
Human Gut Microbiome and Liver Diseases: From Correlation to Causation



Abstract
1. Introduction
2. Clinical Evidence of Microbiota Alterations in Liver Diseases
2.1. NAFLD

{kind=link}
{kind=link}
{kind=link}
| Study | Sample Types | Disease | Method | Major Findings |
|---|---|---|---|---|
| Mouzaki et al., 2013, Hepatology [30] | Stool | NAFLD | Quantitative real-time PCR | The first study assessing the microbiota in adult human NAFLD at different histological stages. Lower percentage of Bacteroidetes is associated with NASH. |
| Jerome et al., 2016, Hepatology [7] | Stool | NAFLD | 16s sequencing | Gut dysbiosis associates with the severity of NAFLD lesions. Bacteroides associated with NASH and Ruminococcus with fibrosis. |
| Loomba et al., 2017, Cell Metabolism [17] | Stool | NAFLD | Shotgun sequencing | A gut microbiome signature was identified to predict NAFLD with fibrosis. The abundance of Firmicutes and Proteobacteria was higher in mild/moderate fibrosis, and advanced fibrosis, separately. |
| Mutlu et al., 2012, Am J Physiol Gastrointest Liver Physiol [40] | Human sigmoid mucosa biopsy | ALD | Multitag-pyrosequencing | The first study showing the association between alcohol consumption and microbiome using non-culture method in human. Lower abundances of Bacteroidetes and higher ones of Proteobacteria was found in alcoholics. |
| Dubinkina et al., 2017, Microbiome [9] | Stool | ALD | Shotgun sequencing | Alcoholic dependence was inversely associated with Clostridiales. The expansion of Bifidobacterium and Lactobacillus was found in ALD patients, but the species were different between patients with and without cirrhosis. |
| Liu et al., 2004, Hepatology [16] | Stool | Cirrhosis | Quantitative bacteriological Culture | Early evidence of disturbed microecology in cirrhotic patients with MHE, with outgrowth of Escherichia coli and Staphylococcal species. |
| Chen et al., 2011, Hepatology [41] | Stool | Cirrhosis | 454 pyrosequencing 16s | The first major study of microbiome in cirrhosis. Fecal microbial diversity was lower in cirrhotic patients, with higher levels of Enterobacteriaceae and Streptococcaceae, and lower levels of Lachnospiraceae. |
| Bajaj et al. 2014, Journal of Hepatology [42] | Stool | Cirrhosis | Multi-tagged pyrosequencing | The first major study to compare the microbiome in compensated and decompensated cirrhosis. Introduction of CDR, which is associated with disease progression and prognostic. |
| Qin et al., 2014, Nature [43] | Stool | Cirrhosis | Metagenome sequencing | The major study characterized the gut microbiome in liver cirrhosis. Patient-enriched species are of buccal origin, with higher levels of Proteobacteria and Fusobacteria and fewer levels of Bacteroides. |
| Caussy C et al., 2019, Nat Commun. [44] | Stool | Cirrhosis (NAFLD-associated) | 16s sequencing | Bacterial signature was discovered to detect NAFLD-cirrhosis based on a prospective twin and family cohort. |
2.2. ALD
2.3. Cirrhosis
2.4. Liver Cancer
3. Microorganisms from Other Origins and Kingdoms beyond Fecal Bacteria
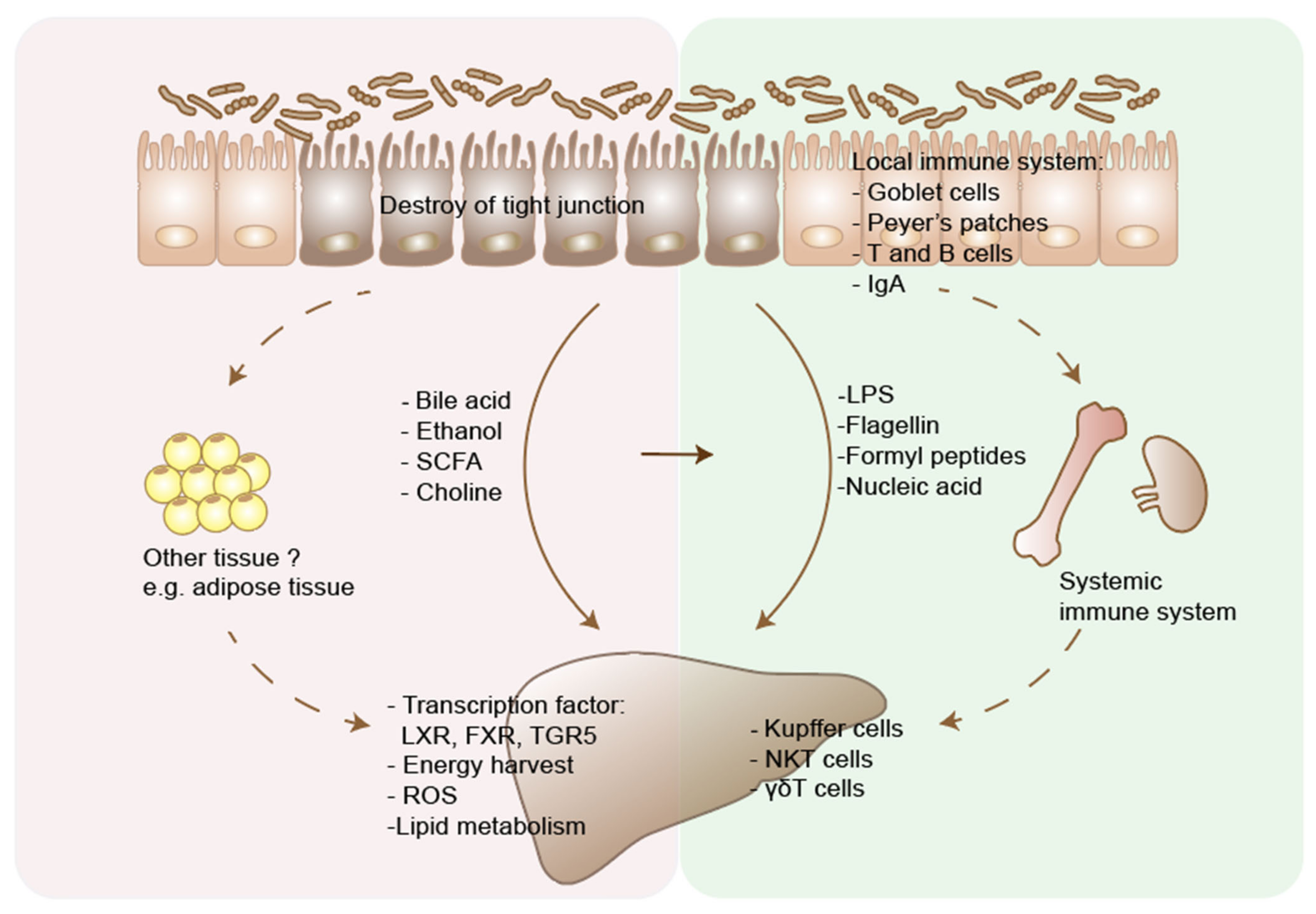
4. Mechanism of Action of Microbiome in Liver Disease Pathogenesis and Development
4.1. Metabolism
4.1.1. Bile Acids
4.1.2. Short Chain Fatty Acids
4.1.3. Ethanol
4.1.4. Choline
4.2. Immunity
4.2.1. The Impact of Commensal Bacterial on Local and Systemic Immunity
4.2.2. The Impact of Gut Microbiome and Its Derivatives on Innate and Adaptive Immunity
