
Global Changes in Asexual Epichloë Transcriptomes during the Early Stages, from Seed to Seedling, of Symbiotum Establishment


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and Seed Germination
2.2. RNA Extraction, Library Construction, and Sequencing
2.3. Reference Genomes
2.4. Preprocessing and Mapping of Illumina Reads
2.5. Identification of the Most Highly Expressed Genes—The Top 50
2.6. Differential Gene Expression Analysis
2.7. Enrichment Analysis
2.8. Clustering
2.9. Identification of Unique Genes of Each Endophyte Strain
3. Results and Discussion
3.1. Library Preparation and Mapping Reads to Endophyte Genome
3.2. Insights into the Most Highly Expressed Genes during Early Stages of Symbiotum Establishment—The Top 50
3.2.1. The Most Abundant Endophyte Genes Expressed by SE, NEA11, and NEA12
3.2.2. The Most Abundant Endophyte Genes Common to SE, NEA11, and NEA12
3.2.3. Other Highly Expressed Endophyte Genes
3.3. Identification of Differentially Expressed Genes
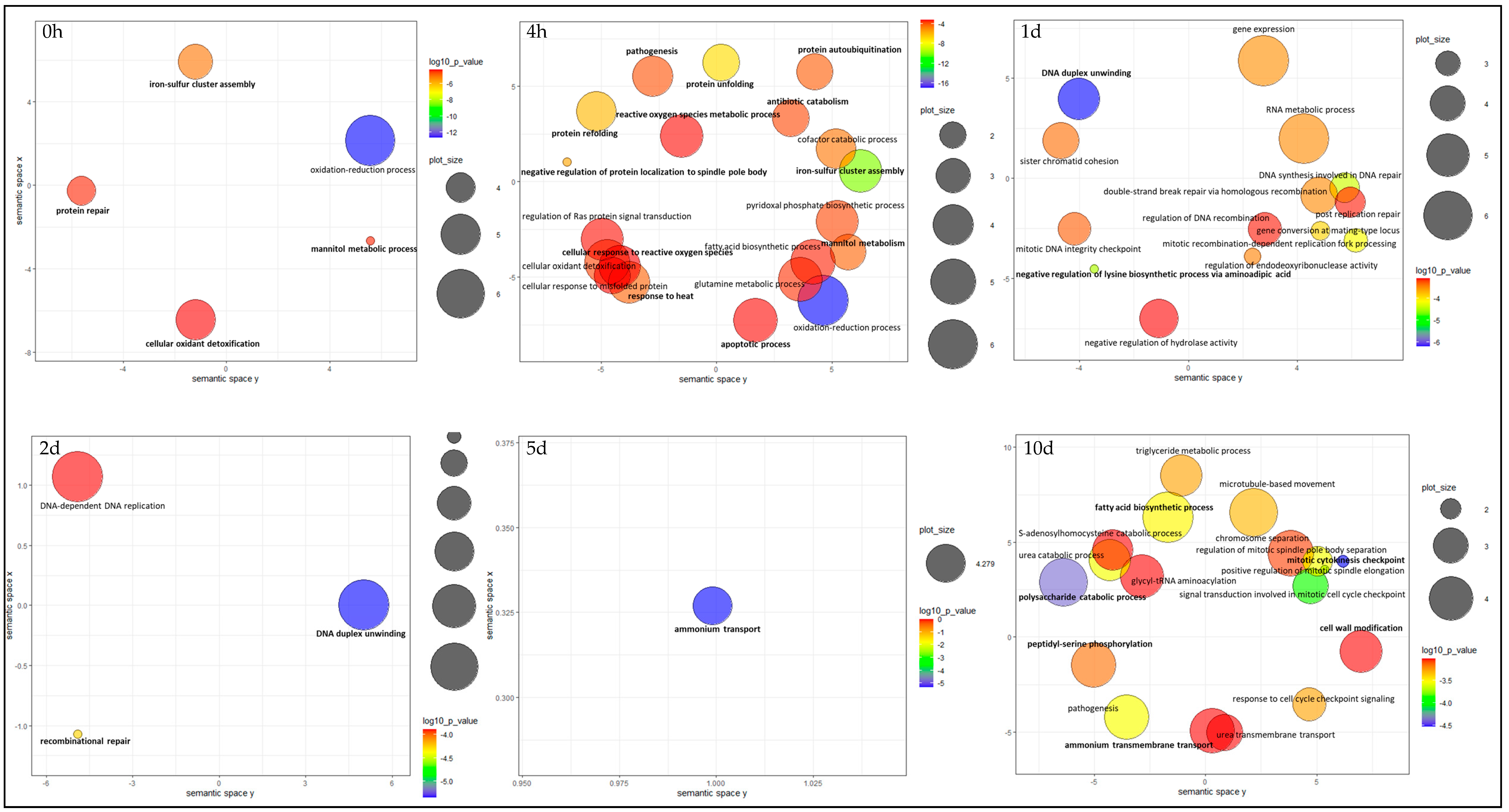
3.4. Gene Ontology (GO) Functional Enrichment Analysis
3.4.1. 0 h and 4 h
3.4.2. Day 1 and 2
3.4.3. Day 5 and 10
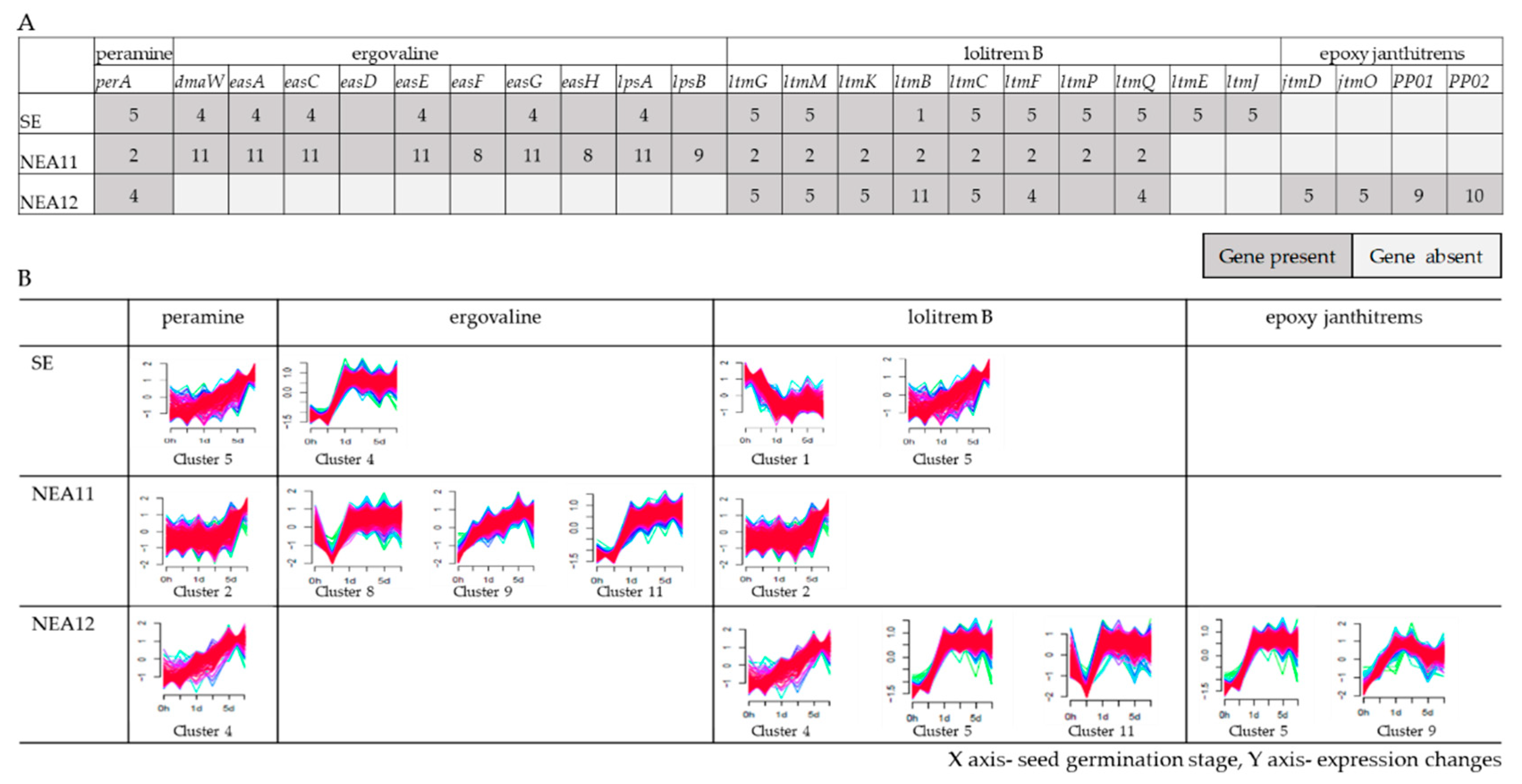
3.5. Gene Expression Profiles of Known Alkaloid Biosynthesis Genes
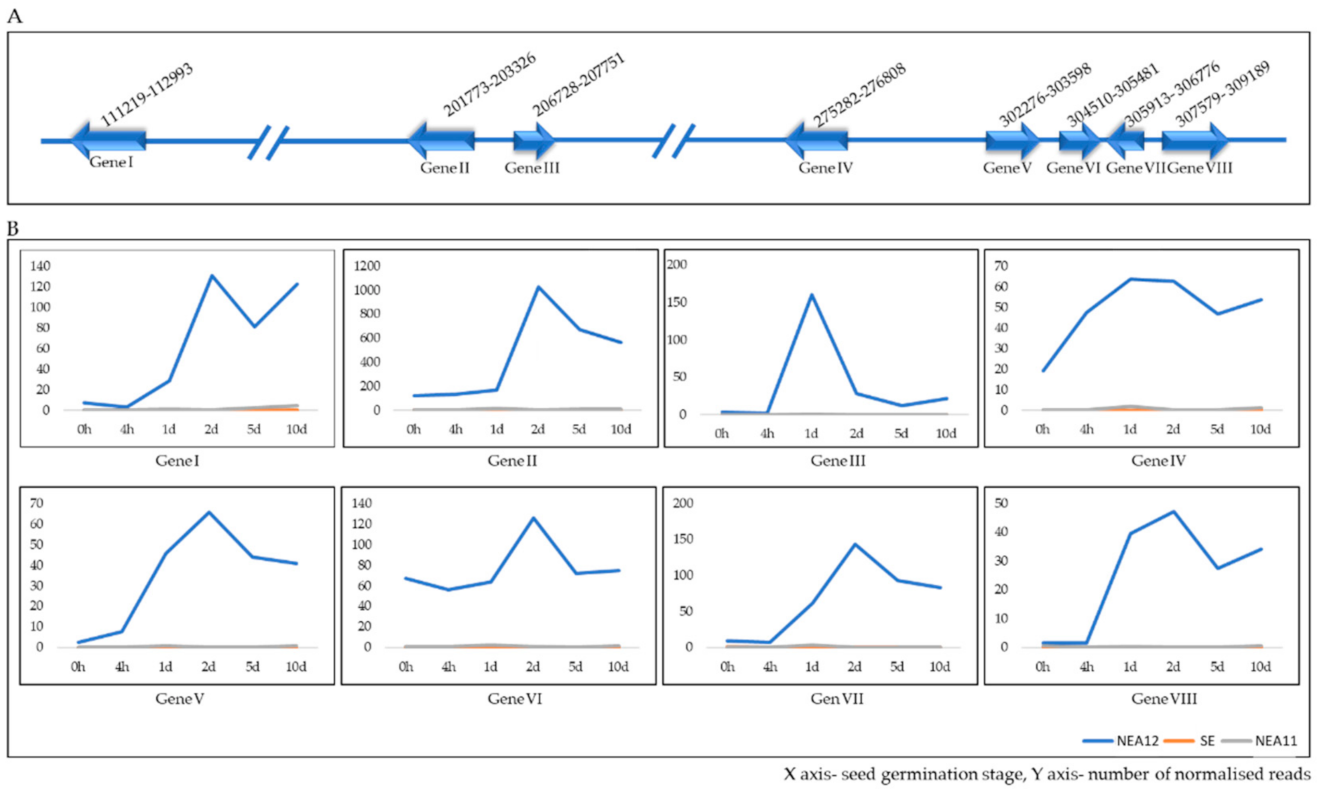
3.6. Identification of Unique Genes of Each Endophyte Strain
3.6.1. Comparative Analysis Between SE and NEA12
3.6.2. Genes Unique to NEA11
3.6.3. Genes Unique to NEA12
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schardl, C.L. The Epichloë, symbionts of the grass subfamily Poöideae. Ann. Mo. Bot. Gard. 2010, 97, 646–665. [Google Scholar] [CrossRef]
- Card, S.D.; Rolston, M.P.; Lloyd-West, C.; Hume, D.E. Novel perennial ryegrass-Neotyphodium endophyte associations: Relationships between seed weight, seedling vigour and endophyte presence. Symbiosis 2014, 62, 51–62. [Google Scholar] [CrossRef]
- Leuchtmann, A.; Bacon, C.W.; Schardl, C.L.; White, J.J.F.; Tadych, M. Nomenclatural realignment of Neotyphodium species with genus Epichloë. Mycolgia 2014, 106, 202–215. [Google Scholar] [CrossRef]
- Schardl, C.L.; Craven, K.D.; Speakman, S.; Stromberg, A.; Lindstrom, A.; Yoshida, R. A novel test for host-symbiont codivergence indicates ancient origin of fungal endophytes in grasses. Syst. Biol. 2008, 57, 483–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schardl, C.L.; Leuchtmann, A. The Epichloë Endophytes of Grasses and the Symbiotic Continuum; Boca Raton Publishers: Boca Raton, FL, USA, 2005; pp. 475–503. [Google Scholar]
- Förster, L.; Grant, J.; Michel, T.; Ng, C.; Barth, S. Growth under cold conditions in a wide perennial ryegrass panel is under tight physiological control. PeerJ 2018, 6, e5520. [Google Scholar] [CrossRef]
- Li, T.; Blande, J.D.; Gundel, P.E.; Helander, M.; Saikkonen, K. Epichloë endophytes alter inducible indirect defences in host grasses. PLoS ONE 2014, 9, e101331. [Google Scholar] [CrossRef] [Green Version]
- Clay, K. Fungal endophytes of grasses: A defensive mutualism between plants and fungi. Ecology 1988, 69, 10–16. [Google Scholar] [CrossRef]
- Clay, K.; Schardl, S. Evolutionary origins and ecological consequences of endophyte symbiosis with grasses. Am. Nat. 2002, 160, S99–S127. [Google Scholar] [CrossRef]
- Xia, C.; Li, N.; Zhang, Y.; Li, C.; Zhang, X.; Nan, Z. Role of Epichloë endophytes in defense responses of cool-season grasses to pathogens: A review. Plant Dis. 2018, 102, 2061–2073. [Google Scholar] [CrossRef] [Green Version]
- Zabalgogeazcoa, I.; de Aldana, B.R.V.; Ciudad, A.G.; Criado, B.G. Fungal endophytes in grasses from semi-arid permanent grasslands of western Spain. Grass Forage Sci. 2003, 58, 94–97. [Google Scholar] [CrossRef]
- Schardl, C.L.; Leuchtmann, A.; Spiering, M.J. Symbioses of grasses with seedborne fungal endophytes. Annu. Rev. Plant Biol. 2004, 55, 315–340. [Google Scholar] [CrossRef]
- Fernando, K.; Reddy, P.; Hettiarachchige, I.K.; Spangenberg, G.C.; Rochfort, S.J.; Guthridge, K.M. Novel antifungal activity of Lolium-associated Epichloë endophytes. Microorganisms 2020, 8, 955. [Google Scholar] [CrossRef] [PubMed]
- Scott, B. Epichloë endophytes: Fungal symbionts of grasses. Curr. Opin. Microbiol. 2001, 4, 393–398. [Google Scholar] [CrossRef]
- Young, C.; Tapper, B.; May, K.; Moon, C.; Schardl, C.; Scott, B. Indole-diterpene biosynthetic capability of Epichloë endophytes as predicted by ltm gene analysis. Appl. Environ. Microbiol. 2009, 75, 2200–2211. [Google Scholar] [CrossRef] [Green Version]
