taiwanensis |
| KACC 18962 | 1 | - | - | - | 1 | ND |
| AU0032 | 1 | - | - | - | 1 | ND |
| AU26456 | 2 | - | - | - | 1,1 | ND |
| AU29541 | - | - | - | - | - | - |
| AU30473 | 2 | - | - | - | 1,1 | ND |
| Coa14 | 3 | Type I-F | 6 | Cas1,Cas3-Cas2,Csy1,Csy2,Csy3,Cas6 | 1,1,39 | Streptomyces spp. b Ralstonia solanacearum |
| MSMB1756 | 2 | Type I-F | 6 | Cas1,Cas3-Cas2,Csy1,Csy2,Csy3,Cas6 | 1,46 | Streptomyces spp. Ralstonia solanacearum |
| FDAARGOS_390 | - | - | - | - | - | - |
| FDAARGOS_391 | - | - | - | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.-H.; Park, J.; Jung, H.; Seo, Y.-S. Pan-Genome Analysis Reveals Host-Specific Functional Divergences in Burkholderia gladioli. Microorganisms 2021, 9, 1123. https://doi.org/10.3390/microorganisms9061123
Lee H-H, Park J, Jung H, Seo Y-S. Pan-Genome Analysis Reveals Host-Specific Functional Divergences in Burkholderia gladioli. Microorganisms. 2021; 9(6):1123. https://doi.org/10.3390/microorganisms9061123
Chicago/Turabian StyleLee, Hyun-Hee, Jungwook Park, Hyejung Jung, and Young-Su Seo. 2021. "Pan-Genome Analysis Reveals Host-Specific Functional Divergences in Burkholderia gladioli" Microorganisms 9, no. 6: 1123. https://doi.org/10.3390/microorganisms9061123
APA StyleLee, H.-H., Park, J., Jung, H., & Seo, Y.-S. (2021). Pan-Genome Analysis Reveals Host-Specific Functional Divergences in Burkholderia gladioli. Microorganisms, 9(6), 1123. https://doi.org/10.3390/microorganisms9061123