Innate Immunity
Adaptive Immunity
5. Gut Microbial Molecules in Liver Immune Modulation
6. Microbiota in Therapeutics of Liver Diseases
7. Potential Impact of Microbiota on Liver Injury in COVID-19 Patients
8. Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tripathi, A.; Debelius, J.; Brenner, D.A.; Karin, M.; Loomba, R.; Schnabl, B.; Knight, R. The gut-liver axis and the intersection with the microbiome. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Schnabl, B.; Brenner, D.A. Interactions between the intestinal microbiome and liver diseases. Gastroenterology 2014, 146, 1513–1524. [Google Scholar] [CrossRef]
- Tilg, H.; Cani, P.D.; Mayer, E.A. Gut microbiome and liver diseases. Gut 2016, 65, 2035–2044. [Google Scholar] [CrossRef]
- Marchesi, J.R.; Adams, D.H.; Fava, F.; Hermes, G.D.; Hirschfield, G.M.; Hold, G.; Quraishi, M.N.; Kinross, J.; Smidt, H.; Tuohy, K.M.; et al. The gut microbiota and host health: A new clinical frontier. Gut 2016, 65, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Dapito, D.H.; Mencin, A.; Gwak, G.Y.; Pradere, J.P.; Jang, M.K.; Mederacke, I.; Caviglia, J.M.; Khiabanian, H.; Adeyemi, A.; Bataller, R.; et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell 2012, 21, 504–516. [Google Scholar] [CrossRef]
- Boursier, J.; Mueller, O.; Barret, M.; Machado, M.; Fizanne, L.; Araujo-Perez, F.; Guy, C.D.; Seed, P.C.; Rawls, J.F.; David, L.A.; et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 2016, 63, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Ponziani, F.R.; Bhoori, S.; Castelli, C.; Putignani, L.; Rivoltini, L.; Del Chierico, F.; Sanguinetti, M.; Morelli, D.; Paroni Sterbini, F.; Petito, V.; et al. Hepatocellular Carcinoma Is Associated With Gut Microbiota Profile and Inflammation in Nonalcoholic Fatty Liver Disease. Hepatology 2019, 69, 107–120. [Google Scholar] [CrossRef]
- Dubinkina, V.B.; Tyakht, A.V.; Odintsova, V.Y.; Yarygin, K.S.; Kovarsky, B.A.; Pavlenko, A.V.; Ischenko, D.S.; Popenko, A.S.; Alexeev, D.G.; Taraskina, A.Y.; et al. Links of gut microbiota composition with alcohol dependence syndrome and alcoholic liver disease. Microbiome 2017, 5, 141. [Google Scholar] [CrossRef]
- Hoyles, L.; Fernandez-Real, J.M.; Federici, M.; Serino, M.; Abbott, J.; Charpentier, J.; Heymes, C.; Luque, J.L.; Anthony, E.; Barton, R.H.; et al. Molecular phenomics and metagenomics of hepatic steatosis in non-diabetic obese women. Nat. Med. 2018, 24, 1070–1080. [Google Scholar] [CrossRef]
- Ma, C.; Han, M.; Heinrich, B.; Fu, Q.; Zhang, Q.; Sandhu, M.; Agdashian, D.; Terabe, M.; Berzofsky, J.A.; Fako, V.; et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science 2018, 360. [Google Scholar] [CrossRef]
- Csak, T.; Ganz, M.; Pespisa, J.; Kodys, K.; Dolganiuc, A.; Szabo, G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology 2011, 54, 133–144. [Google Scholar] [CrossRef]
- Zuo, T.; Zhang, F.; Lui, G.C.Y.; Yeoh, Y.K.; Li, A.Y.L.; Zhan, H.; Wan, Y.; Chung, A.; Cheung, C.P.; Chen, N.; et al. Alterations in Gut Microbiota of Patients With COVID-19 During Time of Hospitalization. Gastroenterology 2020. [Google Scholar] [CrossRef] [PubMed]
- Phipps, M.M.; Barraza, L.H.; LaSota, E.D.; Sobieszczyk, M.E.; Pereira, M.R.; Zheng, E.X.; Fox, A.N.; Zucker, J.; Verna, E.C. Acute Liver Injury in COVID-19: Prevalence and Association with Clinical Outcomes in a Large US Cohort. Hepatology 2020. [Google Scholar] [CrossRef]