- Ludlow, E.; Vassiliadis, S.; Ekanayake, P.N.; Hettiarachchige, I.K.; Reddy, P.; Sawbridge, T.I.; Rochfort, S.J.; Spangenberg, G.C.; Guthridge, K.M. Analysis of the indole diterpene gene cluster for biosynthesis of the epoxy-janthitrems in Epichloë endophytes. Microorganisms 2019, 7, 560. [Google Scholar] [CrossRef] [Green Version]
- Spiering, M.J.; Moon, C.D.; Wilkinson, H.H.; Schardl, C.L. Gene clusters for insecticidal loline alkaloids in the grass-endophytic fungus Neotyphodium uncinatum. Genetics 2005, 169, 1403–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, A.; Tapper, B.A.; Popay, A.; Parker, E.J.; Scott, B. A symbiosis expressed non-ribosomal peptide synthetase from a mutualistic fungal endophyte of perennial ryegrass confers protection to the symbiotum from insect herbivory. Mol. Microbiol. 2005, 57, 1036–1050. [Google Scholar] [CrossRef]
- Wang, J.H.; Machado, C.; Panaccione, D.G.; Tsai, H.F.; Schardl, C.L. The determinant step in ergot alkaloid biosynthesis by an endophyte of perennial ryegrass. Fungal Genet. Biol. 2004, 41, 189–198. [Google Scholar] [CrossRef]
- Damrongkool, P.; Sedlock, A.B.; Young, C.A.; Johnson, R.D.; Goetz, K.E.; Scott, B.; Schardl, C.L.; Panaccione, D.G. Structural analysis of a peptide synthetase gene required for ergopeptine production in the endophytic fungus Neotyphodium lolii. DNA Seq. 2005, 16, 379–385. [Google Scholar] [CrossRef]
- Bush, L.P.; Wilkinson, H.H.; Schardl, C.L. Bioprotective alkaloids of grass-fungal endphyte sysmbioses. Plant Physiol. 1997, 114, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, M.R.; Latch, G.C.M.; Bush, L.P.; Fannin, F.F.; Rowan, D.D.; Tapper, B.A.; Bacon, C.W.; Johnson, M.C. Fungal endophyte-infected grasses: Alkaloid accumulation and aphid response. J. Chem. Ecol. 1990, 16, 3301–3315. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.; Guthridge, K.; Vassiliadis, S.; Hemsworth, J.; Hettiarachchige, I.; Spangenberg, G.; Rochfort, S. Tremorgenic mycotoxins: Structure diversity and biological activity. Toxins 2019, 11, 302. [Google Scholar] [CrossRef] [Green Version]
- Schardl, C.L.; Young, C.A.; Hesse, U.; Amyotte, S.G.; Andreeva, K.; Calie, P.J.; Fleetwood, D.J.; Haws, D.C.; Moore, N.; Oeser, B.; et al. Plant-symbiotic fungi as chemical engineers: Multi-genome analysis of the Clavicipitaceae reveals dynamics of alkaloid loci. PLoS Genet. 2013, 9, e1003323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babu, J.; Popay, A.; Miles, C.; Wilkins, A.; Menna, M.; Finch, S. Identification and structure elucidation of Janthitrems A and D from Penicillium janthinellum and determination of the tremorgenic and anti-Insect activity of Janthitrems A and B. J. Agric. Food Chem. 2018, 66. [Google Scholar] [CrossRef]
- Gagic, M.; Faville, M.J.; Zhang, W.; Forester, N.T.; Rolston, M.P.; Johnson, R.D.; Ganesh, S.; Koolaard, J.P.; Easton, H.S.; Hudson, D.; et al. Seed transmission of Epichloë endophytes in Lolium perenne is heavily influenced by host genetics. Front. Plant Sci. 2018, 9, 1580. [Google Scholar] [CrossRef] [Green Version]
- Kauppinen, M.; Saikkonen, K.; Helander, M.; Pirttilä, A.M.; Wäli, P. Epichloë grass endophytes in sustainable agriculture. Nat. Plants 2016, 2, 15224. [Google Scholar] [CrossRef] [PubMed]
- Eaton, C.J.; Cox, M.P.; Ambrose, B.; Becker, M.; Hesse, U.; Schardl, C.L.; Scott, B. Disruption of signaling in a fungal-grass symbiosis leads to pathogenesis. Plant Physiol. 2010, 153, 1780–1794. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.Y.; Spiering, M.J.; Scott, V.; Lane, G.A.; Christensen, M.J.; Schmid, J. In planta regulation of extension of an endophytic fungus and maintenance of high metabolic rates in its mycelium in the absence of apical extension. Appl. Environ. Microbiol. 2001, 67, 5377–5383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Card, S.; Johnson, L.; Teasdale, S.; Caradus, J. Deciphering endophyte behaviour: The link between endophyte biology and efficacious biological control agents. FEMS Microbiol. Ecol. 2016, 92. [Google Scholar] [CrossRef] [Green Version]
- Majewska-Sawka, A.; Nakashima, H. Endophyte transmission via seeds of Lolium perenne L.: Immunodetection of fungal antigens. Fungal Genet. Biol. 2004, 41, 534–541. [Google Scholar] [CrossRef]
