
Systematic Review: The Gut Microbiome and Its Potential Clinical Application in Inflammatory Bowel Disease



Abstract
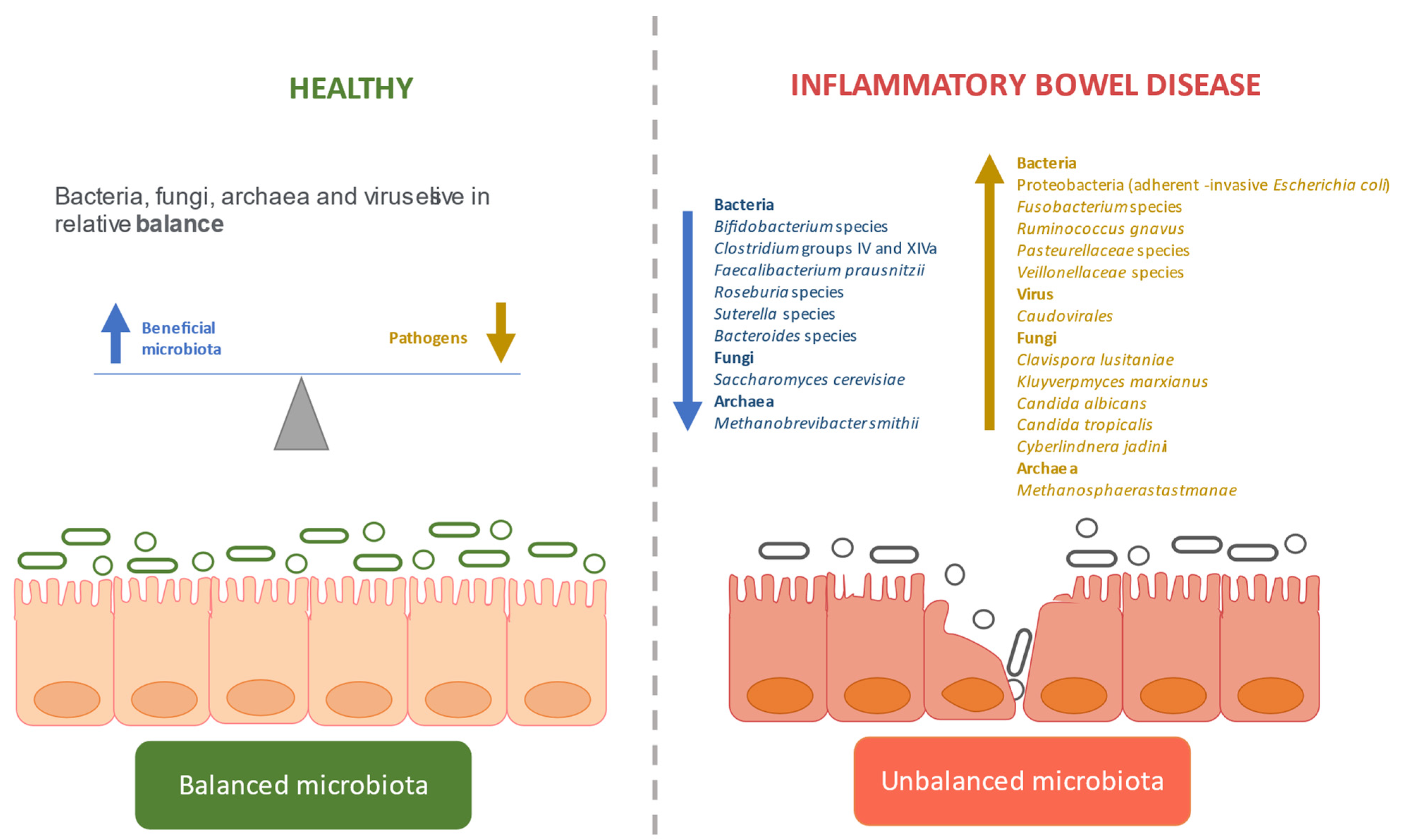
1. Introduction
2. Methodology
2.1. Search Strategy
2.2. Eligibility Criteria
3. Results
3.1. Gut Microbiome Studies in IBD: Methodologic Aspects
3.1.1. Study Design
3.1.2. Microbiome Analysis Methods
3.1.3. Sample Type and Site
3.1.4. Structural and Functional Analysis
3.2. Dysbiosis in IBD
3.2.1. Defining the Gut Microbiome in IBD
Bacterial Dysbiosis
Fungal Dysbiosis
Viral Dysbiosis
Archaeal Dysbiosis
Disease Activity and Severity
3.3. Gut Microbiome-Based Biomarkers in IBD
4. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
| Section/Topic | # | Checklist Item | Reported on Page # |
|---|---|---|---|
| TITLE | |||
| Title | 1 | Identify the report as a systematic review, meta-analysis, or both. | 1 |
| ABSTRACT | |||
| Structured summary | 2 | Provide a structured summary including, as applicable: background; objectives; data sources; study eligibility criteria, participants, and interventions; study appraisal and synthesis methods; results; limitations; conclusions and implications of key findings; systematic review registration number. | 1 |
| INTRODUCTION | |||
| Rationale | 3 | Describe the rationale for the review in the context of what is already known. | 1–2 |
| Objectives | 4 | Provide an explicit statement of questions being addressed with reference to participants, interventions, comparisons, outcomes, and study design (PICOS). | 2 |
| METHODS | |||
| Protocol and registration | 5 | Indicate if a review protocol exists, if and where it can be accessed (e.g., Web address), and, if available, provide registration information including registration number. | 2 |
| Eligibility criteria | 6 | Specify study characteristics (e.g., PICOS, length of follow-up) and report characteristics (e.g., years considered, language, publication status) used as criteria for eligibility, giving rationale. | 2 |
| Information sources | 7 | Describe all information sources (e.g., databases with dates of coverage, contact with study authors to identify additional studies) in the search and date last searched. | 2 |
| Search | 8 | Present full electronic search strategy for at least one database, including any limits used, such that it could be repeated. | 2 |
| Study selection | 9 | State the process for selecting studies (i.e., screening, eligibility, included in systematic review, and, if applicable, included in the meta-analysis). | 2 |
| Data collection process | 10 | Describe method of data extraction from reports (e.g., piloted forms, independently, in duplicate) and any processes for obtaining and confirming data from investigators. | 2 |
| Section/topic | # | Checklist item | Reported on page # |
| Data items | 11 | List and define all variables for which data were sought (e.g., PICOS, funding sources) and any assumptions and simplifications made. | N/A |
| Risk of bias in individual studies | 12 | Describe methods used for assessing risk of bias of individual studies (including specification of whether this was done at the study or outcome level), and how this information is to be used in any data synthesis. | N/A |
| Summary measures | 13 | State the principal summary measures (e.g., risk ratio, difference in means). | N/A |
| Synthesis of results | 14 | Describe the methods of handling data and combining results of studies, if done, including measures of consistency (e.g., I2) for each meta-analysis. | N/A |
| Risk of bias across studies | 15 | Specify any assessment of risk of bias that may affect the cumulative evidence (e.g., publication bias, selective reporting within studies). | N/A |
| Additional analyses | 16 | Describe methods of additional analyses (e.g., sensitivity or subgroup analyses, meta-regression), if done, indicating which were pre-specified. | N/A |
| RESULTS | |||
| Study selection | 17 | Give numbers of studies screened, assessed for eligibility, and included in the review, with reasons for exclusions at each stage, ideally with a flow diagram. | 2 |
| Study characteristics | 18 | For each study, present characteristics for which data were extracted (e.g., study size, PICOS, follow-up period) and provide the citations. | 3–8 |
| Risk of bias within studies | 19 | Present data on risk of bias of each study and, if available, any outcome level assessment (see item 12). | N/A |
| Results of individual studies | 20 | For all outcomes considered (benefits or harms), present, for each study: (a) simple summary data for each intervention group (b) effect estimates and confidence intervals, ideally with a forest plot. | N/A |
| Synthesis of results | 21 | Present results of each meta-analysis done, including confidence intervals and measures of consistency. | N/A |
| Risk of bias across studies | 22 | Present results of any assessment of risk of bias across studies (see Item 15). | N/A |
| Additional analysis | 23 | Give results of additional analyses, if done (e.g., sensitivity or subgroup analyses, meta-regression [see Item 16]). | N/A |
| Section/topic | # | Checklist item | Reported on page # |
| DISCUSSION | |||
| Summary of evidence | 24 | Summarize the main findings including the strength of evidence for each main outcome; consider their relevance to key groups (e.g., healthcare providers, users, and policy makers). | N/A |
| Limitations | 25 | Discuss limitations at study and outcome level (e.g., risk of bias), and at review-level (e.g., incomplete retrieval of identified research, reporting bias). | N/A |
| Conclusions | 26 | Provide a general interpretation of the results in the context of other evidence, and implications for future research. | 8–9 |
| FUNDING | |||
| Funding | 27 | Describe sources of funding for the systematic review and other support (e.g., supply of data); role of funders for the systematic review. | 9 |
References
- Hoyles, L.; Swann, J. Influence of the human gut microbiome on the metabolic phenotype. In The Handbook of Metabolic Phenotyping; Elsevier Inc.: Amsterdam, The Netherlands, 2018; pp. 535–560. ISBN 9780128122938. [Google Scholar]
- Rajilic-Stojanovic, M.; Figueiredo, C.; Smet, A.; Hansen, R.; Kupcinskas, J.; Rokkas, T.; Andersen, L.; Machado, J.C.; Ianiro, G.; Gasbarrini, A.; et al. Systematic review: Gastric microbiota in health and disease. Aliment. Pharmacol. Ther. 2020, 51, 582–602. [Google Scholar] [CrossRef] [PubMed]
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef]
- Pittayanon, R.; Lau, J.T.; Leontiadis, G.I.; Tse, F.; Yuan, Y.; Surette, M.; Moayyedi, P. Differences in gut microbiota in patients with vs without inflammatory bowel diseases: A systematic review. Gastroenterology 2020, 158, 930–946e1. [Google Scholar] [CrossRef]
- Medina Benítez, E.; Fuentes Lugo, D.; Suárez Cortina, L.; Prieto Bozano, G. Enfermedad inflamatoria intestinal. In Protocolos Diagnóstico-Terapéuticos de Gastroenterología, Hepatología y Nutrición Pediátrica SEGHNP-AEP; Ediciones Ergon: Madrid, Spain, 2010; pp. 151–160. [Google Scholar]
- Khan, I.; Ullah, N.; Zha, L.; Bai, Y.; Khan, A.; Zhao, T.; Che, T.; Zhang, C. Alteration of gut microbiota in inflammatory bowel disease (IBD): Cause or consequence? IBD treatment targeting the gut microbiome. Pathogens 2019, 8, 126. [Google Scholar] [CrossRef]
- Ni, J.; Wu, G.D.; Albenberg, L.; Tomov, V.T. Gut microbiota and IBD: Causation or correlation? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; He, J.; Shen, Y.; Zhang, C.; Wang, J.; Chen, Y. New frontiers in genetics, gut microbiota, and immunity: A rosetta stone for the pathogenesis of inflammatory bowel disease. BioMed Res. Int. 2017, 2017, 8201672. [Google Scholar] [CrossRef]
- Zuo, T.; Ng, S.C. The gut microbiota in the pathogenesis and therapeutics of inflammatory bowel disease. Front. Microbiol. 2018, 9, 2247. [Google Scholar] [CrossRef]
- Glassner, K.L.; Abraham, B.P.; Quigley, E.M.M. The microbiome and inflammatory bowel disease. J. Allergy Clin. Immunol. 2020, 145, 16–27. [Google Scholar] [CrossRef]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; Altman, D.; Antes, G.; Atkins, D.; Barbour, V.; Barrowman, N.; Berlin, J.A.; et al. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. PLoS Med. 2009, 6, e1000097. [Google Scholar] [CrossRef]
- Macfarlane, S.; Furrie, E.; Cummings, J.H.; Macfarlane, G.T. Chemotaxonomic Analysis of Bacterial Populations Colonizing the Rectal Mucosa in Patients with Ulcerative Colitis. Clin. Infect. Dis. 2004, 38, 1690–1699. [Google Scholar]
- Lepage, P.; Seksik, P.; Sutren, M.; de la Cochetière, M.F.; Jian, R.; Marteau, P.; Doré, J. Biodiversity of the mucosa-associated microbiota is stable along the distal digestive tract in healthy individuals and patients with IBD. Inflamm. Bowel Dis. 2005, 11, 473–480. [Google Scholar] [CrossRef]
- Manichanh, C.; Bonnaud, E.; Gloux, K.; Pelletier, E.; Frangeul, L.; Nalin, R.; Jarrin, C.; Chardon, P.; Marteau, P.; Roca, J.; et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut 2006, 55, 205–211. [Google Scholar] [CrossRef]
- Bibiloni, R.; Mangold, M.; Madsen, K.L.; Fedorak, R.N.; Tannock, G.W. The bacteriology of biopsies differs between newly diagnosed, untreated, Crohn’s disease and ulcerative colitis patients. J. Med. Microbiol. 2006, 55, 1141–1149. [Google Scholar] [CrossRef]
- Sokol, H.; Lepage, P.; Seksik, P.; Dore, J.; Marteau, P. Temperature Gradient Gel Electrophoresis of Fecal 16S rRNA Reveals Active Escherichia coli in the Microbiota of Patients with Ulcerative Colitis. J. Clin. Microbiol. 2006, 44, 3172–3177. [Google Scholar] [CrossRef]
- Gophna, U.; Sommerfeld, K.; Gophna, S.; Doolittle, W.F.; Zanten, S.J.O.V. Van Differences between Tissue-Associated Intestinal Microfloras of Patients with Crohn ’ s Disease and Ulcerative Colitis. J. Clin. Microbiol. 2006, 44, 4136–4141. [Google Scholar] [CrossRef]