- Ferrere, G.; Wrzosek, L.; Cailleux, F.; Turpin, W.; Puchois, V.; Spatz, M.; Ciocan, D.; Rainteau, D.; Humbert, L.; Hugot, C.; et al. Fecal microbiota manipulation prevents dysbiosis and alcohol-induced liver injury in mice. J. Hepatol. 2017, 66, 806–815. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Duan, Z.P.; Ha, D.K.; Bengmark, S.; Kurtovic, J.; Riordan, S.M. Synbiotic modulation of gut flora: Effect on minimal hepatic encephalopathy in patients with cirrhosis. Hepatology 2004, 39, 1441–1449. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Seguritan, V.; Li, W.; Long, T.; Klitgord, N.; Bhatt, A.; Dulai, P.S.; Caussy, C.; Bettencourt, R.; Highlander, S.K.; et al. Gut Microbiome-Based Metagenomic Signature for Non-invasive Detection of Advanced Fibrosis in Human Nonalcoholic Fatty Liver Disease. Cell Metab. 2017, 25, 1054–1062.e1055. [Google Scholar] [CrossRef]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef]
- Wieland, A.; Frank, D.N.; Harnke, B.; Bambha, K. Systematic review: Microbial dysbiosis and nonalcoholic fatty liver disease. Aliment Pharmacol. Ther. 2015, 42, 1051–1063. [Google Scholar] [CrossRef]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef]
- Perry, R.J.; Peng, L.; Barry, N.A.; Cline, G.W.; Zhang, D.; Cardone, R.L.; Petersen, K.F.; Kibbey, R.G.; Goodman, A.L.; Shulman, G.I. Acetate mediates a microbiome-brain-beta-cell axis to promote metabolic syndrome. Nature 2016, 534, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Forslund, K.; Hildebrand, F.; Nielsen, T.; Falony, G.; Le Chatelier, E.; Sunagawa, S.; Prifti, E.; Vieira-Silva, S.; Gudmundsdottir, V.; Pedersen, H.K.; et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 2015, 528, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Schwiertz, A.; Taras, D.; Schafer, K.; Beijer, S.; Bos, N.A.; Donus, C.; Hardt, P.D. Microbiota and SCFA in lean and overweight healthy subjects. Obesity 2010, 18, 190–195. [Google Scholar] [CrossRef]
- Pedersen, H.K.; Gudmundsdottir, V.; Nielsen, H.B.; Hyotylainen, T.; Nielsen, T.; Jensen, B.A.; Forslund, K.; Hildebrand, F.; Prifti, E.; Falony, G.; et al. Human gut microbes impact host serum metabolome and insulin sensitivity. Nature 2016, 535, 376–381. [Google Scholar] [CrossRef]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef]
- Mouzaki, M.; Comelli, E.M.; Arendt, B.M.; Bonengel, J.; Fung, S.K.; Fischer, S.E.; McGilvray, I.D.; Allard, J.P. Intestinal microbiota in patients with nonalcoholic fatty liver disease. Hepatology 2013, 58, 120–127. [Google Scholar] [CrossRef]
- Wong, V.W.; Tse, C.H.; Lam, T.T.; Wong, G.L.; Chim, A.M.; Chu, W.C.; Yeung, D.K.; Law, P.T.; Kwan, H.S.; Yu, J.; et al. Molecular characterization of the fecal microbiota in patients with nonalcoholic steatohepatitis—A longitudinal study. PLoS ONE 2013, 8, e62885. [Google Scholar] [CrossRef]
- Yuan, J.; Chen, C.; Cui, J.; Lu, J.; Yan, C.; Wei, X.; Zhao, X.; Li, N.; Li, S.; Xue, G.; et al. Fatty Liver Disease Caused by High-Alcohol-Producing Klebsiella pneumoniae. Cell Metab. 2019, 30, 1172. [Google Scholar] [CrossRef] [PubMed]
- Spencer, M.D.; Hamp, T.J.; Reid, R.W.; Fischer, L.M.; Zeisel, S.H.; Fodor, A.A. Association between composition of the human gastrointestinal microbiome and development of fatty liver with choline deficiency. Gastroenterology 2011, 140, 976–986. [Google Scholar] [CrossRef]