- Philipson, M.N.; Christey, M.C. The relationship of host and endophyte during flowering, seed formation and germination of Lolium perenne. N. Z. J. Bot. 1986, 24, 125–134. [Google Scholar] [CrossRef]
- Christensen, M.J.; Bennett, R.J.; Ansari, H.A.; Koga, H.; Johnson, R.D.; Bryan, G.T.; Simpson, W.R.; Koolaard, J.P.; Nickless, E.M.; Voisey, C.R. Epichloë endophytes grow by intercalary hyphal extension in elongating grass leaves. Fungal Genet. Biol. 2008, 45, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, K.V.; Belanger, F.C. SOLiD-SAGE of endophyte-infected red fescue reveals numerous effects on host transcriptome and an abundance of highly expressed fungal secreted proteins. PLoS ONE 2012, 7, e53214. [Google Scholar] [CrossRef] [Green Version]
- Voisey, C.R.; Christensen, M.T.; Johnson, L.J.; Forester, N.T.; Gagic, M.; Bryan, G.T.; Simpson, W.R.; Fleetwood, D.J.; Card, S.D.; Koolaard, J.P.; et al. cAMP Signaling regulates synchronised growth of symbiotic Epichloë fungi with the host grass Lolium perenne. Front. Plant Sci. 2016, 7, 546. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Nagabhyru, P.; Schardl, C. Epichloë festucae endophytic growth in florets, seeds, and seedlings of perennial ryegrass (Lolium perenne). Mycologia 2017, 109, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, K.; Ohkubo, H.; Yamashita, M.; Mikoshiba, Y. Flowers for Neotyphodium endophytes detection: A new observation method using flowers of host grasses. Mycoscience 2004, 45, 222–226. [Google Scholar] [CrossRef]
- Green, K.A.; Berry, D.; Feussner, K.; Eaton, C.J.; Ram, A.; Mesarich, C.H.; Solomon, P.; Feussner, I.; Scott, B. Lolium perenne apoplast metabolomics for identification of novel metabolites produced by the symbiotic fungus Epichloë festucae. New Phytol. 2020, 227, 559–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, S.; Parsons, A.J.; Fraser, K.; Xue, H.; Newman, J.A. Metabolic profiles of Lolium perenne are differentially affected by nitrogen supply, carbohydrate content, and fungal endophyte infection. Plant Physiol. 2008, 146, 1440–1453. [Google Scholar] [CrossRef] [Green Version]
- Dupont, P.-Y.; Eaton, C.; Wargent, J.; Fechtner, S.; Solomon, P.; Schmid, J.; Day, R.; Scott, B.; Cox, M. Fungal endophyte infection of ryegrass reprograms host metabolism and alters development. New Phytol. 2015, 208. [Google Scholar] [CrossRef] [PubMed]
- Nagabhyru, P.; Dinkins, R.D.; Schardl, C.L. Transcriptomics of Epichloë-grass symbioses in host vegetative and reproductive stages. Mol. Plant Microbe Interact. 2019, 32, 194–207. [Google Scholar] [CrossRef] [Green Version]
- Rahnama, M.; Maclean, P.; Fleetwood, D.J.; Johnson, R.D. VelA and LaeA are key regulators of Epichloë festucae transcriptomic response during symbiosis with perennial ryegrass. Microorganisms 2020, 8, 33. [Google Scholar] [CrossRef] [Green Version]
- Dinkins, R.D.; Nagabhyru, P.; Graham, M.A.; Boykin, D.; Schardl, C.L. Transcriptome response of Lolium arundinaceum to its fungal endophyte Epichloë coenophiala. New Phytol. 2017, 213, 324–337. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.J.; Johnson, R.D.; Schardl, C.L.; Pinaccione, D.G. Identification of differentially expressed genes in the mutualistic association of tall fescue with Netyphodim coenophialum. Physiol. Mol. Plant Pathol. 2003, 63, 305–317. [Google Scholar] [CrossRef]
- Khan, A.; Bassett, S.; Voisey, C.; Gaborit, C.; Johnson, L.; Christensen, M.; McCulloch, A.; Bryan, G.; Johnson, R. Gene expression profiling of the endophytic fungus Neotyphodium lolii in association with its host plant perennial ryegrass. Australas. Plant Pathol. 2010, 39, 467–476. [Google Scholar] [CrossRef]
- Wang, R.; Clarke, B.B.; Belanger, F.C. Transcriptome analysis of choke stroma and asymptomatic inflorescence tissues reveals changes in gene expression in both Epichloë festucae and its host plant Festuca rubra subsp. rubra. Microorganisms 2019, 7, 567. [Google Scholar] [CrossRef] [Green Version]
- Hettiarachchige, I.K.; Elkins, A.C.; Reddy, P.; Mann, R.C.; Guthridge, K.M.; Sawbridge, T.I.; Forster, J.W.; Spangenberg, G.C. Genetic modification of asexual Epichloë endophytes with the perA gene for peramine biosynthesis. Mol. Genet. Genom. 2019, 294, 315–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hettiarachchige, I.K.; Ekanayake, P.N.; Mann, R.C.; Guthridge, K.M.; Sawbridge, T.I.; Spangenberg, G.C.; Forster, J.W. Phylogenomics of asexual Epichloë fungal endophytes forming associations with perennial ryegrass. BMC Evol. Biol. 2015, 15, 72. [Google Scholar] [CrossRef] [Green Version]