- Scanlan, P.D.; Shanahan, F.; Mahony, C.O.; Marchesi, J.R. Culture-Independent Analyses of Temporal Variation of the Dominant Fecal Microbiota and Targeted Bacterial Subgroups in Crohn ’ s Disease. J. Clin. Microbiol. 2006, 44, 3980–3988. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, B.; Zhang, Y.; Wei, H.; Lei, Y.; Zhao, L. Structural shifts of mucosa-associated lactobacilli and Clostridium leptum subgroup in patients with ulcerative colitis. J. Clin. Microbiol. 2007, 45, 496–500. [Google Scholar] [CrossRef]
- Sepehri, S.; Kotlowski, R.; Bernstein, C.N.; Krause, D.O. Microbial diversity of inflamed and noninflamed gut biopsy tissues in inflammatory bowel disease. Inflamm. Bowel Dis. 2007, 13, 675–683. [Google Scholar] [CrossRef]
- Andoh, A.; Sakata, S.; Koizumi, Y.; Mitsuyama, K.; Fujiyama, Y.; Benno, Y. Terminal restriction fragment length polymorphism analysis of the diversity of fecal microbiota in patients with ulcerative colitis. Inflamm. Bowel Dis. 2007, 13, 955–962. [Google Scholar] [CrossRef]
- Frank, D.N.; Amand, A.L.S.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef]
- Ott, S.J.; Plamondon, S.; Hart, A.; Begun, A.; Rehman, A.; Kamm, M.A.; Schreiber, S. Dynamics of the mucosa-associated flora in ulcerative colitis patients during remission and clinical relapse. J. Clin. Microbiol. 2008, 46, 3510–3513. [Google Scholar] [CrossRef]
- Ott, S.J.; Kühbacher, T.; Musfeldt, M.; Rosenstiel, P.; Hellmig, S.; Rehman, A.; Drews, O.; Weichert, W.; Timmis, K.N.; Schreiber, S. Fungi and inflammatory bowel diseases: Alterations of composition and diversity. Scand. J. Gastroenterol. 2008, 43, 831–841. [Google Scholar] [CrossRef]
- Martinez, C.; Antolin, M.; Santos, J.; Torrejon, A.; Casellas, F.; Borruel, N.; Guarner, F.; Malagelada, J.R. Unstable composition of the fecal microbiota in ulcerative colitis during clinical remission. Am. J. Gastroenterol. 2008, 103, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Dicksved, J.; Halfvarson, J.; Rosenquist, M.; Järnerot, G.; Tysk, C.; Apajalahti, J.; Engstrand, L.; Jansson, J.K. Molecular analysis of the gut microbiota of identical twins with Crohn’s disease. ISME J. 2008, 2, 716–727. [Google Scholar] [CrossRef]
- Kuehbacher, T.; Rehman, A.; Lepage, P.; Hellmig, S.; Fölsch, U.R.; Schreiber, S.; Ott, S.J. Intestinal TM7 bacterial phylogenies in active inflammatory bowel disease. J. Med. Microbiol. 2008, 57, 1569–1576. [Google Scholar] [CrossRef]
- Andoh, A.; Tsujikawa, T.; Sasaki, M.; Mitsuyama, K.; Suzuki, Y.; Matsui, T.; Matsumoto, T.; Benno, Y.; Fujiyama, Y. Faecal microbiota profile of Crohn’s disease determined by terminal restriction fragment length polymorphism analysis. Aliment. Pharmacol. Ther. 2008, 29, 75–82. [Google Scholar] [CrossRef]
- Nishikawa, J.; Kudo, T.; Sakata, S.; Benno, Y.; Sugiyama, T. Diversity of mucosa-associated microbiota in active and inactive ulcerative colitis. Scand. J. Gastroenterol. 2009, 44, 180–186. [Google Scholar] [CrossRef]
- Willing, B.; Halfvarson, J.; Dicksved, J.; Rosenquist, M.; Järnerot, G.; Engstrand, L.; Tysk, C.; Jansson, J.K. Twin studies reveal specific imbalances in the mucosa-associated microbiota of patients with ileal Crohn’s disease. Inflamm. Bowel Dis. 2009, 15, 653–660. [Google Scholar] [CrossRef]
- Andoh, A.; Ida, S.; Tsujikawa, T.; Benno, Y.; Fujiyama, Y. Terminal restriction fragment polymorphism analyses of fecal microbiota in five siblings including two with ulcerative colitis. Clin. J. Gastroenterol. 2009, 2, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Gillevet, P.; Sikaroodi, M.; Keshavarzian, A.; Mutlu, E.A. Quantitative Assessment of the Human Gut Microbiome using Multitag Pyrosequencing. Chem. Biodivers. 2010, 7, 1065–1075. [Google Scholar] [CrossRef]
- Rehman, A.; Lepage, P.; Nolte, A.; Hellmig, S.; Schreiber, S.; Ott, S.J. Transcriptional activity of the dominant gut mucosal microbiota in chronic inflammatory bowel disease patients. J. Med. Microbiol. 2010, 59, 1114–1122. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Denman, S.E.; Morrison, M.; Yu, Z.; Dore, J.; Leclerc, M.; McSweeney, C.S. Dysbiosis of fecal microbiota in Crohn’s disease patients as revealed by a custom phylogenetic microarray. Inflamm. Bowel Dis. 2010, 16, 2034–2042. [Google Scholar] [CrossRef]
- Rowan, F.; Docherty, N.G.; Murphy, M.; Murphy, B.; Coffey, J.C.; O’Connell, P.R. Desulfovibrio Bacterial Species Are Increased in Ulcerative Colitis. Dis. Colon Rectum 2010, 53, 1530–1536. [Google Scholar] [CrossRef]
- Andoh, A.; Imaeda, H.; Aomatsu, T.; Inatomi, O.; Bamba, S.; Sasaki, M.; Saito, Y.; Tsujikawa, T.; Fujiyama, Y. Comparison of the fecal microbiota profiles between ulcerative colitis and Crohn’s disease using terminal restriction fragment length polymorphism analysis. J. Gastroenterol. 2011, 46, 479–486. [Google Scholar] [CrossRef]
- Mondot, S.; Kang, S.; Furet, J.P.; Aguirre De Carcer, D.; McSweeney, C.; Morrison, M.; Marteau, P.; Doré, J.; Leclerc, M. Highlighting new phylogenetic specificities of Crohn’s disease microbiota. Inflamm. Bowel Dis. 2011, 17, 185–192. [Google Scholar] [CrossRef]
- Joossens, M.; Huys, G.; Cnockaert, M.; De Preter, V.; Verbeke, K.; Rutgeerts, P.; Vandamme, P.; Vermeire, S. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut 2011, 60, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Lepage, P.; Hösler, R.; Spehlmann, M.E.; Rehman, A.; Zvirbliene, A.; Begun, A.; Ott, S.; Kupcinskas, L.; Doré, J.; Raedler, A.; et al. Twin study indicates loss of interaction between microbiota and mucosa of patients with ulcerative colitis. Gastroenterology 2011, 141, 227–236. [Google Scholar] [CrossRef]
- Benjamin, J.L.; Hedin, C.R.H.; Koutsoumpas, A.; Ng, S.C.; McCarthy, N.E.; Prescott, N.J.; Pessoa-Lopes, P.; Mathew, C.G.; Sanderson, J.; Hart, A.L.; et al. Smokers with active Crohn’s disease have a clinically relevant dysbiosis of the gastrointestinal microbiota. Inflamm. Bowel Dis. 2012, 18, 1092–1100. [Google Scholar] [CrossRef]
- Hotte, N.S.C.; Salim, S.Y.; Tso, R.H.; Albert, E.J.; Bach, P.; Walker, J.; Dieleman, L.A.; Fedorak, R.N.; Madsen, K.L. Patients with inflammatory bowel disease exhibit dysregulated responses to microbial DNA. PLoS ONE 2012, 7, e37932. [Google Scholar] [CrossRef]
- Pistone, D.; Marone, P.; Pajoro, M.; Fabbi, M.; Vicari, N.; Daffara, S.; Dalla Valle, C.; Gabba, S.; Sassera, D.; Verri, A.; et al. Mycobacterium avium paratuberculosis in Italy: Commensal or emerging human pathogen? Dig. Liver Dis. 2012, 44, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Andoh, A.; Kuzuoka, H.; Tsujikawa, T.; Nakamura, S. Multicenter analysis of fecal microbiota profiles in Japanese patients with Crohn ’ s disease. J Gastroenterol. 2012, 47, 1298–1307. [Google Scholar] [CrossRef]
- Li, Q.; Wang, C.; Tang, C.; Li, N.; Li, J. Molecular-phylogenetic characterization of the microbiota in ulcerated and non-ulcerated regions in the patients with crohn’s disease. PLoS ONE 2012, 7, e34939. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, H.; Kataoka, K.; Ishikawa, H.; Ikata, K.; Arimochi, H.; Iwasaki, T.; Ohnishi, Y.; Kuwahara, T.; Yasutomo, K. Reduced diversity and imbalance of fecal microbiota in patients with ulcerative colitis. Dig. Dis. Sci. 2012, 57, 2955–2964. [Google Scholar] [CrossRef]
- Vigsnæs, L.K.; Brynskov, J.; Steenholdt, C.; Wilcks, A.; Licht, T.R. Gram-negative bacteria account for main differences between faecal microbiota from patients with ulcerative colitis and healthy controls. Benef. Microbes 2012, 3, 287–297. [Google Scholar] [CrossRef]
- De Souza, H.L.; De Carvalho, V.R.; Romeiro, F.G.; Sassaki, L.Y.; Keller, R.; Rodrigues, J. Mucosa-associated but not luminal Escherichia coli is augmented in Crohn’s disease and ulcerative colitis. Gut Pathog. 2012, 4, 1–8. [Google Scholar] [CrossRef]
- Duboc, H.; Rajca, S.; Rainteau, D.; Benarous, D.; Maubert, M.A.; Quervain, E.; Thomas, G.; Barbu, V.; Humbert, L.; Despras, G.; et al. Connecting dysbiosis, bile-acid dysmetabolism and Gut inflammation in inflammatory bowel diseases. Gut 2012, 62, 531–539. [Google Scholar] [CrossRef]
- Sha, S.; Xu, B.; Wang, X.; Zhang, Y.; Wang, H.; Kong, X.; Zhu, H.; Wu, K. The biodiversity and composition of the dominant fecal microbiota in patients with inflammatory bowel disease. Diagn. Microbiol. Infect. Dis. 2013, 75, 245–251. [Google Scholar] [CrossRef]
- Kabeerdoss, J.; Sankaran, V.; Pugazhendhi, S.; Ramakrishna, B.S. Clostridium leptum group bacteria abundance and diversity in the fecal microbiota of patients with inflammatory bowel disease: A case – control study in India. BMC Gastroenterol. 2013, 13, 1. [Google Scholar] [CrossRef]
- Varela, E.; Manichanh, C.; Gallart, M.; Torrejon, A.; Borruel, N.; Casellas, F.; Guarner, F.; Antolin, M. Colonisation by Faecalibacterium prausnitzii and maintenance of clinical remission in patients with ulcerative colitis. Aliment. Pharmacol. Ther. 2013, 38, 151–161. [Google Scholar] [CrossRef]
- Midtvedt, T.; Zabarovsky, E.; Norin, E.; Bark, J.; Gizatullin, R.; Kashuba, V.; Ljungqvist, O.; Zabarovska, V.; Möllby, R.; Ernberg, I. Increase of Faecal Tryptic Activity Relates to Changes in the Intestinal Microbiome: Analysis of Crohn’s Disease with a Multidisciplinary Platform. PLoS ONE 2013, 8, e66074. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, T.; Imaeda, H.; Takahashi, K.; Kasumi, E.; Bamba, S.; Fujiyama, Y.; Andoh, A. Decreased abundance of Faecalibacterium prausnitzii in the gut microbiota of Crohn’s disease. J. Gastroenterol. Hepatol. 2013, 28, 613–619. [Google Scholar] [CrossRef]
- Fite, A.; Macfarlane, S.; Furrie, E.; Bahrami, B.; Cummings, J.H.; Steinke, D.T.; MacFarlane, G.T. Longitudinal analyses of gut mucosal microbiotas in ulcerative colitis in relation to patient age and disease severity and duration. J. Clin. Microbiol. 2013, 51, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Rajilić-Stojanović, M.; Shanahan, F.; Guarner, F.; De Vos, W.M. Phylogenetic analysis of dysbiosis in ulcerative colitis during remission. Inflamm. Bowel Dis. 2013, 19, 481–488. [Google Scholar] [CrossRef]
- Kumari, R.; Ahuja, V.; Paul, J. Fluctuations in butyrate-producing bacteria in ulcerative colitis patients of North India. World J. Gastroenterol. 2013, 19, 3404–3414. [Google Scholar] [CrossRef]