- Raman, M.; Ahmed, I.; Gillevet, P.M.; Probert, C.S.; Ratcliffe, N.M.; Smith, S.; Greenwood, R.; Sikaroodi, M.; Lam, V.; Crotty, P.; et al. Fecal microbiome and volatile organic compound metabolome in obese humans with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2013, 11, 868.e861–875.e863. [Google Scholar] [CrossRef]
- Backhed, F.; Manchester, J.K.; Semenkovich, C.F.; Gordon, J.I. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc. Natl. Acad. Sci. USA 2007, 104, 979–984. [Google Scholar] [CrossRef]
- Rabot, S.; Membrez, M.; Bruneau, A.; Gerard, P.; Harach, T.; Moser, M.; Raymond, F.; Mansourian, R.; Chou, C.J. Germ-free C57BL/6J. mice are resistant to high-fat-diet-induced insulin resistance and have altered cholesterol metabolism. FASEB J. 2010, 24, 4948–4959. [Google Scholar] [CrossRef] [PubMed]
- Backhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Le Roy, T.; Llopis, M.; Lepage, P.; Bruneau, A.; Rabot, S.; Bevilacqua, C.; Martin, P.; Philippe, C.; Walker, F.; Bado, A.; et al. Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut 2013, 62, 1787–1794. [Google Scholar] [CrossRef]
- Mutlu, E.A.; Gillevet, P.M.; Rangwala, H.; Sikaroodi, M.; Naqvi, A.; Engen, P.A.; Kwasny, M.; Lau, C.K.; Keshavarzian, A. Colonic microbiome is altered in alcoholism. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G966–G978. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, F.; Lu, H.; Wang, B.; Chen, Y.; Lei, D.; Wang, Y.; Zhu, B.; Li, L. Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology 2011, 54, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, J.S.; Heuman, D.M.; Hylemon, P.B.; Sanyal, A.J.; White, M.B.; Monteith, P.; Noble, N.A.; Unser, A.B.; Daita, K.; Fisher, A.R.; et al. Altered profile of human gut microbiome is associated with cirrhosis and its complications. J. Hepatol. 2014, 60, 940–947. [Google Scholar] [CrossRef] [PubMed]
- Qin, N.; Yang, F.; Li, A.; Prifti, E.; Chen, Y.; Shao, L.; Guo, J.; Le Chatelier, E.; Yao, J.; Wu, L.; et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 2014, 513, 59–64. [Google Scholar] [CrossRef]
- Caussy, C.; Tripathi, A.; Humphrey, G.; Bassirian, S.; Singh, S.; Faulkner, C.; Bettencourt, R.; Rizo, E.; Richards, L.; Xu, Z.Z.; et al. A gut microbiome signature for cirrhosis due to nonalcoholic fatty liver disease. Nat. Commun. 2019, 10, 1406. [Google Scholar] [CrossRef]
- Seitz, H.K.; Bataller, R.; Cortez-Pinto, H.; Gao, B.; Gual, A.; Lackner, C.; Mathurin, P.; Mueller, S.; Szabo, G.; Tsukamoto, H. Alcoholic liver disease. Nat. Rev. Dis. Primers 2018, 4, 16. [Google Scholar] [CrossRef]
- Vassallo, G.; Mirijello, A.; Ferrulli, A.; Antonelli, M.; Landolfi, R.; Gasbarrini, A.; Addolorato, G. Review article: Alcohol and gut microbiota—The possible role of gut microbiota modulation in the treatment of alcoholic liver disease. Aliment Pharmacol. Ther. 2015, 41, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Bode, J.C.; Bode, C.; Heidelbach, R.; Durr, H.K.; Martini, G.A. Jejunal microflora in patients with chronic alcohol abuse. Hepatogastroenterology 1984, 31, 30–34. [Google Scholar]
- Tuomisto, S.; Pessi, T.; Collin, P.; Vuento, R.; Aittoniemi, J.; Karhunen, P.J. Changes in gut bacterial populations and their translocation into liver and ascites in alcoholic liver cirrhotics. BMC Gastroenterol. 2014, 14, 40. [Google Scholar] [CrossRef]
- Yan, A.W.; Fouts, D.E.; Brandl, J.; Starkel, P.; Torralba, M.; Schott, E.; Tsukamoto, H.; Nelson, K.E.; Brenner, D.A.; Schnabl, B. Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology 2011, 53, 96–105. [Google Scholar] [CrossRef]