- Kaur, J.; Ekanayake, P.; Tian, P.; van Zijll de Jong, E.; Dobrowolski, M.P.; Rochfort, S.J.; Mann, R.; Smith, K.F.; Forster, J.W.; Guthridge, K.M.; et al. Discovery and characterisation of novel asexual Epichloë endophytes from perennial ryegrass (Lolium perenne L.). Crop Pasture Sci. 2015, 66, 1058–1070. [Google Scholar] [CrossRef]
- Chang, S.; Puryear, J.; Cairney, J. A simple and efficient method for isolating RNA from pine trees. Plant Mol. Biol. Rep. 1993, 11, 113–116. [Google Scholar] [CrossRef]
- Stanke, M.; Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 2003, 19, ii215–ii225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conesa, A.; Götz, S.; García-Gómez, J.; Terol, J.; Talon, M.; Robles, M. BLAST2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [Green Version]
- Shinozuka, H.; Cogan, N.; Spangenberg, G.; Forster, J. Reference transcriptome assembly and annotation for perennial ryegrass. Genome 2017, 60, 1086–1088. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [Green Version]
- Kumar, L.; E Futschik, M. Mfuzz: A software package for soft clustering of microarray data. Bioinformation 2007, 2, 5–7. [Google Scholar] [CrossRef]
- Pfeifer, M.; Martis, M.; Asp, T.; Mayer, K.; Lübberstedt, T.; Byrne, S.; Frei, U.; Studer, B. The perennial ryegrass genome zippe—Targeted use of genome resources for comparative grass genomics. Plant Physiol. 2012, 161. [Google Scholar] [CrossRef] [Green Version]
- Kuldau, G.A.; Tsai, H.F.; Schardl, C.L. Genome sizes of Epichloë species and anamorphic hybrids. Mycologia 1999, 91, 776–782. [Google Scholar]
- Winter, D.J.; Ganley, A.R.D.; Young, C.A.; Liachko, I.; Schardl, C.L.; Dupont, P.-Y.; Berry, D.; Ram, A.; Scott, B.; Cox, M.P. Repeat elements organise 3D genome structure and mediate transcription in the filamentous fungus Epichloë festucae. PLoS Genet. 2018, 14, e1007467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, A. Small heat shock proteins (HSP12, HSP20 and HSP30) play a role in Ustilago maydis pathogenesis. FEMS Microbiol. Lett. 2014, 361, 17–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, C.-J.; Seo, Y.-S. Heat shock proteins: A review of the molecular chaperones for plant immunity. Plant Pathol. J. 2015, 31, 323–333. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, S.; Thakur, R.; Shankar, J. Role of heat-shock proteins in cellular function and in the biology of fungi. Biotechnol. Res. Int. 2015, 2015, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Hasan, M.K.; Cheng, Y.; Kanwar, M.K.; Chu, X.-Y.; Ahammed, G.J.; Qi, Z.-Y. Responses of plant proteins to heavy metal stress—A review. Front. Plant Sci. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akcapinar, G.B.; Kappel, L.; Sezerman, O.U.; Seidl-Seiboth, V. Molecular diversity of LysM carbohydrate-binding motifs in fungi. Curr. Genet. 2015, 61, 103–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, M.; Glenn, A.; Blacutt, A.; Gold, S. Fungal lactamases: Their occurrence and function. Front. Microbiol. 2017, 8, 1775. [Google Scholar] [CrossRef] [PubMed]
- Hassing, B.; Winter, D.; Becker, Y.; Mesarich, C.H.; Eaton, C.J.; Scott, B. Analysis of Epichloë festucae small secreted proteins in the interaction with Lolium perenne. PLoS ONE 2019, 14, e0209463. [Google Scholar] [CrossRef] [Green Version]
- Bassett, S.A.; Johnson, R.D.; Simpson, W.R.; Laugraud, A.; Jordan, T.W.; Bryan, G.T. Identification of a gene involved in the regulation of hyphal growth of Epichloë festucae during symbiosis. FEMS Microbiol. Lett. 2016, 363. [Google Scholar] [CrossRef]
- Suhandono, S.; Apriyanto, A.; Ihsani, N. Isolation and characterization of three cassava elongation factor 1 alpha (MeEF1A) promoters. PLoS ONE 2014, 9, e84692. [Google Scholar] [CrossRef] [Green Version]
- Queitsch, C.; Hong, S.W.; Vierling, E.; Lindquist, S. Heat shock protein 101 plays a crucial role in thermotolerance in Arabidopsis. Plant Cell 2000, 12, 479–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, S.; Huang, Z.; Li, H.; Bashir, M.H.; Ren, S. Antioxidant enzyme influences germination, stress tolerance, and virulence of Isaria fumosorosea. J. Basic Microbiol. 2013, 53, 489–497. [Google Scholar] [CrossRef]