- Hedin, C.R.; McCarthy, N.E.; Louis, P.; Farquharson, F.M.; McCartney, S.; Taylor, K.; Prescott, N.J.; Murrells, T.; Stagg, A.J.; Whelan, K.; et al. Altered intestinal microbiota and blood T cell phenotype are shared by patients with Crohn’s disease and their unaffected siblings. Gut 2014, 63, 1578–1586. [Google Scholar] [CrossRef] [PubMed]
- Lennon, G.; Balfe, Á.; Bambury, N.; Lavelle, A.; Maguire, A.; Docherty, N.G.; Coffey, J.C.; Winter, D.C.; Sheahan, K.; O’Connell, P.R. Correlations between colonic crypt mucin chemotype, inflammatory grade and Desulfovibrio species in ulcerative colitis. Color. Dis. 2014, 16, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Machiels, K.; Joossens, M.; Sabino, J.; De Preter, V.; Arijs, I.; Eeckhaut, V.; Ballet, V.; Claes, K.; Van Immerseel, F.; Verbeke, K.; et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut 2014, 63, 1275–1283. [Google Scholar] [CrossRef]
- Wang, W.; Chen, L.; Zhou, R.; Wang, X.; Song, L.; Huang, S.; Wang, G.; Xia, B. Increased Proportions of Bifidobacterium and the Lactobacillus Group and Loss of Butyrate-Producing Bacteria in Inflammatory Bowel. J. Clin. Microbiol. 2014, 52, 398–406. [Google Scholar] [CrossRef]
- Blais Lecours, P.; Marsolais, D.; Cormier, Y.; Berberi, M.; Hache, C.; Bourdages, R.; Duchaine, C. Increased Prevalence of Methanosphaera stadtmanae in Inflammatory Bowel Diseases. PLoS ONE 2014, 9, e87734. [Google Scholar] [CrossRef]
- Fukuda, K.; Fujita, Y. Determination of the discriminant score of intestinal microbiota as a biomarker of disease activity in patients with ulcerative colitis. BMC Gastroenterol. 2014, 14, 49. [Google Scholar] [CrossRef]
- Li, Q.; Wang, C.; Tang, C.; He, Q.; Li, N.; Li, J. Dysbiosis of gut fungal microbiota is associated with mucosal inflammation in crohn’s disease. J. Clin. Gastroenterol. 2014, 48, 513–523. [Google Scholar] [CrossRef]
- Andoh, A.; Kobayashi, T.; Kuzuoka, H.; Tsujikawa, T.; Suzuki, Y.; Hirai, F.; Matsui, T.; Nakamura, S.; Matsumoto, T.; Fujiyama, Y. Characterization of gut microbiota profiles by disease activity in patients with Crohn’s disease using data mining analysis of terminal restriction fragment length polymorphisms. Biomed. Rep. 2014, 2, 370–373. [Google Scholar] [CrossRef][Green Version]
- Wisittipanit, N.; Rangwala, H.; Sikaroodi, M.; Keshavarzian, A.; Mutlu, E.A.; Gillevet, P. Classification methods for the analysis of LH-PCR data associated with inflammatory bowel disease patients. Int. J. Bioinform. Res. Appl. 2015, 11, 111–129. [Google Scholar] [CrossRef]
- Kabeerdoss, J.; Jayakanthan, P.; Pugazhendhi, S.; Ramakrishna, B.S. Alterations of mucosal microbiota in the colon of patients with inflammatory bowel disease revealed by real time polymerase chain reaction amplification of 16S ribosomal ribonucleic acid. Indian J. Med. Res. 2015, 23–33. [Google Scholar]
- Takeshita, K.; Mizuno, S.; Mikami, Y.; Sujino, T.; Saigusa, K.; Matsuoka, K.; Naganuma, M.; Sato, T.; Takada, T.; Tsuji, H.; et al. A single species of clostridium Subcluster XIVa decreased in ulcerative colitis patients. Inflamm. Bowel Dis. 2016, 22, 2802–2810. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, S.L.; Li, L.B. Correlation between intestinal flora and serum inflammatory factors in patients with Crohn’s disease. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 4913–4917. [Google Scholar]
- Vrakas, S.; Mountzouris, K.C.; Michalopoulos, G.; Karamanolis, G.; Papatheodoridis, G.; Tzathas, C.; Gazouli, M. Intestinal bacteria composition and translocation of bacteria in inflammatory bowel disease. PLoS ONE 2017, 12, e0170034. [Google Scholar] [CrossRef]
- Zamani, S.; Hesam Shariati, S.; Zali, M.R.; Asadzadeh Aghdaei, H.; Sarabi Asiabar, A.; Bokaie, S.; Nomanpour, B.; Sechi, L.A.; Feizabadi, M.M. Detection of enterotoxigenic Bacteroides fragilis in patients with ulcerative colitis. Gut Pathog. 2017, 9, 53. [Google Scholar] [CrossRef]
- Ghavami, S.B.; Rostami, E.; Sephay, A.A.; Shahrokh, S.; Balaii, H.; Aghdaei, H.A.; Zali, M.R. Alterations of the human gut Methanobrevibacter smithii as a biomarker for inflammatory bowel diseases. Microb. Pathog. 2018, 117, 285–289. [Google Scholar] [CrossRef]
- Le Baut, G.; O’brien, C.; Pavli, P.; Roy, M.; Seksik, P.; Tréton, X.; Nancey, S.; Barnich, N.; Bezault, M.; Auzolle, C.; et al. Prevalence of Yersinia Species in the Ileum of Crohn’s Disease Patients and Controls. Front. Cell. Infect. Microbiol. 2018, 8, 336. [Google Scholar] [CrossRef]
- Al-Bayati, L.; Fasaei, B.N.; Merat, S.; Bahonar, A. Longitudinal analyses of Gut-associated bacterial microbiota in ulcerative colitis patients. Arch. Iran. Med. 2018, 21, 578–584. [Google Scholar] [PubMed]
- Heidarian, F.; Alebouyeh, M.; Shahrokh, S.; Balaii, H.; Zali, M.R. Altered fecal bacterial composition correlates with disease activity in inflammatory bowel disease and the extent of IL8 induction. Curr. Res. Transl. Med. 2019, 67, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Vatn, S.; Carstens, A.; Kristoffersen, A.B.; Bergemalm, D.; Casén, C.; Moen, A.E.F.; Tannaes, T.M.; Lindstrøm, J.; Detlie, T.E.; Olbjørn, C.; et al. Faecal microbiota signatures of IBD and their relation to diagnosis, disease phenotype, inflammation, treatment escalation and anti-TNF response in a European Multicentre Study (IBD-Character). Scand. J. Gastroenterol. 2020, 55, 1146–1156. [Google Scholar] [CrossRef]
- Willing, B.P.; Dicksved, J.; Halfvarson, J.; Andersson, A.F.; Lucio, M.; Zheng, Z.; Järnerot, G.; Tysk, C.; Jansson, J.K.; Engstrand, L. A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology 2010, 139, 1844–1854. [Google Scholar] [CrossRef]
- Rausch, P.; Rehman, A.; Künzel, S.; Häsler, R.; Ott, S.J.; Schreiber, S.; Rosenstiel, P.; Franke, A.; Baines, J.F. Colonic mucosa-associated microbiota is influenced by an interaction of crohn disease and FUT2 (Secretor) genotype. Proc. Natl. Acad. Sci. USA 2011, 108, 19030–19035. [Google Scholar] [CrossRef]
- Walker, A.W.; Sanderson, J.D.; Churcher, C.; Parkes, G.C.; Hudspith, B.N.; Rayment, N.; Brostoff, J.; Parkhill, J.; Dougan, G.; Petrovska, L. High-throughput clone library analysis of the mucosa-associated microbiota reveals dysbiosis and differences between inflamed and non-inflamed regions of the intestine in inflammatory bowel disease. BMC Microbiol. 2011, 11, 7. [Google Scholar] [CrossRef]
- Erickson, A.R.; Cantarel, B.L.; Lamendella, R.; Darzi, Y.; Mongodin, E.F.; Pan, C.; Shah, M.; Halfvarson, J.; Tysk, C.; Henrissat, B.; et al. Integrated Metagenomics/Metaproteomics Reveals Human Host-Microbiota Signatures of Crohn’s Disease. PLoS ONE 2012, 7, e49138. [Google Scholar] [CrossRef] [PubMed]
- Morgan, X.C.; Tickle, T.L.; Sokol, H.; Gevers, D.; Devaney, K.L.; Ward, D.V.; Reyes, J.A.; Shah, S.A.; Leleiko, N.; Snapper, S.B.; et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012, 13, R79. [Google Scholar] [CrossRef] [PubMed]
- Ricanek, P.; Lothe, S.M.; Frye, S.A.; Rydning, A.; Vatn, M.H. Gut bacterial profile in patients newly diagnosed with treatment-naïve Crohn ’ s disease. Clin. Exp. Gastroenterol. 2012, 5, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Hamm, C.M.; Gulati, A.S.; Sartor, R.B.; Chen, H.; Wu, X.; Zhang, T.; Rohlf, F.J.; Zhu, W.; Gu, C.; et al. Inflammatory bowel diseases phenotype, C. difficile and NOD2 genotype are associated with shifts in human ileum associated microbial composition. PLoS ONE 2012, 7, e26284. [Google Scholar] [CrossRef] [PubMed]
- Tong, M.; Li, X.; Parfrey, L.W.; Roth, B.; Ippoliti, A.; Wei, B.; Borneman, J.; McGovern, D.P.B.; Frank, D.N.; Li, E.; et al. A modular organization of the human intestinal mucosal microbiota and its association with inflammatory bowel disease. PLoS ONE 2013, 8, e80702. [Google Scholar]
- Thorkildsen, L.T.; Nwosu, F.C.; Avershina, E.; Ricanek, P.; Perminow, G.; Brackmann, S.; Vatn, M.H.; Rudi, K. Dominant Fecal Microbiota in Newly Diagnosed Untreated Inflammatory Bowel Disease Patients. Gastroenterol. Res. Pract. 2013, 636785. [Google Scholar] [CrossRef]
- Prideaux, L.; Kang, S.; Wagner, J.; Buckley, M.; Mahar, J.E.; De Cruz, P.; Wen, Z.; Chen, L.; Xia, B.; Van Langenberg, D.R.; et al. Impact of ethnicity, geography, and disease on the microbiota in health and inflammatory bowel disease. Inflamm. Bowel Dis. 2013, 19, 2906–2918. [Google Scholar] [CrossRef]
- Chiodini, R.J.; Dowd, S.E.; Davis, B.; Galandiuk, S.; Chamberlin, W.M.; Kuenstner, J.T.; McCallum, R.W.; Zhang, J. Crohn’s disease may be differentiated into 2 distinct biotypes based on the detection of bacterial genomic sequences and virulence genes within submucosal tissues. J. Clin. Gastroenterol. 2013, 47, 612–620. [Google Scholar] [CrossRef]
- Pérez-Brocal, V.; García-López, R.; Vázquez-Castellanos, J.F.; Nos, P.; Beltrán, B.; Latorre, A.; Moya, A. Study of the viral and microbial communities associated with Crohn’s disease: A metagenomic approach. Clin. Transl. Gastroenterol. 2013, 4, e36. [Google Scholar] [CrossRef]
- Davenport, M.; Poles, J.; Leung, J.M.; Wolff, M.J.; Abidi, W.M.; Ullman, T.; Mayer, L.; Cho, I.; Loke, P. Metabolic alterations to the mucosal microbiota in inflammatory bowel disease. Inflamm. Bowel Dis. 2014, 20, 723–731. [Google Scholar] [CrossRef]
- Chen, L.; Wang, W.; Zhou, R.; Ng, S.C.; Li, J.; Huang, M.; Zhou, F.; Wang, X.; Shen, B.; Kamm, M.A.; et al. Characteristics of fecal and mucosa-associated microbiota in chinese patients with inflammatory bowel disease. Medicine 2014, 93, e51. [Google Scholar] [CrossRef]
- Wang, W.; Jovel, J.; Halloran, B.; Wine, E.; Patterson, J.; Ford, G.; O’Keefe, S.; Meng, B.; Song, D.; Zhang, Y.; et al. Metagenomic analysis of microbiome in colon tissue from subjects with inflammatory bowel diseases reveals interplay of viruses and bacteria. Inflamm. Bowel Dis. 2015, 21, 1419–1427. [Google Scholar] [CrossRef]