- Canesso, M.C.; Lacerda, N.L.; Ferreira, C.M.; Gonçalves, J.L.; Almeida, D.; Gamba, C.; Cassali, G.; Pedroso, S.H.; Moreira, C.; Martins, F.S.; et al. Comparing the effects of acute alcohol consumption in germ-free and conventional mice: The role of the gut microbiota. BMC Microbiol. 2014, 14, 240. [Google Scholar] [CrossRef]
- Chen, P.; Schnabl, B. Host-microbiome interactions in alcoholic liver disease. Gut Liver 2014, 8, 237–241. [Google Scholar] [CrossRef]
- Bjarnason, I.; Peters, T.J.; Wise, R.J. The leaky gut of alcoholism: Possible route of entry for toxic compounds. Lancet 1984, 1, 179–182. [Google Scholar] [CrossRef]
- Keshavarzian, A.; Holmes, E.W.; Patel, M.; Iber, F.; Fields, J.Z.; Pethkar, S. Leaky gut in alcoholic cirrhosis: A possible mechanism for alcohol-induced liver damage. Am. J. Gastroenterol. 1999, 94, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Rao, R. Endotoxemia and gut barrier dysfunction in alcoholic liver disease. Hepatology 2009, 50, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Ferrier, L.; Berard, F.; Debrauwer, L.; Chabo, C.; Langella, P.; Bueno, L.; Fioramonti, J. Impairment of the intestinal barrier by ethanol involves enteric microflora and mast cell activation in rodents. Am. J. Pathol. 2006, 168, 1148–1154. [Google Scholar] [CrossRef] [PubMed]
- Keshavarzian, A.; Choudhary, S.; Holmes, E.W.; Yong, S.; Banan, A.; Jakate, S.; Fields, J.Z. Preventing gut leakiness by oats supplementation ameliorates alcohol-induced liver damage in rats. J. Pharmacol. Exp. Ther. 2001, 299, 442–448. [Google Scholar]
- Keshavarzian, A.; Farhadi, A.; Forsyth, C.B.; Rangan, J.; Jakate, S.; Shaikh, M.; Banan, A.; Fields, J.Z. Evidence that chronic alcohol exposure promotes intestinal oxidative stress, intestinal hyperpermeability and endotoxemia prior to development of alcoholic steatohepatitis in rats. J. Hepatol. 2009, 50, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Kirpich, I.A.; Solovieva, N.V.; Leikhter, S.N.; Shidakova, N.A.; Lebedeva, O.V.; Sidorov, P.I.; Bazhukova, T.A.; Soloviev, A.G.; Barve, S.S.; McClain, C.J.; et al. Probiotics restore bowel flora and improve liver enzymes in human alcohol-induced liver injury: A pilot study. Alcohol 2008, 42, 675–682. [Google Scholar] [CrossRef]
- Philips, C.A.; Pande, A.; Shasthry, S.M.; Jamwal, K.D.; Khillan, V.; Chandel, S.S.; Kumar, G.; Sharma, M.K.; Maiwall, R.; Jindal, A.; et al. Healthy Donor Fecal Microbiota Transplantation in Steroid-Ineligible Severe Alcoholic Hepatitis: A Pilot Study. Clin. Gastroenterol. Hepatol. 2017, 15, 600–602. [Google Scholar] [CrossRef] [PubMed]
- Grander, C.; Adolph, T.E.; Wieser, V.; Lowe, P.; Wrzosek, L.; Gyongyosi, B.; Ward, D.V.; Grabherr, F.; Gerner, R.R.; Pfister, A.; et al. Recovery of ethanol-induced Akkermansia muciniphila depletion ameliorates alcoholic liver disease. Gut 2018, 67, 891–901. [Google Scholar] [CrossRef]
- Ahluwalia, V.; Betrapally, N.S.; Hylemon, P.B.; White, M.B.; Gillevet, P.M.; Unser, A.B.; Fagan, A.; Daita, K.; Heuman, D.M.; Zhou, H.; et al. Impaired Gut-Liver-Brain Axis in Patients with Cirrhosis. Sci. Rep. 2016, 6, 26800. [Google Scholar] [CrossRef] [PubMed]
- Yanguas, S.C.; Cogliati, B.; Willebrords, J.; Maes, M.; Colle, I.; van den Bossche, B.; de Oliveira, C.; Andraus, W.; Alves, V.A.F.; Leclercq, I.; et al. Experimental models of liver fibrosis. Arch. Toxicol. 2016, 90, 1025–1048. [Google Scholar] [CrossRef] [PubMed]