- Pradhan, A.; Herrero-de-Dios, C.; Belmonte, R.; Budge, S.; Lopez Garcia, A.; Kolmogorova, A.; Lee, K.K.; Martin, B.D.; Ribeiro, A.; Bebes, A.; et al. Elevated catalase expression in a fungal pathogen is a double-edged sword of iron. PLoS Pathog. 2017, 13, e1006405. [Google Scholar] [CrossRef] [PubMed]
- Rittenour, W.R.; Harris, S.D. Glycosylphosphatidylinositol-anchored proteins in Fusarium graminearum: Inventory, variability, and virulence. PLoS ONE 2013, 8, e81603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, R.D.; Lane, G.A.; Koulman, A.; Cao, M.; Fraser, K.; Fleetwood, D.J.; Voisey, C.R.; Dyer, J.M.; Pratt, J.; Christensen, M.; et al. A novel family of cyclic oligopeptides derived from ribosomal peptide synthesis of an in planta-induced gene, gigA, in Epichloë endophytes of grasses. Fungal Genet. Biol. 2015, 85, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Marcos, C.M.; de Oliveira, H.C.; da Silva, J.F.; Assato, P.A.; Yamazaki, D.S.; da Silva, R.A.M.; Santos, C.T.; Santos-Filho, N.A.; Portuondo, D.L.; Mendes-Giannini, M.J.S.; et al. Identification and characterisation of elongation factor Tu, a novel protein involved in Paracoccidioides brasiliensis–host interaction. FEMS Yeast Res. 2016, 16. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Liu, H.; Wang, C.; Xu, J.-R. Comparative analysis of fungal genomes reveals different plant cell wall degrading capacity in fungi. BMC Genom. 2013, 14, 274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panagiotou, G.; Andersen, M.R.; Grotkjær, T.; Regueira, T.B.; Hofmann, G.; Nielsen, J.; Olsson, L. Systems analysis unfolds the relationship between the phosphoketolase pathway and growth in Aspergillus nidulans. PLoS ONE 2008, 3, e3847. [Google Scholar] [CrossRef] [Green Version]
- Duan, Z.; Shang, Y.; Gao, Q.; Zheng, P.; Wang, C. A phosphoketolase Mpk1 of bacterial origin is adaptively required for full virulence in the insect-pathogenic fungus Metarhizium anisopliae. Environ. Microbiol. 2009, 11, 2351–2360. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Rubio, R.; de Oliveira, H.C.; Rivera, J.; Trevijano-Contador, N. The fungal cell wall: Candida, Cryptococcus, and Aspergillus Species. Front. Microbiol. 2020, 10. [Google Scholar] [CrossRef]
- Baccelli, I. Cerato-platanin family proteins: One function for multiple biological roles? Front. Plant Sci. 2015, 5, 769. [Google Scholar] [CrossRef]
- López-García, B.; Gandía, M.; Muñoz, A.; Carmona, L.; Marcos, J.F. A genomic approach highlights common and diverse effects and determinants of susceptibility on the yeast Saccharomyces cerevisiae exposed to distinct antimicrobial peptides. BMC Microbiol. 2010, 10, 289. [Google Scholar] [CrossRef] [Green Version]
- Pennerman, K.K.; Yin, G.; Bennett, J.W.; Hua, S.-S.T. Aspergillus flavus NRRL 35739, a poor biocontrol agent, may have increased relative expression of stress response genes. J. Fungi 2019, 5, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, A.; Kögl, S.A.; Jung, K. Time-dependent proteome alterations under osmotic stress during aerobic and anaerobic growth in Escherichia coli. J. Bacteriol. 2006, 188, 7165–7175. [Google Scholar] [CrossRef] [Green Version]
- Heberle, H.; Meirelles, G.V.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 2015, 16, 169. [Google Scholar] [CrossRef]
- Barrett, B.A.; Faville, M.J.; Nichols, S.N.; Simpson, W.R.; Bryan, G.T.; Conner, A.J. Breaking through the feed barrier: Options for improving forage genetics. Anim. Prod. Sci. 2015, 55, 883–892. [Google Scholar] [CrossRef] [Green Version]
- Van Zijll de Jong, E.; Dobrowolski, M.P.; Sandford, A.; Smith, K.F.; Willocks, M.J.; Spangenberg, G.C.; Forster, J.W. Detection and characterisation of novel fungal endophyte genotypic variation in cultivars of perennial ryegrass (Lolium perenne L.). Aust. J. Agric. Res. 2008, 59, 214–221. [Google Scholar] [CrossRef]
- Tian, P.; Le, T.-N.; Ludlow, E.J.; Smith, K.F.; Forster, J.W.; Guthridge, K.M.; Spangenberg, G.C. Characterisation of novel perennial ryegrass host–Neotyphodium endophyte associations. Crop Pasture Sci. 2013, 64, 716–725. [Google Scholar] [CrossRef]
- Scott, B.; Takemoto, D.; Tanaka, A. Fungal endophyte production of reactive oxygen species is critical for maintaining the mutualistic symbiotic interaction between Epichloë festucae and perennial ryegrass. Plant Signal Behav. 2007, 2, 171–173. [Google Scholar] [CrossRef] [Green Version]