- Lavelle, A.; Lennon, G.; O’Sullivan, O.; Docherty, N.; Balfe, A.; Maguire, A.; Mulcahy, H.E.; Doherty, G.; O’Donoghue, D.; Hyland, J.; et al. Spatial variation of the colonic microbiota in patients with ulcerative colitis and control volunteers. Gut 2015, 64, 1553–1561. [Google Scholar] [CrossRef] [PubMed]
- Chiodini, R.J.; Dowd, S.E.; Chamberlin, W.M.; Galandiuk, S.; Davis, B.; Glassing, A. Microbial population differentials between mucosal and submucosal intestinal tissues in advanced Crohn’s disease of the ileum. PLoS ONE 2015, 10, e0134382. [Google Scholar]
- Pérez-Brocal, V.; García-López, R.; Nos, P.; Beltrán, B.; Moret, I.; Moya, A. Metagenomic analysis of Crohn’s disease patients identifies changes in the virome and microbiome related to disease status and therapy, and detects potential interactions and biomarkers. Inflamm. Bowel Dis. 2015, 21, 2515–2532. [Google Scholar] [CrossRef]
- Vidal, R.; Ginard, D.; Khorrami, S.; Mora-Ruiz, M.; Munoz, R.; Hermoso, M.; Díaz, S.; Cifuentes, A.; Orfila, A.; Rosselló-Móra, R. Crohn associated microbial communities associated to colonic mucosal biopsies in patients of the western Mediterranean. Syst. Appl. Microbiol. 2015, 38, 442–452. [Google Scholar] [CrossRef]
- Norman, J.M.; Handley, S.A.; Baldridge, M.T.; Droit, L.; Catherine, Y.; Keller, B.C.; Kambal, A.; Zhao, G.; Stappenbeck, T.S.; Mcgovern, D.P.B.; et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 2015, 160, 447–460. [Google Scholar] [CrossRef]
- Eun, C.S.; Kwak, M.J.; Han, D.S.; Lee, A.R.; Park, D.I.; Yang, S.K.; Kim, Y.S.; Kim, J.F. Does the intestinal microbial community of Korean Crohn’s disease patients differ from that of western patients? BMC Gastroenterol. 2016, 16, 28. [Google Scholar] [CrossRef] [PubMed]
- Chiodini, R.J.; Dowd, S.E.; Galandiuk, S.; Davis, B.; Glassing, A. The predominant site of bacterial translocation across the intestinal mucosal barrier occurs at the advancing disease margin in Crohn’s disease. Microbiology 2016, 162, 1608–1619. [Google Scholar] [CrossRef]
- Rehman, A.; Rausch, P.; Wang, J.; Skieceviciene, J.; Kiudelis, G.; Bhagalia, K.; Amarapurkar, D.; Kupcinskas, L.; Schreiber, S.; Rosenstiel, P.; et al. Geographical patterns of the standing and active human gut microbiome in health and IBD. Gut 2016, 65, 238–248. [Google Scholar] [CrossRef]
- Takahashi, K.; Nishida, A.; Fujimoto, T.; Fujii, M.; Shioya, M.; Imaeda, H.; Inatomi, O.; Bamba, S.; Andoh, A.; Sugimoto, M. Reduced Abundance of Butyrate-Producing Bacteria Species in the Fecal Microbial Community in Crohn’s Disease. Digestion 2016, 93, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.D.; Van Domselaar, G.; Bernstein, C.N. Microbiome survey of the inflamed and noninflamed gut at different compartments within the gastrointestinal tract of inflammatory bowel disease patients. Inflamm. Bowel Dis. 2016, 22, 817–825. [Google Scholar] [CrossRef]
- Liguori, G.; Lamas, B.; Richard, M.L.; Brandi, G.; da Costa, G.; Hoffmann, T.W.; Di Simone, M.P.; Calabrese, C.; Poggioli, G.; Langella, P.; et al. Fungal dysbiosis in mucosa-associated microbiota of Crohn’s disease patients. J. Crohn’s Colitis 2016, 10, 296–305. [Google Scholar] [CrossRef]
- Mar, J.S.; Lamere, B.J.; Lin, D.L.; Levan, S.; Nazareth, M.; Mahadevan, U.; Lynch, S.V. Disease severity and immune activity relate to distinct interkingdom gut microbiome states in ethnically distinct ulcerative colitis patients. MBio 2016, 7, e01072-16. [Google Scholar] [CrossRef]
- Hoarau, G.; Mukherjee, P.K.; Gower-Rousseau, C.; Hager, C.; Chandra, J.; Retuerto, M.A.; Neut, C.; Vermeire, S.; Clemente, J.; Colombel, J.F.; et al. Bacteriome and mycobiome interactions underscore microbial dysbiosis in familial Crohn’s disease. MBio 2016, 7, e01250-16. [Google Scholar] [CrossRef]
- Hedin, C.; Van Der Gast, C.J.; Rogers, G.B.; Cuthbertson, L.; McCartney, S.; Stagg, A.J.; Lindsay, J.O.; Whelan, K. Siblings of patients with Crohn’s disease exhibit a biologically relevant dysbiosis in mucosal microbial metacommunities. Gut 2016, 65, 944–953. [Google Scholar] [CrossRef]
- Naftali, T.; Reshef, L.; Kovacs, A.; Porat, R.; Amir, I.; Konikoff, F.M.; Gophna, U. Distinct Microbiotas are Associated with Ileum-Restricted and Colon-Involving Crohn’s Disease. Inflamm. Bowel Dis. 2016, 22, 293–302. [Google Scholar] [CrossRef]
- Pedamallu, C.S.; Bhatt, A.S.; Bullman, S.; Fowler, S.; Freeman, S.S.; Durand, J.; Jung, J.; Duke, F.; Manzo, V.; Cai, D.; et al. Metagenomic Characterization of Microbial Communities In Situ Within the Deeper Layers of the Ileum in Crohn’s Disease. Cmgh 2016, 2, 563–566.e5. [Google Scholar] [CrossRef]
- Sokol, H.; Leducq, V.; Aschard, H.; Pham, H.P.; Jegou, S.; Landman, C.; Cohen, D.; Liguori, G.; Bourrier, A.; Nion-Larmurier, I.; et al. Fungal microbiota dysbiosis in IBD. Gut 2016, 66, 1039–1048. [Google Scholar] [CrossRef]
- Santoru, M.L.; Piras, C.; Murgia, A.; Palmas, V.; Camboni, T.; Liggi, S.; Ibba, I.; Lai, M.A.; Orrù, S.; Blois, S.; et al. Cross sectional evaluation of the gut-microbiome metabolome axis in an Italian cohort of IBD patients. Sci. Rep. 2017, 7, 9523. [Google Scholar] [CrossRef] [PubMed]
- Pascal, V.; Pozuelo, M.; Borruel, N.; Casellas, F.; Campos, D.; Santiago, A.; Martinez, X.; Varela, E.; Sarrabayrouse, G.; Machiels, K.; et al. A microbial signature for Crohn’s disease. Gut 2017, 66, 813–822. [Google Scholar] [CrossRef]
- Chen, G.; Zhang, Y.; Wang, W.; Ji, X.; Meng, F.; Xu, P.; Yang, N.; Ye, F.; Bo, X. Partners of patients with ulcerative colitis exhibit a biologically relevant dysbiosis in fecal microbial metacommunities. World J. Gastroenterol. 2017, 23, 4624–4631. [Google Scholar] [CrossRef]
- Hall, A.B.; Yassour, M.; Sauk, J.; Garner, A.; Jiang, X.; Arthur, T.; Lagoudas, G.K.; Vatanen, T.; Fornelos, N.; Wilson, R.; et al. A novel Ruminococcus gnavus clade enriched in inflammatory bowel disease patients. Genome Med. 2017, 9, 103. [Google Scholar] [CrossRef]
- Qiu, X.; Ma, J.; Jiao, C.; Mao, X.; Zhao, X.; Lu, M.; Wang, K.; Zhang, H. Alterations in the mucosa-associated fungal microbiota in patients with ulcerative colitis. Oncotarget 2017, 8, 107577–107588. [Google Scholar] [CrossRef]
- Kennedy, N.A.; Lamb, C.A.; Berry, S.H.; Walker, A.W.; Mansfield, J.; Parkes, M.; Simpkins, R.; Tremelling, M.; Nutland, S.; Parkhill, J.; et al. The impact of NOD2 variants on fecal microbiota in Crohn’s disease and controls without gastrointestinal disease. Inflamm. Bowel Dis. 2018, 24, 583–592. [Google Scholar] [CrossRef]
- Ji, Y.; Li, X.; Zhu, Y.; Li, N.; Zhang, N.; Niu, M. Faecal microRNA as a biomarker of the activity and prognosis of inflammatory bowel diseases. Biochem. Biophys. Res. Commun. 2018, 503, 2443–2450. [Google Scholar] [CrossRef]
- Imhann, F.; Vila, A.V.; Bonder, M.J.; Fu, J.; Gevers, D.; Visschedijk, M.C.; Spekhorst, L.M.; Alberts, R.; Franke, L.; van Dullemen, H.M.; et al. Interplay of host genetics and gut microbiota underlying the onset and clinical presentation of inflammatory bowel disease. Gut 2018, 67, 108–119. [Google Scholar] [CrossRef]
- Nishino, K.; Nishida, A.; Inoue, R.; Kawada, Y.; Ohno, M.; Sakai, S.; Inatomi, O.; Bamba, S.; Sugimoto, M.; Kawahara, M.; et al. Analysis of endoscopic brush samples identified mucosa-associated dysbiosis in inflammatory bowel disease. J. Gastroenterol. 2018, 53, 95–106. [Google Scholar] [CrossRef]
- Rojas-feria, M.; Romero-garcía, T.; Caballero-rico, J.Á.F.; Ramírez, H.P.; Avilés-recio, M.; Castro-fernandez, M.; Porcuna, N.C.; Romero-gόmez, M.; García, F.; Grande, L.; et al. Modulation of faecal metagenome in Crohn’ s disease: Role of microRNAs as biomarkers. World J. Gastroenterol. 2018, 24, 5223–5233. [Google Scholar] [CrossRef]
- Schirmer, M.; Franzosa, E.A.; Lloyd-Price, J.; Mciver, L.J.; Schwager, R.; Poon, T.W.; Ananthakrishnan, A.N.; Andrews, E.; Barron, G.; Lake, K.; et al. Dynamics of metatranscription in the inflammatory bowel disease gut microbiome. Nat. Microbiol. 2018, 3, 337–346. [Google Scholar] [CrossRef]
- Chiodini, R.J.; Dowd, S.E.; Barron, J.N.; Galandiuk, S.; Davis, B.; Glassing, A. Transitional and temporal changes in the mucosal and submucosal intestinal microbiota in advanced crohn’s disease of the terminal ileum. J. Med. Microbiol. 2018, 67, 549–559. [Google Scholar] [CrossRef]
- Hirano, A.; Umeno, J.; Okamoto, Y.; Shibata, H.; Ogura, Y.; Moriyama, T.; Torisu, T.; Fujioka, S.; Fuyuno, Y.; Kawarabayasi, Y.; et al. Comparison of the microbial community structure between inflamed and non-inflamed sites in patients with ulcerative colitis. J. Gastroenterol. Hepatol. 2018, 33, 1590–1597. [Google Scholar] [CrossRef]
- Ma, H.Q.; Yu, T.T.; Zhao, X.J.; Zhang, Y.; Zhang, H.J. Fecal microbial dysbiosis in Chinese patients with inflammatory bowel disease. World J. Gastroenterol. 2018, 24, 1464–1477. [Google Scholar] [CrossRef]
- Walujkar, S.A.; Kumbhare, S.V.; Marathe, N.P.; Patangia, D.V.; Lawate, P.S.; Bharadwaj, R.S.; Shouche, Y.S. Molecular profiling of mucosal tissue associated microbiota in patients manifesting acute exacerbations and remission stage of ulcerative colitis. World J. Microbiol. Biotechnol. 2018, 34, 76. [Google Scholar] [CrossRef]
- Moen, A.E.F.; Lindstrøm, J.C.; Tannæs, T.M.; Vatn, S.; Ricanek, P.; Vatn, M.H.; Jahnsen, J.; Frengen, A.B.; Dahl, F.A.; You, P.; et al. The prevalence and transcriptional activity of the mucosal microbiota of ulcerative colitis patients. Sci. Rep. 2018, 8, 17278. [Google Scholar] [CrossRef]
- Laserna-Mendieta, E.J.; Clooney, A.G.; Carretero-Gomez, J.F.; Moran, C.; Sheehan, D.; Nolan, J.A.; Hill, C.; Gahan, C.G.M.; Joyce, S.A.; Shanahan, F.; et al. Determinants of reduced genetic capacity for butyrate synthesis by the gut microbiome in Crohn’s disease and ulcerative colitis. J. Crohn’s Colitis 2018, 12, 204–216. [Google Scholar] [CrossRef]
- Libertucci, J.; Dutta, U.; Kaur, S.; Jury, J.; Rossi, L.; Fontes, M.E.; Shajib, M.S.; Khan, W.I.; Surette, M.G.; Verdu, E.F.; et al. Inflammation-related differences in mucosa-associated microbiota and intestinal barrier function in colonic Crohn’s disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G420–G431. [Google Scholar] [CrossRef]