- Fouts, D.E.; Torralba, M.; Nelson, K.E.; Brenner, D.A.; Schnabl, B. Bacterial translocation and changes in the intestinal microbiome in mouse models of liver disease. J. Hepatol. 2012, 56, 1283–1292. [Google Scholar] [CrossRef]
- Brenner, D.A.; Paik, Y.H.; Schnabl, B. Role of Gut Microbiota in Liver Disease. J. Clin. Gastroenterol. 2015, 49 (Suppl. 1), S25–S27. [Google Scholar] [CrossRef]
- Hugenholtz, F.; de Vos, W.M. Mouse models for human intestinal microbiota research: A critical evaluation. Cell Mol. Life Sci. 2018, 75, 149–160. [Google Scholar] [CrossRef]
- Walter, J.; Armet, A.M.; Finlay, B.B.; Shanahan, F. Establishing or Exaggerating Causality for the Gut Microbiome: Lessons from Human Microbiota-Associated Rodents. Cell 2020, 180, 221–232. [Google Scholar] [CrossRef]
- Fessler, J.; Matson, V.; Gajewski, T.F. Exploring the emerging role of the microbiome in cancer immunotherapy. J. Immunother. Cancer 2019, 7, 108. [Google Scholar] [CrossRef] [PubMed]
- Grat, M.; Wronka, K.M.; Krasnodebski, M.; Masior, L.; Lewandowski, Z.; Kosinska, I.; Grat, K.; Stypulkowski, J.; Rejowski, S.; Wasilewicz, M.; et al. Profile of Gut Microbiota Associated With the Presence of Hepatocellular Cancer in Patients With Liver Cirrhosis. Transpl. Proc. 2016, 48, 1687–1691. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Fan, X.G.; Wang, Z.M.; Zhou, J.H.; Tian, X.F.; Li, N. Identification of helicobacter species in human liver samples from patients with primary hepatocellular carcinoma. J. Clin. Pathol. 2004, 57, 1273–1277. [Google Scholar] [CrossRef]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef]
- Viaud, S.; Saccheri, F.; Mignot, G.; Yamazaki, T.; Daillere, R.; Hannani, D.; Enot, D.P.; Pfirschke, C.; Engblom, C.; Pittet, M.J.; et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science 2013, 342, 971–976. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.X.; Schwabe, R.F. The gut microbiome and liver cancer: Mechanisms and clinical translation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, J.S.; Betrapally, N.S.; Hylemon, P.B.; Heuman, D.M.; Daita, K.; White, M.B.; Unser, A.; Thacker, L.R.; Sanyal, A.J.; Kang, D.J.; et al. Salivary microbiota reflects changes in gut microbiota in cirrhosis with hepatic encephalopathy. Hepatology 2015, 62, 1260–1271. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Hylemon, P.B.; Ridlon, J.M.; Heuman, D.M.; Daita, K.; White, M.B.; Monteith, P.; Noble, N.A.; Sikaroodi, M.; Gillevet, P.M. Colonic mucosal microbiome differs from stool microbiome in cirrhosis and hepatic encephalopathy and is linked to cognition and inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G675–G685. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, J.S.; Betrapally, N.S.; Hylemon, P.B.; Thacker, L.R.; Daita, K.; Kang, D.J.; White, M.B.; Unser, A.B.; Fagan, A.; Gavis, E.A.; et al. Gut Microbiota Alterations can predict Hospitalizations in Cirrhosis Independent of Diabetes Mellitus. Sci. Rep. 2015, 5, 18559. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ji, F.; Guo, J.; Shi, D.; Fang, D.; Li, L. Dysbiosis of small intestinal microbiota in liver cirrhosis and its association with etiology. Sci. Rep. 2016, 6, 34055. [Google Scholar] [CrossRef]
- Lloyd-Price, J.; Abu-Ali, G.; Huttenhower, C. The healthy human microbiome. Genome Med. 2016, 8, 51. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Liu, E.J.; Kheradman, R.; Fagan, A.; Heuman, D.M.; White, M.; Gavis, E.A.; Hylemon, P.; Sikaroodi, M.; Gillevet, P.M. Fungal dysbiosis in cirrhosis. Gut 2018, 67, 1146–1154. [Google Scholar] [CrossRef]