- Cartwright, G.M.; Scott, B. Redox regulation of an AP-1-like transcription factor, YapA, in the fungal symbiont Epichloe festucae. Eukaryot Cell 2013, 12, 1335–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, J.F.; Kingsley, K.L.; Zhang, Q.; Verma, R.; Obi, N.; Dvinskikh, S.; Elmore, M.T.; Verma, S.K.; Gond, S.K.; Kowalski, K.P. Review: Endophytic microbes and their potential applications in crop management. Pest Manag. Sci. 2019, 75, 2558–2565. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Zhang, S.; Borchert, S.; Richardson, K.; Schmid, J. High levels of a fungal superoxide dismutase and increased concentration of a PR-10 plant protein in associations between the endophytic fungus Neotyphodium lolii and ryegrass. Mol. Plant Microbe Interact. 2011, 24, 984–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, A.; Christensen, M.J.; Takemoto, D.; Park, P.; Scott, B. Reactive oxygen species play a role in regulating a fungus–perennial ryegrass mutualistic interaction. Plant Cell 2006, 18, 1052–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanda, A.K.; Andrio, E.; Marino, D.; Pauly, N.; Dunand, C. Reactive oxygen species during plant-microorganism early interactions. J. Integr. Plant Biol. 2010, 52, 195–204. [Google Scholar] [CrossRef]
- White, J.F.; Torres, M.S. Is plant endophyte-mediated defensive mutualism the result of oxidative stress protection? Physiol. Plant 2010, 138, 440–446. [Google Scholar] [CrossRef]
- Patel, T.; Williamson, J. Mannitol in plants, fungi, and plant–fungal interactions. Trends Plant Sci. 2016, 21. [Google Scholar] [CrossRef]
- Meena, M.; Prasad, V.; Zehra, A.; Gupta, V.K.; Upadhyay, R.S. Mannitol metabolism during pathogenic fungal-host interactions under stressed conditions. Front. Microbiol. 2015, 6, 1019. [Google Scholar] [CrossRef] [Green Version]
- Gerwien, F.; Skrahina, V.; Kasper, L.; Hube, B.; Brunke, S. Metals in fungal virulence. FEMS Microbiol Rev. 2018, 42, fux050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reininger, V.; Schlegel, M. Analysis of the Phialocephala subalpina transcriptome during colonization of Its host plant Picea abies. PLoS ONE 2016, 11, e0150591. [Google Scholar] [CrossRef]
- Johnson, L.J.; Koulman, A.; Christensen, M.; Lane, G.A.; Fraser, K.; Forester, N.; Johnson, R.D.; Bryan, G.T.; Rasmussen, S. An extracellular siderophore is required to maintain the mutualistic interaction of Epichloë festucae with Lolium perenne. PLoS Pathog. 2013, 9, e1003332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braymer, J.J.; Lill, R. Iron-sulfur cluster biogenesis and trafficking in mitochondria. J. Biol. Chem. 2017, 292, 12754–12763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez, M.; Pérez-Gallardo, R.V.; Sánchez, L.A.; Díaz-Pérez, A.L.; Cortés-Rojo, C.; Meza Carmen, V.; Saavedra-Molina, A.; Lara-Romero, J.; Jiménez-Sandoval, S.; Rodríguez, F.; et al. Malfunctioning of the iron-sulfur cluster assembly machinery in Saccharomyces cerevisiae produces oxidative stress via an iron-dependent mechanism, causing dysfunction in respiratory complexes. PLoS ONE 2014, 9, e111585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Wu, X.-M.; Liu, C.-H.; Shan, J.-Y.; Gao, F.; Guo, H.-S. Verticillium dahliae chromatin remodeling facilitates the DNA damage repair in response to plant ROS stress. PLoS Pathog. 2020, 16, e1008481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dantas, A.S.; Day, A.; Ikeh, M.; Kos, I.; Achan, B.; Quinn, J. Oxidative stress responses in the human fungal pathogen, Candida albicans. Biomolecules 2015, 5, 142–165. [Google Scholar] [CrossRef] [Green Version]
- Knoll, A.; Puchta, H. The role of DNA helicases and their interaction partners in genome stability and meiotic recombination in plants. J. Exp. Bot. 2011, 62, 1565–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tivey, T.R.; Parkinson, J.E.; Weis, V.M. Host and symbiont cell cycle coordination is mediated by symbiotic state, nutrition, and partner Identity in a model Cnidarian-Dinoflagellate symbiosis. MBio 2020, 11, e02626-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, M.; Becker, Y.; Green, K.; Scott, B. The endophytic symbiont Epichloë festucae establishes an epiphyllous net on the surface of Lolium perenne leaves by development of an expressorium, an appressorium-like leaf exit structure. New Phytol. 2016, 211, 240–254. [Google Scholar] [CrossRef] [Green Version]