- Moustafa, A.; Li, W.; Anderson, E.L.; Wong, E.H.M.; Dulai, P.S.; Sandborn, W.J.; Biggs, W.; Yooseph, S.; Jones, M.B.; Venter, J.C.; et al. Genetic risk, dysbiosis, and treatment stratification using host genome and gut microbiome in inflammatory bowel disease. Clin. Transl. Gastroenterol. 2018, 9, e132. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.L.; Kiely, C.J.; Pavli, P. The microbiome of Crohn’s disease aphthous ulcers. Gut Pathog. 2018, 10, 44. [Google Scholar] [CrossRef]
- Zakrzewski, M.; Simms, L.A.; Brown, A.; Appleyard, M.; Irwin, J.; Waddell, N.; Radford-Smith, G.L. IL23R-Protective Coding Variant Promotes Beneficial Bacteria and Diversity in the Ileal Microbiome in Healthy Individuals Without Inflammatory Bowel Disease. J. Crohn’s Colitis 2019, 13, 451–461. [Google Scholar] [CrossRef]
- Zuo, T.; Lu, X.J.; Zhang, Y.; Cheung, C.P.; Lam, S.; Zhang, F.; Tang, W.; Ching, J.Y.L.; Zhao, R.; Chan, P.K.S.; et al. Gut mucosal virome alterations in ulcerative colitis. Gut 2019, 68, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Altomare, A.; Putignani, L.; Del Chierico, F.; Cocca, S.; Angeletti, S.; Ciccozzi, M.; Tripiciano, C.; Dalla Piccola, B.; Cicala, M.; Guarino, M.P.L. Gut mucosal-associated microbiota better discloses inflammatory bowel disease differential patterns than faecal microbiota. Dig. Liver Dis. 2019, 51, 648–656. [Google Scholar] [CrossRef]
- Franzosa, E.A.; Sirota-madi, A.; Avila-pacheco, J.; Fornelos, N.; Haiser, H.J.; Reinker, S.; Vatanen, T.; Hall, A.B.; Mallick, H.; Mciver, L.J.; et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 2019, 4, 293–305. [Google Scholar] [PubMed]
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E.; Ajami, N.J.; Bonham, K.S.; Brislawn, C.J.; et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Imai, T.; Inoue, R.; Kawada, Y.; Morita, Y.; Inatomi, O.; Nishida, A.; Bamba, S.; Kawahara, M.; Andoh, A. Characterization of fungal dysbiosis in Japanese patients with inflammatory bowel disease. J. Gastroenterol. 2019, 54, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Zhang, Y.; Tian, X.; Wang, X.; Gathungu, G.; Wolber, A.; Shiekh, S.S.; Sartor, R.B.; Davidson, N.O.; Ciorba, M.A.; et al. Influence of Crohn’s disease related polymorphisms in innate immune function on ileal microbiome. PLoS ONE 2019, 14, e0213108. [Google Scholar] [CrossRef] [PubMed]
- Vester-Andersen, M.K.; Mirsepasi-Lauridsen, H.C.; Prosberg, M.V.; Mortensen, C.O.; Träger, C.; Skovsen, K.; Thorkilgaard, T.; Nøjgaard, C.; Vind, I.; Krogfelt, K.A.; et al. Increased abundance of proteobacteria in aggressive Crohn’s disease seven years after diagnosis. Sci. Rep. 2019, 9, 13473. [Google Scholar] [CrossRef]
- Clooney, A.G.; Sutton, T.D.S.; Shkoporov, A.N.; Holohan, R.K.; Daly, K.M.; O’Regan, O.; Ryan, F.J.; Draper, L.A.; Plevy, S.E.; Ross, R.P.; et al. Whole-Virome Analysis Sheds Light on Viral Dark Matter in Inflammatory Bowel Disease. Cell Host Microbe 2019, 26, 764–778.e5. [Google Scholar] [CrossRef]
- Braun, T.; Di Segni, A.; Benshoshan, M.; Neuman, S.; Levhar, N.; Bubis, M.; Picard, O.; Sosnovski, K.; Efroni, G.; Farage Barhom, S.; et al. Individualized Dynamics in the Gut Microbiota Precede Crohn’s Disease Flares. Am. J. Gastroenterol. 2019, 114, 1142–1151. [Google Scholar] [CrossRef]
- Galazzo, G.; Tedjo, D.I.; Wintjens, D.S.J.; Savelkoul, P.H.M.; Masclee, A.A.M.; Bodelier, A.G.L.; Pierik, M.J.; Jonkers, D.M.A.E.; Penders, J. Faecal Microbiota Dynamics and their Relation to Disease Course in Crohn’s Disease. J. Crohn’s Colitis 2019, 13, 1273–1282. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Du, B.; Shi, Y.; Lu, Y.; Zhou, Y.; Liu, B. Combined signature of the fecal microbiome and plasma metabolome in patients with ulcerative colitis. Med. Sci. Monit. 2019, 25, 3303–3315. [Google Scholar] [CrossRef]
- Yilmaz, B.; Juillerat, P.; Øyås, O.; Ramon, C.; Bravo, F.D.; Franc, Y.; Fournier, N.; Michetti, P.; Mueller, C.; Geuking, M.; et al. Microbial network disturbances in relapsing refractory Crohn’s disease. Nat. Med. 2019, 25, 323–336. [Google Scholar] [CrossRef]
- Magro, D.O.; Santos, A.; Guadagnini, D.; de Godoy, F.M.; Silva, S.H.M.; Lemos, W.J.F.; Vitulo, N.; Torriani, S.; Pinheiro, L.V.; Martinez, C.A.R.; et al. Remission in Crohn’s disease is accompanied by alterations in the gut microbiota and mucins production. Sci. Rep. 2019, 9, 53. [Google Scholar] [CrossRef]
- Zhang, Y.-L.; Cai, L.-T.; Qi, J.-Y.; Lin, Y.-Z.; Dai, Y.-C.; Jiao, N.; Chen, Y.-L.; Zheng, L.; Wang, B.-B.; Zhu, L.-X.; et al. Gut microbiota contributes to the distinction between two traditional Chinese medicine syndromes of ulcerative colitis. World J. Gastroenterol. 2019, 25, 3108–3282. [Google Scholar] [CrossRef]
- Alam, M.T.; Amos, G.C.A.; Murphy, A.R.J.; Murch, S.; Wellington, E.M.H.; Arasaradnam, R.P. Microbial imbalance in inflammatory bowel disease patients at different taxonomic levels. Gut Pathog. 2020, 12, 1. [Google Scholar] [CrossRef] [PubMed]
- Ryan, F.J.; Ahern, A.M.; Fitzgerald, R.S.; Laserna-Mendieta, E.J.; Power, E.M.; Clooney, A.G.; O’Donoghue, K.W.; McMurdie, P.J.; Iwai, S.; Crits-Christoph, A.; et al. Colonic microbiota is associated with inflammation and host epigenomic alterations in inflammatory bowel disease. Nat. Commun. 2020, 11, 1512. [Google Scholar] [CrossRef] [PubMed]
- Butera, A.; Di Paola, M.; Vitali, F.; De Nitto, D.; Covotta, F.; Borrini, F.; Pica, R.; De Filippo, C.; Cavalieri, D.; Giuliani, A.; et al. IL-13 mRNA Tissue Content Identifies Two Subsets of Adult Ulcerative Colitis Patients with Different Clinical and Mucosa-Associated Microbiota Profiles. J. Crohn’s Colitis 2020, 14, 369–380. [Google Scholar] [CrossRef]
- Boland, K.; Bedrani, L.; Turpin, W.; Kabakchiev, B.; Stempak, J.; Borowski, K.; Nguyen, G.; Steinhart, A.H.; Smith, M.I.; Croitoru, K.; et al. Persistent Diarrhea in Patients With Crohn’s Disease After Mucosal Healing Is Associated With Lower Diversity of the Intestinal Microbiome and Increased Dysbiosis. Clin. Gastroenterol. Hepatol. 2020, 19, 296–304.e3. [Google Scholar] [CrossRef] [PubMed]
- Olaisen, M.; Flatberg, A.; van Beelen Granlund, A.; Røyset, E.S.; Martinsen, T.C.; Sandvik, A.K.; Fossmark, R. Bacterial mucosa-associated microbiome in inflamed and proximal noninflamed ileum of patients with Crohn’s disease. Inflamm. Bowel Dis. 2020, 27, 12–24. [Google Scholar] [CrossRef]
- Shahir, N.M.; Wang, J.R.; Wolber, E.A.; Schaner, M.S.; Frank, D.N.; Ir, D.; Robertson, C.E.; Chaumont, N.; Sadiq, T.S.; Koruda, M.J.; et al. Crohn’s Disease Differentially Affects Region-Specific Composition and Aerotolerance Profiles of Mucosally Adherent Bacteria. Inflamm. Bowel Dis. 2020, 26, 1843–1855. [Google Scholar] [CrossRef]
- Park, S.; Kim, H.-N.; Choi, C.H.; Im, J.P.; Cha, J.M.; Eun, C.S.; Kim, T.-O.; Kang, S.-B.; Bang, K.B.; Kim, H.G.; et al. Differentially Abundant Bacterial Taxa Associated with Prognostic Variables of Crohn’s Disease: Results from the IMPACT Study. J. Clin. Med. 2020, 9, 1748. [Google Scholar] [CrossRef] [PubMed]
- Clooney, A.G.; Eckenberger, J.; Laserna-Mendieta, E.; Sexton, K.A.; Bernstein, M.T.; Vagianos, K.; Sargent, M.; Ryan, F.J.; Moran, C.; Sheehan, D.; et al. Ranking microbiome variance in inflammatory bowel disease: A large longitudinal intercontinental study. Gut 2020, 70, 499–510. [Google Scholar] [CrossRef]
- Park, Y.M.; Ha, E.; Gu, K.-N.; Shin, G.Y.; Lee, C.K.; Kim, K.; Kim, H.J. Host Genetic and Gut Microbial Signatures in Familial Inflammatory Bowel Disease. Clin. Transl. Gastroenterol. 2020, 11, e00213. [Google Scholar] [CrossRef]
- Lo Sasso, G.; Khachatryan, L.; Kondylis, A.; Battey, J.N.D.; Solovyeva, V.V.; Garanina, E.E.; Kitaeva, K.V.; Ivanov, K.Y. Inflammatory bowel disease—Associated changes in the gut: Focus on Kazan patients. Inflamm. Bowel Dis. 2020, 27, 418–433. [Google Scholar] [CrossRef]
- Borren, N.Z.; Plichta, D.; Joshi, A.D.; Bonilla, G.; Sadreyev, R.; Vlamakis, H.; Xavier, R.J.; Ananthakrishnan, A.N. Multi-“-Omics” Profiling in Patients With Quiescent Inflammatory Bowel Disease Identifies Biomarkers Predicting Relapse. Inflamm. Bowel Dis. 2020, 26, 1524–1532. [Google Scholar] [CrossRef]
- Rubbens, P.; Props, R.; Kerckhof, F.M.; Boon, N.; Waegeman, W. Cytometric fingerprints of gut microbiota predict Crohn’s disease state. ISME J. 2021, 15, 354–358. [Google Scholar] [CrossRef]
- Halfvarson, J.; Brislawn, C.J.; Lamendella, R.; Vázquez-Baeza, Y.; Walters, W.A.; Bramer, L.M.; D’Amato, M.; Bonfiglio, F.; McDonald, D.; Gonzalez, A.; et al. Dynamics of the human gut microbiome in Inflammatory Bowel Disease. Nat. Microbiol. 2017, 2, 17004. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Xu, Z.Z.; He, Y.; Yang, Y.; Liu, L.; Lin, Q.; Nie, Y.; Li, M.; Zhi, F.; Liu, S.; et al. Gut Microbiota Offers Universal Biomarkers across Ethnicity in Inflammatory Bowel Disease Diagnosis and Infliximab Response Prediction. mSystems 2018, 3, e00188-17. [Google Scholar] [CrossRef]
- Feng, T.; Wang, L.; Schoeb, T.R.; Elson, C.O.; Cong, Y. Microbiota innate stimulation is a prerequisite for T cell spontaneous proliferation and induction of experimental colitis. J. Exp. Med. 2010, 207, 1321–1332. [Google Scholar] [CrossRef] [PubMed]
- Nagao-Kitamoto, H.; Shreiner, A.B.; Gillilland, M.G.; Kitamoto, S.; Ishii, C.; Hirayama, A.; Kuffa, P.; El-Zaatari, M.; Grasberger, H.; Seekatz, A.M.; et al. Functional Characterization of Inflammatory Bowel Disease-Associated Gut Dysbiosis in Gnotobiotic Mice. Cmgh 2016, 2, 468–481. [Google Scholar] [CrossRef] [PubMed]
- Ward, N.L.; Phillips, C.D.; Nguyen, D.D.; Shanmugam, N.K.N.; Song, Y.; Hodin, R.; Shi, H.N.; Cherayil, B.J.; Goldstein, A.M. Antibiotic Treatment Induces Long-lasting Changes in the Fecal Microbiota that Protect Against Colitis. Inflamm. Bowel Dis. 2016, 22, 2328–2340. [Google Scholar] [CrossRef]
- Khan, K.J.; Ullman, T.A.; Ford, A.C.; Abreu, M.T.; Abadir, A.; Marshall, J.K.; Talley, N.J.; Moayyedi, P. Antibiotic therapy in inflammatory bowel disease: A systematic review and meta-analysis. Am. J. Gastroenterol. 2011, 106, 661–673. [Google Scholar] [CrossRef]