- Yang, A.M.; Inamine, T.; Hochrath, K.; Chen, P.; Wang, L.; Llorente, C.; Bluemel, S.; Hartmann, P.; Xu, J.; Koyama, Y.; et al. Intestinal fungi contribute to development of alcoholic liver disease. J. Clin. Investig. 2017, 127, 2829–2841. [Google Scholar] [CrossRef]
- Lang, S.; Demir, M.; Martin, A.; Jiang, L.; Zhang, X.; Duan, Y.; Gao, B.; Wisplinghoff, H.; Kasper, P.; Roderburg, C.; et al. Intestinal Virome Signature Associated With Severity of Nonalcoholic Fatty Liver Disease. Gastroenterology 2020, 159, 1839–1852. [Google Scholar] [CrossRef]
- Jiang, L.; Lang, S.; Duan, Y.; Zhang, X.; Gao, B.; Chopyk, J.; Schwanemann, L.K.; Ventura-Cots, M.; Bataller, R.; Bosques-Padilla, F.; et al. Intestinal Virome in Patients With Alcoholic Hepatitis. Hepatology 2020, 72, 2182–2196. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Sikaroodi, M.; Shamsaddini, A.; Henseler, Z.; Santiago-Rodriguez, T.; Acharya, C.; Fagan, A.; Hylemon, P.B.; Fuchs, M.; Gavis, E.; et al. Interaction of bacterial metagenome and virome in patients with cirrhosis and hepatic encephalopathy. Gut 2020. [Google Scholar] [CrossRef]
- Duan, Y.; Llorente, C.; Lang, S.; Brandl, K.; Chu, H.; Jiang, L.; White, R.C.; Clarke, T.H.; Nguyen, K.; Torralba, M.; et al. Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature 2019, 575, 505–511. [Google Scholar] [CrossRef]
- Aguilar-Toalá, J.; Garcia-Varela, R.; Garcia, H.; Mata-Haro, V.; González-Córdova, A.; Vallejo-Cordoba, B.; Hernández-Mendoza, A. Postbiotics: An evolving term within the functional foods field. Trends Food Sci. Technol. 2018, 75, 105–114. [Google Scholar] [CrossRef]
- Haileselassie, Y.; Navis, M.; Vu, N.; Qazi, K.R.; Rethi, B.; Sverremark-Ekström, E. Postbiotic modulation of retinoic acid imprinted mucosal-like dendritic cells by probiotic Lactobacillus reuteri 17938 in vitro. Front. Immunol. 2016, 7, 96. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Guzmán, M.; Toral, M.; Romero, M.; Jiménez, R.; Galindo, P.; Sánchez, M.; Zarzuelo, M.J.; Olivares, M.; Gálvez, J.; Duarte, J. Antihypertensive effects of probiotics Lactobacillus strains in spontaneously hypertensive rats. Mol. Nutr. Food Res. 2015, 59, 2326–2336. [Google Scholar] [CrossRef]
- Xu, R.; Shang, N.; Li, P. In vitro and in vivo antioxidant activity of exopolysaccharide fractions from Bifidobacterium animalis RH. Anaerobe 2011, 17, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Jensen, B.A.H.; Holm, J.B.; Larsen, I.S.; von Burg, N.; Derer, S.; Sonne, S.B.; Paerregaard, S.I.; Damgaard, M.V.; Indrelid, S.A.; Rivollier, A.; et al. Lysates of Methylococcus capsulatus Bath induce a lean-like microbiota, intestinal FoxP3(+)RORgammat(+)IL-17(+) Tregs and improve metabolism. Nat. Commun. 2021, 12, 1093. [Google Scholar] [CrossRef] [PubMed]
- Russo, E.; Giudici, F.; Fiorindi, C.; Ficari, F.; Scaringi, S.; Amedei, A. Immunomodulating Activity and Therapeutic Effects of Short Chain Fatty Acids and Tryptophan Post-biotics in Inflammatory Bowel Disease. Front. Immunol. 2019, 10, 2754. [Google Scholar] [CrossRef]
- Homayouni Rad, A.; Aghebati Maleki, L.; Samadi Kafil, H.; Fathi Zavoshti, H.; Abbasi, A. Postbiotics as Promising Tools for Cancer Adjuvant Therapy. Adv. Pharm. Bull. 2021, 11, 1–5. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Bajaj, J.S. The human gut sterolbiome: Bile acid-microbiome endocrine aspects and therapeutics. Acta Pharm. Sin. B 2015, 5, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Abu-Shanab, A.; Quigley, E.M. The role of the gut microbiota in nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 691–701. [Google Scholar] [CrossRef] [PubMed]