- Christensen, M.J.; Voissey, C.R. The biology of the endophyte/grass partnership. In Proceedings of the 6th International Symposium on Fungal Endophytes of Grasses, Christchurch, New Zealand, 25–28 March 2006; Ag Research Grasslands: Palmerston North, New Zealand, 2007; pp. 123–133. [Google Scholar]
- Van den Berg, B.; Lister, S.; Rutherford, J.C. Ammonium transceptors: Novel regulators of fungal development. PLoS Pathog. 2019, 15, e1008059. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, K.; Park, P.; Nakayashiki, H. Cell biology in phytopathogenic fungi during host infection: Commonalities and differences. J. Gen. Plant Pathol. 2019, 85, 163–173. [Google Scholar] [CrossRef]
- Miao, Y.; Tenor, J.L.; Toffaletti, D.L.; Washington, E.J.; Liu, J.; Shadrick, W.R.; Schumacher, M.A.; Lee, R.E.; Perfect, J.R.; Brennan, R.G. Structures of trehalose-6-phosphate phosphatase from pathogenic fungi reveal the mechanisms of substrate recognition and catalysis. Proc. Natl. Acad. Sci. USA 2016, 113, 7148–7153. [Google Scholar] [CrossRef] [Green Version]
- Thammahong, A.; Puttikamonkul, S.; Perfect, J.R.; Brennan, R.G.; Cramer, R.A. Central role of the trehalose biosynthesis pathway in the pathogenesis of human fungal Infections: Opportunities and challenges for therapeutic development. Microbiol. Mol. Biol. Rev. 2017, 81, e00053-16. [Google Scholar] [CrossRef] [Green Version]
- Seifbarghi, S.; Borhan, M.H.; Wei, Y.; Coutu, C.; Robinson, S.J.; Hegedus, D.D. Changes in the Sclerotinia sclerotiorum transcriptome during infection of Brassica napus. BMC Genom. 2017, 18, 266. [Google Scholar] [CrossRef] [Green Version]
- Keyhani, N.O. Lipid biology in fungal stress and virulence: Entomopathogenic fungi. Fungal Biol. 2018, 122, 420–429. [Google Scholar] [CrossRef]
- Saikia, S.; Takemoto, D.; Tapper, B.A.; Lane, G.A.; Fraser, K.; Scott, B. Functional analysis of an indole-diterpene gene cluster for lolitrem B biosynthesis in the grass endosymbiont Epichloë festucae. FEBS Lett. 2012, 586, 2563–2569. [Google Scholar] [CrossRef] [Green Version]
- Reimegård, J.; Kundu, S.; Pendle, A.; Irish, V.F.; Shaw, P.; Nakayama, N.; Sundström, J.F.; Emanuelsson, O. Genome-wide identification of physically clustered genes suggests chromatin-level co-regulation in male reproductive development in Arabidopsis thaliana. Nucleic Acids Res. 2017, 45, 3253–3265. [Google Scholar] [CrossRef] [Green Version]
- Ball, O.J.P.; Prestidge, R.A.; Sprosen, J.M. Interrelationships between Acremonium lolii, peramine, and lolitrem B in perennial ryegrass. Appl. Environ. Microbiol. 1995, 61, 1527–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, R.; Voisey, C.; Johnson, L.; Pratt, J.; Fleetwood, D.; Khan, A.; Bryan, G. Distribution of NRPS gene families within the Neotyphodium/Epichloë complex. Fungal Genet. Biol. 2007, 44, 1180–1190. [Google Scholar] [CrossRef]
- Ishidoh, K.-I.; Kinoshita, H.; Nihira, T. Identification of a gene cluster responsible for the biosynthesis of cyclic lipopeptide verlamelin. Appl. Microbiol. Biotechnol. 2014, 98, 7501–7510. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-C.; Choi, G.; Kim, H.-J.; Kim, H.; Woong, J.; Cho, K. Verlamelin, an antifungal compound produced by a mycoparasite, Acremonium strictum. Plant Pathol. J. 2002, 18. [Google Scholar] [CrossRef]
- Peyyala, R.; Farman, M.L. Magnaporthe oryzae isolates causing gray leaf spot of perennial ryegrass possess a functional copy of the AVR1-CO39 avirulence gene. Mol. Plant Pathol. 2006, 7, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Turrà, D.; Segorbe, D.; Pietro, A. Protein kinases in plant-pathogenic fungi: Conserved regulators of infection. Annu. Rev. Phytopathol. 2014, 52, 267–288. [Google Scholar] [CrossRef]
- Shin, J.; Kim, J.-E.; Lee, Y.-W.; Son, H. Fungal cytochrome P450s and the P450 complement (CYPome) of Fusarium graminearum. Toxins 2018, 10, 112. [Google Scholar] [CrossRef] [Green Version]
- Lundén, K.; Danielsson, M.; Durling, M.B.; Ihrmark, K.; Nemesio Gorriz, M.; Stenlid, J.; Asiegbu, F.O.; Elfstrand, M. Transcriptional responses associated with virulence and defence in the interaction between Heterobasidion annosum s.s. and Norway Spruce. PLoS ONE 2015, 10, e0131182. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Endophyte Strain | SE | NEA11 | NEA12 |
|---|---|---|---|
| Taxon a | LpTG-1 | LpTG-2 | LpTG-3 |
| Ploidy level b | haploid | heteroploid | haploid |