- Donaldson, G.P.; Lee, S.M.; Mazmanian, S.K. Gut biogeography of the bacterial microbiota. Nat. Rev. Microbiol. 2015, 14, 20–32. [Google Scholar] [CrossRef]
- Doolittle, W.F.; Booth, A. It’s the song, not the singer: An exploration of holobiosis and evolutionary theory. Biol. Philos. 2017, 32, 5–24. [Google Scholar] [CrossRef]
- Mobeen, F.; Sharma, V.; Prakash, T. Enterotype Variations of the Healthy Human Gut Microbiome in Different Geographical Regions. Bioinformation 2018, 14, 560–573. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Fraser, C.M.; Ringel, Y.; Sanders, M.E.; Sartor, R.B.; Sherman, P.M.; Versalovic, J.; Young, V.; Finlay, B.B. Defining a healthy human gut microbiome: Current concepts, future directions, and clinical applications. Cell Host Microbe 2012, 12, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Kleessen, B.; Kroesen, A.J.; Buhr, H.J.; Blaut, M. Mucosal and invading bacteria in patients with inflammatory bowel disease compared with controls. Scand. J. Gastroenterol. 2002, 37, 1034–1041. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.; Seksik, P.; Rigottier-gois, L.; Lay, C.; Lepage, P.; Podglajen, I.; Marteau, P. Specificities of the fecal microbiota in inflammatory bowel disease. Inflamm. Bowel Dis. 2006, 12, 106–111. [Google Scholar] [CrossRef]
- Sokol, H.; Pigneur, B.; Watterlot, L.; Lakhdari, O.; Bermúdez-Humarán, L.G.; Gratadoux, J.J.; Blugeon, S.; Bridonneau, C.; Furet, J.P.; Corthier, G.; et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. USA 2008, 105, 16731–16736. [Google Scholar] [CrossRef]
- Venegas, D.P.; De La Fuente, M.K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short chain fatty acids (SCFAs) mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef]
- Underhill, D.M.; Iliev, I.D. The mycobiota: Interactions between commensal fungi and the host immune system. Nat. Rev. Immunol. 2014, 14, 405–416. [Google Scholar] [CrossRef]
- McKenzie, H.; Main, J.; Pennington, C.R.; Parratt, D. Antibody to selected strains of Sacharomyces cerevisiae (baker’s and brewer’s yeast) and Candida albicans in Crohn’s disease. Gut 1990, 31, 536–538. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, S.; Manichanh, C.; Nielsen, T.; Pons, N.; Yamada, T.; Mende, D.R.; et al. A human gut microbial gene catalog established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef]
- Shkoporov, A.N.; Clooney, A.G.; Sutton, T.D.S.; Ryan, F.J.; Daly, K.M.; Nolan, J.A.; McDonnell, S.A.; Khokhlova, E.V.; Draper, L.A.; Forde, A.; et al. The human gut virome is highly diverse, stable, and individual specific. Cell Host Microbe 2019, 26, 527–541.e5. [Google Scholar] [CrossRef]
- Dridi, B.; Raoult, D.; Drancourt, M. Archaea as emerging organisms in complex human microbiomes. Anaerobe 2011, 17, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Mitsuyama, K.; Niwa, M.; Takedatsu, H.; Yamasaki, H.; Kuwaki, K.; Yoshioka, S.; Yamauchi, R.; Fukunaga, S.; Torimura, T. Antibody markers in the diagnosis of inflammatory bowel disease. World J. Gastroenterol. 2016, 22, 1304–1310. [Google Scholar] [CrossRef] [PubMed]
- Gisbert, J.P.; Gomollón, F.; Maté, J.; Pajares, J.M. Papel de los anticuerpos anticitoplasma de los neutrófilos (ANCA) y anti-Saccharomyces cerevisiae (ASCA) en la enfermedad inflamatoria intestinal. Gastroenterol. Hepatol. 2003, 26, 312–324. [Google Scholar] [CrossRef]
- Li, Y.; Hauenstein, K. New Imaging Techniques in the Diagnosis of Inflammatory Bowel Diseases. Visz. Gastrointest. Med. Surg. 2015, 31, 227–234. [Google Scholar] [CrossRef]
- Lopez, R.N.; Leach, S.T.; Lemberg, D.A.; Duvoisin, G.; Gearry, R.B.; Day, A.S. Fecal biomarkers in inflammatory bowel disease. J. Gastroenterol. Hepatol. 2017, 32, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Tedjo, D.I.; Smolinska, A.; Savelkoul, P.H.; Masclee, A.A.; Van Schooten, F.J.; Pierik, M.J.; Penders, J.; Jonkers, D.M.A.E. The fecal microbiota as a biomarker for disease activity in Crohn’s disease. Sci. Rep. 2016, 6, 35216. [Google Scholar] [CrossRef]

| Reference | Year | Treatment | No. of Participants | Disease State | Specimen | Histology | Design | Microbiome Analysis Method | Focus | Microbiota Findings | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CD | UC | IBD/IBDU | HC/C | ||||||||||
| Macfarlane et al. [12] | 2004 | Not naïve | NA | 9 | NA | 10 | Active | Biopsy | NA | Cross-sectional | Culture, FISH | Bacteria | UC
|
| Lepage et al. [13] | 2005 | Not naïve | 20 | 11 | NA | 4 | Active/Inactive | Stool and biopsy | Non-inflamed | Cross-sectional | TTGE (16S rDNA V6–V8 region) | Bacteria | CD and UC
|
| Manichanh et al. [14] | 2006 | Not naïve | 6 | NA | NA | 6 | Inactive | Stool | NA | Cross-sectional | Cloning, Sequencing (16S rDNA) | Bacteria | CD
|
| Bibiloni et al. [15] | 2006 | Naïve | 20 | 15 | NA | 14 | Active | Biopsy | Inflamed/non-inflamed | Cross-sectional | DGGE (16S rDNA V3 region) and qPCR | Bacteria | CD and UC
|
| Sokol et al. [16] | 2006 | Not naïve | NA | 9 | NA | 9 | Active | Stool | NA | Cross-sectional | TTGE (16S rDNA V6–V8 region) | Bacteria | UC
|
| Gophna et al. [17] | 2006 | Not naïve | 6 | 5 | NA | 5 | Active/Inactive | Biopsy | Inflamed/non-inflamed | Cross-sectional | PCR, cloning, sequencing (16S rDNA) | Bacteria | CD and UC
|
| Scanlan et al. [18] | 2006 | Not naïve | 16 | NA | NA | 6 | Active/Inactive | Stool | NA | Longitudinal | DGGE (16S rDNA) | Bacteria | CD
|
| Zhang et al. [19] | 2007 | Not naïve | NA | 24 | NA | NA | Active | Biopsy | Inflamed/non-inflamed | Cross-sectional | DGGE (16S rDNA V3 region) | Bacteria | UC
|
| Sepehri et al. [20] | 2007 | Not naïve | 10 | 15 | NA | 16 | NA | Biopsy | Inflamed/non-inflamed | Cross-sectional | ARISA, T-RFLP | Bacteria | CD and UC
|
| Andoh et al. [21] | 2007 | Not naïve | NA | 44 | NA | 46 | Active/Inactive | Stool | NA | Cross-sectional | T-RFLP (16S rDNA) | Bacteria | UC
|
| Frank et al. [22] | 2007 | Not naïve | 68 | 61 | NA | 61 | NA | Resected tissue | Inflamed/non-inflamed | Cross-sectional | PCR, cloning, sequencing (16S rDNA) | Bacteria | CD and UC
|
| Ott et al. [23] | 2008 | Not naïve | NA | 13 | NA | 5 | Active/Inactive | Biopsy | NA | Longitudinal | PCR, cloning and sequencing | Bacteria | UC
|
| Ott et al. [24] | 2008 | Not naïve | 31 | 26 | NA | 47 | Active | Biopsy | Inflamed | Cross-sectional | DGGE, clone libraries, sequencing, in situ hybridization (18S rDNA) | Fungi | CD
|
| Martinez et al. [25] | 2008 | Not naïve | NA | 16 | NA | 8 | Inactive | Stool | NA | Longitudinal | DGGE (16S rDNA V6–V8 region) | Bacteria | UC
|
| Dicksved et al. [26] | 2008 | Not naïve | 14 | NA | NA | 6 | Active/Inactive | Stool | NA | Cross-sectional | T-RFLP, cloning and sequencing (16S rDNA) | Bacteria | CD
|
| Kuehbacher et al. [27] | 2008 | Not naïve | 42 | 31 | NA | 33 | Active | Biopsy | Inflamed | Cross-sectional | Clone libraries, sequencing and in situ hybridization (16S rDNA) | Bacteria | CD and UC
|
| Andoh et al. [28] | 2008 | Not naïve | 34 | NA | NA | 30 | Active/Inactive | Stool | NA | Cross-sectional | T-RFLP (16S rDNA) | Bacteria | CD
|
| Nishikawa et al. [29] | 2009 | Not naïve | 9 | NA | NA | 11 | Active/Inactive | Biopsy | Inflamed/non-inflamed | Longitudinal | T-RFLP (16S rDNA) | Bacteria | UC
|
| Willing et al. [30] | 2009 | Not naïve | 14 | NA | NA | 6 | Active/Inactive | Biopsy | Inflamed/non-inflamed | Cross-sectional | T-RFLP, cloning and sequencing, qPCR (16S rDNA) | Bacteria | CD
|
| Andoh et al. [31] | 2009 | Not naïve | NA | 2 | NA | 3Ur | Inactive | Stool | NA | Cross-sectional | T-RFLP (16S rDNA) | Bacteria | UC
|
| Gillevet et al. [32] | 2010 | Not naïve | 4 | 2 | NA | 4 | NA | Stool and biopsy | NA | Cross-sectional | LH- PCR, cloning, sequencing, and multitagged pyrosequencing (16S rDNA) | Bacteria | CD and UC
|
| Rehman et al. [33] | 2010 | Not naïve | 10 | 10 | NA | 10 | Active | Biopsy | Inflamed | Cross-sectional | PCR, cloning, sequencing (16S rDNA) | Bacteria | CD and UC
|
| Kang et al. [34] | 2010 | Not naïve | 6 | NA | NA | 6 | Inactive | Stool | NA | Cross-sectional | Microarray (16S rDNA) | Bacteria | CD.
|
| Rowan et al. [35] | 2010 | Not naïve | NA | 20 | NA | 19 | Active/Inactive | Biopsy | NA | Cross-sectional | PCR, qPCR (16S rDNA) | Bacteria | UC
|
| Andoh et al. [36] | 2011 | Not naïve | 31 | 31 | NA | 30 | Active/Inactive | Stool | NA | Cross-sectional | T-RFLP (16S rDNA V4–V9) | Bacteria | CD and UC.
|
| Mondot et al. [37] | 2011 | Not naïve | 16 | NA | NA | 16 | Active | Stool | NA | Cross-sectional | qPCR, RT qPCR (16S rDNA) | Bacteria | CD
|
| Joossens et al. [38] | 2011 | Not naïve | 68 | NA | NA | 84 Ur + 55 | Inactive | Stool | NA | Cross-sectional | DGGE (16S rDNA V3), qPCR | Bacteria | CD
|
| Lepage et al. [39] | 2011 | Not naïve | NA | 8 | NA | 54 | Active | Biopsy | NA | Cross-sectional | PCR, cloning, sequencing (16S rDNA) | Bacteria | UC.
|
| Benjamin et al. [40] | 2012 | Not naïve | 103 | NA | NA | 66 | Active | Stool | NA | Cross-sectional | FISH (16S rDNA) | Bacteria | CD
|
| Hotte et al. [41] | 2012 | Not naïve | 15 | 14 | NA | 21 | Inactive | Biopsy | Non-inflamed | Cross-sectional | T-RFLP (16S rDNA) | Bacteria | CD and UC
|
| Pistone et al. [42] | 2012 | Not naïve | 35 | 18 | NA | 35 | NA | Biopsy | Inflamed/non-inflamed | Cross-sectional | PCR | Mycobacterium avium subspecies paratuberculosis | CD and UC
|
| Andoh et al. [43] | 2012 | Not naïve | 67 | NA | NA | 121 | Active/Inactive | Stool | NA | Longitudinal | T-RFLP (16S rDNA V1–V9) | Bacteria | CD
|
| Li et al. [44] | 2012 | Not naïve | 18 | NA | NA | 9 | Active | Stool and biopsy | Inflamed/non-inflamed | Cross-sectional | DGGE (16S rDNA V3 region), sequencing | Bacteria | CD
|
| Nemoto et al. [45] | 2012 | Not naïve | NA | 48 | NA | 36 | Active/Inactive | Stool | NA | Cross-sectional | Culture, T-RFLP, qPCR | Bacteria | UC
|
| Vigsnæs et al. [46] | 2012 | Not naïve | NA | 12 | NA | 6 | Active/Inactive | Stool | NA | Cross-sectional | DGGE (16S rDNA, 16S-23S rDNA intergenic spacer region), qPCR | Bacteria | UC.
|
| de Souza et al. [47] | 2012 | Not naïve | 11 | 7 | NA | 14 | NA | Stool and biopsy | Inflamed/non-inflamed | Cross-sectional | Culture | E. coli | CD and UC
|
| Duboc et al. [48] | 2013 | Not naïve | 12 | 30 | NA | 26 | Active/Inactive | Stool | NA | Cross-sectional | PCR (rDNA), culture | Bacteria | CD and UC
|
| Sha et al. [49] | 2013 | Not naïve | 10 | 26 | NA | 14 | Active/Inactive | Stool | NA | Cross-sectional | DGGE (16S rDNA V6–V8 region), qPCR | Bacteria | CD and UC.
|
| Kabeerdoss et al. [50] | 2013 | Not naïve | 20 | 22 | NA | 17 | Active/Inactive | Stool | NA | Cross-sectional | TTGE (16S rDNA V1–V9), qPCR | C. leptum group, F. prausnitzii | CD and UC
|
| Varela et al. [51] | 2013 | Not naïve | NA | 116 | NA | 29 Ur + 31 | Inactive | Stool | NA | Cross-sectional and longitudinal | PCR (16S rDNA), qPCR | F. prausnitzii | UC
|
| Midtvedt et al. [52] | 2013 | Not naïve | 4 | NA | NA | 5 | Active | Stool and biopsy | Inflamed | Cross-sectional | Microarray | Bacteria | CD
|
| Fujimoto et al. [53] | 2013 | Not naïve | 47 | NA | NA | 20 | Active/Inactive | Stool | NA | Cross-sectional | qPCR, PCR (16S rDNA V4–V9), T-RFLP | F. prausnitzii and Bilophila wadsworthia | CD
|
| Fite et al. [54] | 2013 | Not naïve | NA | 33 | NA | 18 | Active | Biopsy | Inflamed | Longitudinal | qPCR | Bacteria | UC
|
| Rajilic-Stojanovic et al. [55] | 2013 | Not naïve | NA | 15 | NA | 15 | Inactive | Stool | NA | Longitudinal | Microarray | Bacteria | UC
|
| Kumari et al. [56] | 2013 | Not naïve | NA | 26 | NA | 14 | Active/Inactive | Stool | NA | Cross-sectional | FISH, flow cytometry, qPCR (16S rDNA) | Bacteria | UC
|
| Hedin et al. [57] | 2014 | Not naïve | 22 | NA | NA | 25 + 21Ur | Inactive | Stool | NA | Cross-sectional | qPCR (16S rDNA) | Bacteria | CD
|
| Lennon et al. [58] | 2014 | Not naïve | NA | 19 | NA | 34 | Active | Biopsy | NA | Cross-sectional | qPCR (16S rDNA) | Desulfovibrio species | UC
|
| Machiels et al. [59] | 2014 | Not naïve | NA | 127 | NA | 447 | Active/Inactive | Stool | NA | Cross-sectional | PCR (16S rDNA V3 region) DGGE, sequencing, qPCR | Bacteria | UC
|
| Wang et al. [60] | 2014 | Not naïve | 21 | 34 | NA | 21 | Active/Inactive | Stool and biopsy | Inflamed/non-inflamed | Cross-sectional | qPCR (16S rDNA) | Bacteria | CD and UC
|
| Blais Lecours et al. [61] | 2014 | Not naïve | 18 | 11 | NA | 29 | Active/Inactive | Stool | NA | Cross-sectional | qPCR | Archaea and bacteria | CD and UC
|
| Fukuda et al. [62] | 2014 | Not naïve | NA | 69 | NA | 80Ur | Active/Inactive | Stool | NA | Cross-sectional | PCR (16S rDNA, V4–V9 region), T-RFLP | Bacteria | UC
|
| Li et al. [63] | 2014 | Not naïve | 19 | NA | NA | 7 | Active | Stool and biopsy | Inflamed/non-inflamed | Cross-sectional | DGGE (18S rDNA), cloning, sequencing | Fungi | CD
|
| Andoh et al. [64] | 2014 | Not naïve | 160 | NA | NA | 121 | Active/Inactive | Stool | NA | Longitudinal | T-RFLP (16S rDNA V1–V9) | Bacteria | CD
|
| Wisittipanit et al. [65] | 2015 | Not naïve | 101 | 89 | NA | 235 | Active/Inactive | Biopsy and lumen aspiration | NA | Cross-sectional | LH-PCR (16S rDNA V1–V2 region) | Bacteria |
|
| Kabeerdoss et al. [66] | 2015 | Naïve and not naïve | 28 | 32 | NA | 30 | NA | Biopsy | Inflamed/non-inflamed | Cross-sectional | RT-qPCR (16S rDNA) | Bacteria | CD and UC
|
| Takeshita et al. [67] | 2016 | Not naïve | NA | 48 | NA | 34 | Active/Inactive | Stool | NA | Cross-sectional | RT-qPCR | Bacteria | UC
|
| Zhang et al. [68] | 2017 | Not naïve | 132 | NA | NA | 71 | Active/Inactive | Stool | NA | Cross-sectional | Culture | Bacteria | CD
|
| Vrakas et al. [69] | 2017 | Naïve and not naïve | 12 | 20 | NA | 20 | Active/Inactive | Biopsy | Inflamed | Cross-sectional | RT-qPCR (16S rDNA) | Bacteria | CD and UC
|
| Zamani et al. [70] | 2017 | Not naïve | NA | 35 | NA | 60 | Active | Biopsy | Inflamed | Cross-sectional | Culture, qPCR | Bacteria | UC
|
| Ghavami et al. [71] | 2018 | Not naïve | 9 | 45 | NA | 47 | Active/Inactive | Stool | NA | Cross-sectional | PCR, qPCR (16S rDNA) | Bacteria and Methanobrevibacter smithii (Archaea) | CD and UC
|
| Le Baut et al. [72] | 2018 | Not naïve | 262 | NA | NA | 76 | NA | Resected tissue and biopsy | Inflamed/non-inflamed | Cross-sectional | PCR | Yersinia Species | CD
|
| Al-Bayati et al. [73] | 2018 | Not naïve | NA | 40 | NA | 40 | NA | Biopsy | Inflamed | Cross-sectional | Culture, PCR (16S rDNA) | Bacteria | UC
|
| Heidarian et al. [74] | 2019 | Not naïve | 7 | 22 | NA | 29 | Active/Inactive | Stool | NA | Cross-sectional | qPCR | Bacteria | CD and UC
|
| Vatn et al. [75] | 2020 | Naïve and not naïve | 68 | 84 | 12 | 160 | Active/Inactive | Stool | NA | Cross-sectional | GA-map™ (16S rDNA V3–V9 region) | Bacteria | CD and UC
|
| Reference | Year | Treatment | No. of Participants | Disease State | Specimen | Histology | Design | Microbiome Analysis Method | Focus | Microbiota Findings | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CD | UC | IBD/IBDU | HC/C | ||||||||||
| Willing et al. [76] | 2010 | Not naïve | 29 | 16 | NA | 35 | Active/Inactive | Stool and biopsy | Non-inflamed | Cross-sectional | 16S rDNA sequencing | Bacteria | CD and UC
|
| Rausch et al. [77] | 2011 | Not naïve | 29 | NA | NA | 18 | Inactive | Biopsy | Non-inflamed | Cross-sectional | 16S rDNA V1–V2 region sequencing | Bacteria | CD
|
| Walker et al. [78] | 2011 | Not naïve | 6 | 6 | NA | 5 | Active | Biopsy | Inflamed/non-inflamed | Cross-sectional | 16S rDNA V1–V8 region sequencing | Bacteria | CD and UC
|
| Erickson et al. [79] | 2012 | Not naïve | 8 | NA | NA | 4 | Active/Inactive | Stool | NA | Cross-sectional | 16S rDNA V1–V2 region and WGS | Bacteria | CD
|
| Morgan et al. [80] | 2012 | Not naïve | 121 | 75 | 8 | 27 | Active/Inactive | Stool and biopsy | NA | Cross-sectional | 16S rDNA V3–V5 region and WGS | Bacteria | CD and UC
|
| Ricanek et al. [81] | 2012 | Naïve | 4 | NA | NA | 1 | Active | Biopsy | Inflamed | Cross-sectional | 16S rDNA sequencing | Bacteria | CD
|
| Li et al. [82] | 2012 | Not naïve | 52 | 58 | NA | 60 | NA | Biopsy | Non-inflamed | Cross-sectional | 16S rDNA V1–V3 and V3–V5 regions sequencing and qPCR | Bacteria | CD and UC
|
| Tong et al. [83] | 2013 | Not naïve | 16 | 16 | NA | 32 | Inactive | Mucosal lavage | Non-inflamed | Cross-sectional | 16S rDNA V4 region sequencing | Bacteria | CD and UC
|
| Thorkildsen et al. [84] | 2013 | Naïve | 30 | 33 | 3 | 34 | Active | Stool | NA | Cross-sectional | 16S rDNA (all regions) sequencing | Bacteria | CD and UC
|
| Prideaux et al. [85] | 2013 | Not naïve | 22 | 30 | NA | 29 +6Ur (CD) | NA | Biopsy | Inflamed/non-inflamed | Cross-sectional | Microarray, 16S rDNA V1–V3 region sequencing | Bacteria | CD and UC
|
| Chiodini et al. [86] | 2013 | Not naïve | 14 | NA | NA | 6 | NA | Resected tissue | NA | Cross-sectional | 16S rDNA V3–V6 region sequencing | Bacteria | CD
|
| Pérez-Brocal et al. [87] | 2013 | Naïve and not naïve | 11 | NA | NA | 8 | NA | Stool | NA | Cross-sectional | Viral DNA and 16S rDNA V1–V3 region sequencing | Bacteria and viruses | CD
|
| Davenport et al. [88] | 2014 | Not naïve | 13 | 14 | NA | 27 | NA | Biopsy | Inflamed | Cross-sectional | 16S rDNA V4 region sequencing | Bacteria | CD and UC
|
| Chen et al. [89] | 2014 | Not naïve | 26 | 41 | NA | 21 | Active/Inactive | Stool and biopsy | Inflamed/non-inflamed | Cross-sectional | 16S rDNA V1–V3 region sequencing | Bacteria | CD and UC
|
| Wang et al. [90] | 2015 | Not naïve | 6 | 4 | NA | 5 | NA | Biopsy | NA | Cross-sectional | RNA sequencing | Bacteria and viruses | CD and UC
|
| Lavelle et al. [91] | 2015 | Not naïve | NA | 9 | NA | 4 | NA | Luminal brush, mucosal biopsy, mucus gel layer | Inflamed/non-inflamed | Cross-sectional | 16S rDNA V4 region sequencing | Bacteria | UC
|
| Chiodini et al. [92] | 2015 | Not naïve | 20 | NA | NA | 15 | NA | Biopsy | Inflamed | Cross-sectional | 16S rDNA V4 region sequencing | Bacteria | CD
|
| Pérez-Brocal et al. [93] | 2015 | Naïve and not naïve | 20 | NA | NA | 20 | Active | Stool and biopsy | Inflamed/non-inflamed | Cross-sectional | 16S rDNA V1–V3 region and viral DNA/RNA sequencing | Bacteria and viruses | CD
|
| Vidal et al. [94] | 2015 | Not naïve | 13 | NA | NA | 7 | Active/Inactive | Biopsy | Non-inflamed | Cross-sectional | 16S rDNA V1–V5 region sequencing | Bacteria | CD
|
| Norman et al. [95] | 2015 | Not naïve | 18 | 42 | NA | 12 | Active/Inactive | Stool | NA | Cross-sectional | VLP DNA sequencing | Viruses | CD and UC
|
| Eun et al. [96] | 2016 | Not naïve | 35 | NA | NA | 15 | Inactive | Stool and biopsy | NA | Cross-sectional | 16S rDNA V1–V3 region sequencing | Bacteria | CD
|
| Chiodini et al. [97] | 2016 | Not naïve | 20 | NA | NA | 15 | NA | Biopsy | Inflamed | Cross-sectional | 16S rDNA V1–V3 region sequencing | Bacteria | CD
|
| Rehman et al. [98] | 2016 | Not naïve | 28 | 30 | NA | 30 | Inactive | Biopsy | NA | Cross-sectional | 16S rDNA V1–V2 region sequencing | Bacteria | CD and UC
|
| Takahashi et al. [99] | 2016 | Not naïve | 68 | NA | NA | 10 | Active/Inactive | Stool | NA | Cross-sectional | qPCR and 16S rDNA V3–V4 region sequencing | Bacteria | CD
|
| Forbes et al. [100] | 2016 | Not naïve | 15 | 21 | NA | 7 | NA | Biopsy | Inflamed/non-inflamed | Cross-sectional | 16S rDNA V6 region sequencing | Bacteria | CD and UC
|
| Liguori et al. [101] | 2016 | Not naïve | 23 | NA | NA | 10 | Active/Inactive | Biopsy | Inflamed/non-inflamed | Cross-sectional | qPCR (16S or 18S rDNA) 16S rDNA V3–V4 region and ITS2 sequencing | Bacteria and fungi | CD and UC
|
| Mar et al. [102] | 2016 | Not naïve | NA | 30 | NA | 13 | NA | Stool | NA | Cross-sectional | 16S rDNA V3–V4 region and ITS2 sequencing | Bacteria and fungi | UC
|