| Alkaloid biosynthesis profile c | peramine lolitrem B ergovaline | peramine ergovaline | epoxy-janthitrems |
| Growth rate d | Moderate | Fast | Slow |
| Sequence Name | Sequence Description | Function in Fungi/ Epichloë | |
|---|---|---|---|
| SE and NEA11 | NEA12 | ||
| Ef_C09_g12381 | NEA12_g08100 | HSP 30 | Conservation of energy in cells by inhibiting ATPase during stress conditions [63]. |
| Ef_C09_g08349 | NEA12_g01945 | HSP 101 | Involves in heat tolerance [70]. |
| Ef_C09_g12810 | NEA12_g01905 | catalase A | Decomposes hydrogen peroxide to provide defence against oxidative stress and thereby provides a fitness advantage to pathogenic fungi in the presence of stress [41,71,72]. |
| Ef_C09_g08943 | NEA12_g03844 | GPI anchored serine-rich protein | Play crucial roles in various plant–fungus interaction processes, including attachment of hyphae to surfaces, cell wall integrity and modification, virulence and degradation of host tissues [46,67,73]. |
| Ef_C09_g11992 | NEA12_g01900 | grass induced protein | Suggested to play an important role in symbiosis, since it is present in high abundance in a wide range of Epichloë-grass associations examined thus far [34,44,74]. |
| Ef_C09_g10329 | NEA12_g04886 | translation elongation factor 1-alpha | The main role is in translation. TefA is also involved in signal transduction, virus infection, nuclear export of proteins, mitochondrial tRNA import, virulence, adhesion, invasion and regulation of the immune system [69,75]. |
| Ef_C09_g00763 | NEA12_g07612 | glycoside hydrolase family 10 protein | Plant cell wall degrading CAZYmes, involved in polysaccharide degradation, are particularly important for fungal pathogens due to their involvement during penetration and successful infection of their hosts [76]. Furthermore, carbohydrates released from plant cell walls are a source of nutrition for the growth of fungi [76]. |
| Ef_C09_g02703 | NEA12_g01373 | glycoside hydrolase, superfamily | |
| Ef_C09_g10132 | NEA12_g03401 | glycosyltransferase family 90 protein | |
| Ef_C09_g07063 | NEA12_g05681 | endochitinase B1 | |
| Ef_C09_g07193/Et_E8_g4372 | NEA12_g00608 | phosphoketolase | The phosphoketolase pathway plays an important role in central carbon metabolism of fungi and has been identified as required for full virulence of some pathogenic fungi [77,78]. |
| Ef_C09_g11163 | NEA12_g02369 | hypothetical protein MAM_00430 | Not characterised in detail. |
| Ef_C09_g11597 | NEA12_g01730 | cell surface protein, putative | Not characterised in detail. |
| Sequence Name | Sequence Description | Cluster Number * |
|---|---|---|
| NEA12_g03979 | cystathionine gamma-synthase | 5 |
| NEA12_g03980 | clavaminate synthase-like protein | 5 |
| NEA12_g03981 | HpcH/HpaI aldolase/citrate lyase family protein | 11 |
| NEA12_g03982 | putative efflux pump antibiotic resistance protein | 9 |
| NEA12_g03983 | aspartate aminotransferase | 5 |
| NEA12_g03984 | flavin-nucleotide-binding protein | |
| NEA12_g03985 | 7alpha-cephem-methoxylase P8 chain related protein | 5 |
| NEA12_g03986 | putative D-aminoacylase | 5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hettiarachchige, I.K.; Vander Jagt, C.J.; Mann, R.C.; Sawbridge, T.I.; Spangenberg, G.C.; Guthridge, K.M. Global Changes in Asexual Epichloë Transcriptomes during the Early Stages, from Seed to Seedling, of Symbiotum Establishment. Microorganisms 2021, 9, 991. https://doi.org/10.3390/microorganisms9050991
Hettiarachchige IK, Vander Jagt CJ, Mann RC, Sawbridge TI, Spangenberg GC, Guthridge KM. Global Changes in Asexual Epichloë Transcriptomes during the Early Stages, from Seed to Seedling, of Symbiotum Establishment. Microorganisms. 2021; 9(5):991. https://doi.org/10.3390/microorganisms9050991
Chicago/Turabian StyleHettiarachchige, Inoka K., Christy J. Vander Jagt, Ross C. Mann, Timothy I. Sawbridge, German C. Spangenberg, and Kathryn M. Guthridge. 2021. "Global Changes in Asexual Epichloë Transcriptomes during the Early Stages, from Seed to Seedling, of Symbiotum Establishment" Microorganisms 9, no. 5: 991. https://doi.org/10.3390/microorganisms9050991
APA StyleHettiarachchige, I. K., Vander Jagt, C. J., Mann, R. C., Sawbridge, T. I., Spangenberg, G. C., & Guthridge, K. M. (2021). Global Changes in Asexual Epichloë Transcriptomes during the Early Stages, from Seed to Seedling, of Symbiotum Establishment. Microorganisms, 9(5), 991. https://doi.org/10.3390/microorganisms9050991