| Hoarau et al. [103] | 2016 | Not naïve | 20 | NA | NA | 21 + 28Ur | Active/Inactive | Stool | NA | Cross-sectional | 16S rDNA V4 region and ITS1 sequencing | Bacteria and fungi | CD
|
| Hedin et al. [104] | 2016 | Not naïve | 21 | NA | NA | 19+17Ur | Inactive | Biopsy | NA | Cross-sectional | 16S rDNA V1–V3 region sequencing | Bacteria | CD
|
| Naftali et al. [105] | 2016 | Not naïve | 31 | NA | NA | 5 | Active/Inactive | Biopsy | Inflamed | Cross-sectional | 16S rDNA V1–V3 region sequencing | Bacteria | CD
|
| Pedamallu et al. [106] | 2016 | Not naïve | 12 | NA | NA | 12 | NA | Resected tissue | NA | Cross-sectional | WGS | Bacteria | CD
|
| Sokol et al. [107] | 2016 | Not naïve | 149 | 86 | NA | 38 | Active/Inactive | Stool | NA | Cross-sectional | 16S rDNA V3–V5 region and ITS2 sequencing | Bacteria and fungi | CD and UC
|
| Santoru et al. [108] | 2017 | Not naïve | 50 | 82 | NA | 51 | Active/Inactive | Stool | NA | Cross-sectional | 16S rDNA V3–V4 region sequencing, qPCR | Bacteria | CD and UC
|
| Pascal et al. [109] | 2017 | Not naïve | 34 | 33 | NA | 40 + 71Ur | Active/Inactive | Stool | NA | Longitudinal | 16S rDNA V4 region sequencing | Bacteria | CD and UC
|
| Chen et al. [110] | 2017 | Not naïve | NA | 8 | NA | 8 | NA | Stool | NA | Cross-sectional | 16S rDNA V3–V4 region sequencing | Bacteria | UC
|
| Hall et al. [111] | 2017 | Not naïve | 9 | 10 | 1 | 12 | Active/Inactive | Stool | NA | Longitudinal | WGS | Bacteria | CD and UC
|
| Qiu et al. [112] | 2017 | Not naïve | NA | 14 | NA | 15 | Active | Biopsy | Inflamed | Cross-sectional | 18S rDNA sequencing | Fungi | UC
|
| Kennedy et al. [113] | 2018 | Not naïve | 37 | NA | NA | 54 | Inactive | Stool | NA | Cross-sectional | 16S rDNA V1–V2 region sequencing | Bacteria | CD
|
| Ji et al. [114] | 2018 | Not naïve | 51 | 66 | NA | 66 | Active/Inactive | Stool | NA | Cross-sectional | 16S rDNA V4 region sequencing | Bacteria | CD and UC
|
| Imhann et al. [115] | 2018 | Not naïve | 188 | 107 | 18 | 582 | Active/Inactive | Stool | NA | Cross-sectional | 16S rDNA V4 region sequencing | Bacteria | CD and UC
|
| Nishino et al. [116] | 2018 | Not naïve | 26 | 43 | NA | 14 | Active/Inactive | Mucosal brush | Non-inflamed | Cross-sectional | 16S rDNA V3–V4 region sequencing | Bacteria | CD and UC
|
| Rojas-Feria et al. [117] | 2018 | Naïve | 13 | NA | NA | 16 | Onset | Stool | NA | Cross-sectional | 16S rDNA V1–V3 region sequencing | Bacteria | CD
|
| Schirmer et al. [118] | 2018 | Naïve and not naïve | 30 | 21 | NA | 11 | Active/Inactive | Stool | NA | Longitudinal | WGS | Bacteria | CD and UC
|
| Chiodini et al. [119] | 2018 | Not naïve | 20 | NA | NA | 15 | NA | Resected tissue | Inflamed | Cross-sectional | 16S rDNA V1–V3 region sequencing | Bacteria | CD
|
| Hirano et al. [120] | 2018 | Not naïve | NA | 14 | NA | 14 | Active | Biopsy | Inflamed/non-inflamed | Cross-sectional | 16S rDNA V4 region sequencing | Bacteria | UC
|
| Ma et al. [121] | 2018 | Not naïve | 15 | 14 | NA | 13 | Active/Inactive | Stool | NA | Cross-sectional | 16S rDNA V4 region sequencing | Bacteria | CD and UC
|
| Walujkar et al. [122] | 2018 | Not naïve | NA | 12 | NA | 7 | Active | Biopsy | Inflamed | Longitudinal | 16S rDNA V4 region sequencing | Bacteria | UC
|
| Moen et al. [123] | 2018 | Naïve | NA | 44 | NA | 35 | Onset | Biopsy | Inflamed/non-inflamed | Cross-sectional | 16S rDNA V4 region sequencing | Bacteria | UC
|
| Laserna-Mendieta et al. [124] | 2018 | Not naïve | 71 | 58 | NA | 75 | Active/Inactive | Stool | NA | Cross-sectional | 16S rDNA V3–V4 region sequencing | Bacteria | CD and UC
|
| Libertucci et al. [125] | 2018 | Not naïve | 43 | NA | NA | 10 | Active/Inactive | Biopsy | Inflamed/non-inflamed | Cross-sectional | 16S rDNA V3 region and ITS2 sequencing | Bacteria and fungi | CD
|
| Moustafa et al. [126] | 2018 | Not naïve | 45 | 41 | NA | 146 | Active/Inactive | Stool | NA | Cross-sectional | WGS | Bacteria | CD and UC
|
| O’Brien et al. [127] | 2018 | Not naïve | 24 | NA | NA | 17 | NA | Biopsy | Inflamed/non-inflamed | Cross-sectional | 16S rDNA V1–V3 region sequencing | Bacteria | CD
|
| Zakrzewski et al. [128] | 2019 | Not naïve | 15 | NA | NA | 58 | Active | Biopsy | Inflamed/non-inflamed | Cross-sectional | 16S rDNA V3–V4 region sequencing | Bacteria |
|
| Zuo et al. [129] | 2019 | Not naïve | NA | 91 | NA | 76 | Active/Inactive | Biopsy | Inflamed/non-inflamed | Cross-sectional | VLP and 16S rDNA sequencing | Viruses | UC
|
| Altomare et al. [130] | 2019 | Not naïve | 10 | 4 | NA | 11 | Active/Inactive | Stool and biopsy | Inflamed/non-inflamed | Cross-sectional | 16S rDNA V1–V3 region sequencing | Bacteria | CD and UC
|
| Franzosa et al. [131] | 2019 | Not naïve | 68 | 53 | NA | 34 | Active/Inactive | Stool | NA | Cross-sectional | WGS | Bacteria | CD and UC
|
| Lloyd-Price et al. [132] | 2019 | Not naïve | 67 | 38 | NA | 27 | Active/Inactive | Stool and biopsy | NA | Longitudinal | 16S rDNA sequencing and WGS | Bacteria and viruses | CD and UC
|
| Imai et al. [133] | 2019 | Not naïve | 20 | 18 | NA | 20 | Inactive | Stool | NA | Cross-sectional | 16S rDNA V3–V4 region and ITS sequencing | Bacteria and fungi | CD and UC
|
| Li et al. [134] | 2019 | Not naïve | 106 | NA | 88 | 89 | NA | Resected tissue | Inflamed/non-inflamed | Longitudinal | 16S rDNA V3–V5 region sequencing, qPCR | Bacteria | CD
|
| Vester-Andersen et al. [135] | 2019 | Not naïve | 58 | 82 | NA | 30 | Active/Inactive | Stool | NA | Cross-sectional | 16S rDNA V3–V4 region sequencing | Bacteria | CD and UC
|
| Clooney et al. [136] | 2019 | Not naïve | 27 | 82 | NA | 61 | Active/Inactive | Stool | NA | Longitudinal | Whole-virome analysis and 16S rDNA V3–V4 region sequencing | Bacteria and viruses | CD and UC
|
| Braun et al. [137] | 2019 | Not naïve | 45 | NA | NA | 22 | Inactive | Stool | NA | Longitudinal | 16S rDNA V4 region sequencing | Bacteria | CD
|
| Galazzo et al. [138] | 2019 | Not naïve | 57 | NA | NA | 15 | Active/Inactive | Stool | NA | Longitudinal | 16S rDNA V4 region sequencing | Bacteria | CD
|
| Sun et al. [139] | 2019 | Not naïve | NA | 58 | NA | 30 | Active/Inactive | Stool | NA | Cross-sectional | 16S rDNA V3–V4 region sequencing | Bacteria | UC
|
| Yilmaz et al. [140] | 2019 | Not naïve | 270 | 232 | NA | 573 | Active/Inactive | Biopsy | Inflamed/non-inflamed | Longitudinal | 16S rDNA V5–V6 region sequencing | Bacteria | CD and UC
|
| Magro et al. [141] | 2019 | Not naïve | 18 | NA | NA | 18 | Inactive | Stool | NA | Cross-sectional | 16S rDNA V3–V4 region sequencing | Bacteria | UC
|
| Zhang et al. [142] | 2019 | Not naïve | NA | 63 | NA | 30 | Active/Inactive | Stool | NA | Cross-sectional | 16S rDNA V4 region sequencing | Bacteria | UC
|
| Alam et al. [143] | 2020 | Not naïve | 9 | 11 | NA | 10 | NA | Stool | NA | Cross-sectional | 16S rDNA V1–V3 region sequencing | Bacteria | CD and UC
|
| Ryan et al. [144] | 2020 | Not naïve | 80 | 50 | NA | 31 | Active/Inactive | Biopsy | Inflamed/non-inflamed | Cross-sectional | 16S rDNA V3–V4 region sequencing | Bacteria | CD and UC
|
| Butera et al. [145] | 2020 | No naïve | NA | 88 | NA | 24 | Active | Biopsy | Inflamed/non-inflamed | Cross-sectional | 16S rDNA V3–V4 region sequencing | Bacteria | UC
|
| Boland et al. [146] | 2020 | No naïve | 101 | 99 | 15 | 48 | Active/Inactive | Biopsy | NA | Cross-sectional | 16S rDNA V4 region sequencing | Bacteria | CD and UC
|
| Olaisen et al. [147] | 2020 | No naïve | 51 | NA | NA | 40 | Active/Inactive | Biopsy | Inflamed/non-inflamed | Cross-sectional | 16S rDNA V3–V4 region sequencing | Bacteria | CD
|
| Shahir et al. [148] | 2020 | No naïve | 125 | NA | NA | 23 | NA | Biopsy | Inflamed/non-inflamed | Cross-sectional | 16S rDNA V1–V2 region sequencing | Bacteria | CD
|
| Park et al. [149] | 2020 | No naïve | 370 | NA | NA | 740 | Active/Inactive | Stool | NA | Longitudinal | 16S rDNA V3–V4 region sequencing | Bacteria | CD
|
| Clooney et al. [150] | 2020 | No naïve | 303 | 228 | NA | 161 | Active/Inactive | Stool | NA | Longitudinal | 16S rDNA V3–V4 region sequencing | Bacteria | CD and UC
|
| Park et al. [151] | 2020 | No naïve | 10 | 6 | NA | 9Ur | Inactive | Stool | NA | Cross-sectional | 16S rDNA V3–V4 region sequencing | Bacteria | CD and UC
|
| Lo Sasso et al. [152] | 2020 | No naïve | 41 | 43 | NA | 42 | Active | Stool | NA | Cross-sectional | 16S rDNA V4 region and WGS | Bacteria | CD and UC
|
| Borren et al. [153] | 2020 | No naïve | 108 | 56 | NA | NA | Inactive | Stool | NA | Longitudinal | WGS | Bacteria | CD and UC
|
| Rubbens et al. [154] | 2020 | No naïve | 29 | NA | NA | 66 | Inactive | Stool | NA | Cross-sectional | Flow cytometry and 16S rDNA sequencing | Bacteria | CD
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aldars-García, L.; Chaparro, M.; Gisbert, J.P. Systematic Review: The Gut Microbiome and Its Potential Clinical Application in Inflammatory Bowel Disease. Microorganisms 2021, 9, 977. https://doi.org/10.3390/microorganisms9050977
Aldars-García L, Chaparro M, Gisbert JP. Systematic Review: The Gut Microbiome and Its Potential Clinical Application in Inflammatory Bowel Disease. Microorganisms. 2021; 9(5):977. https://doi.org/10.3390/microorganisms9050977
Chicago/Turabian StyleAldars-García, Laila, María Chaparro, and Javier P. Gisbert. 2021. "Systematic Review: The Gut Microbiome and Its Potential Clinical Application in Inflammatory Bowel Disease" Microorganisms 9, no. 5: 977. https://doi.org/10.3390/microorganisms9050977
APA StyleAldars-García, L., Chaparro, M., & Gisbert, J. P. (2021). Systematic Review: The Gut Microbiome and Its Potential Clinical Application in Inflammatory Bowel Disease. Microorganisms, 9(5), 977. https://doi.org/10.3390/microorganisms9050977