- Lambertucci, F.; Arboatti, A.; Sedlmeier, M.G.; Motino, O.; Alvarez, M.L.; Ceballos, M.P.; Villar, S.R.; Roggero, E.; Monti, J.A.; Pisani, G.; et al. Disruption of tumor necrosis factor alpha receptor 1 signaling accelerates NAFLD progression in mice upon a high-fat diet. J. Nutr. Biochem. 2018, 58, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Gorjifard, S.; Goldszmid, R.S. Microbiota-myeloid cell crosstalk beyond the gut. J. Leukoc. Biol. 2016, 100, 865–879. [Google Scholar] [CrossRef]
- Vaikunthanathan, T.; Safinia, N.; Lombardi, G.; Lechler, R.I. Microbiota, immunity and the liver. Immunol. Lett. 2016, 171, 36–49. [Google Scholar] [CrossRef]
- Brestoff, J.R.; Artis, D. Commensal bacteria at the interface of host metabolism and the immune system. Nat. Immunol. 2013, 14, 676–684. [Google Scholar] [CrossRef]
- Zelante, T.; Iannitti, R.G.; Cunha, C.; De Luca, A.; Giovannini, G.; Pieraccini, G.; Zecchi, R.; D’Angelo, C.; Massi-Benedetti, C.; Fallarino, F.; et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 2013, 39, 372–385. [Google Scholar] [CrossRef]
- Sayin, S.I.; Wahlstrom, A.; Felin, J.; Jantti, S.; Marschall, H.U.; Bamberg, K.; Angelin, B.; Hyotylainen, T.; Oresic, M.; Backhed, F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013, 17, 225–235. [Google Scholar] [CrossRef]
- Hylemon, P.B.; Zhou, H.; Pandak, W.M.; Ren, S.; Gil, G.; Dent, P. Bile acids as regulatory molecules. J. Lipid Res. 2009, 50, 1509–1520. [Google Scholar] [CrossRef]
- Clausen, M.R.; Mortensen, P.B. Kinetic studies on colonocyte metabolism of short chain fatty acids and glucose in ulcerative colitis. Gut 1995, 37, 684–689. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Gambino, R.; Cassader, M. Obesity, diabetes, and gut microbiota: The hygiene hypothesis expanded? Diabetes Care 2010, 33, 2277–2284. [Google Scholar] [CrossRef]
- Den Besten, G.; Bleeker, A.; Gerding, A.; van Eunen, K.; Havinga, R.; van Dijk, T.H.; Oosterveer, M.H.; Jonker, J.W.; Groen, A.K.; Reijngoud, D.J.; et al. Short-Chain Fatty Acids Protect Against High-Fat Diet-Induced Obesity via a PPARgamma-Dependent Switch From Lipogenesis to Fat Oxidation. Diabetes 2015, 64, 2398–2408. [Google Scholar] [CrossRef]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef]
- Lieber, C.S. Alcoholic fatty liver: Its pathogenesis and mechanism of progression to inflammation and fibrosis. Alcohol 2004, 34, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Hoek, J.B.; Pastorino, J.G. Ethanol, oxidative stress, and cytokine-induced liver cell injury. Alcohol 2002, 27, 63–68. [Google Scholar] [CrossRef]
- Mehedint, M.G.; Zeisel, S.H. Choline’s role in maintaining liver function: New evidence for epigenetic mechanisms. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.M.; Liu, Y.; Zhou, R.F.; Chen, X.L.; Wang, C.; Tan, X.Y.; Wang, L.J.; Zheng, R.D.; Zhang, H.W.; Ling, W.H.; et al. Associations of gut-flora-dependent metabolite trimethylamine-N-oxide, betaine and choline with non-alcoholic fatty liver disease in adults. Sci. Rep. 2016, 6, 19076. [Google Scholar] [CrossRef]
- Abreu, M.T. Toll-like receptor signalling in the intestinal epithelium: How bacterial recognition shapes intestinal function. Nat. Rev. Immunol. 2010, 10, 131–144. [Google Scholar] [CrossRef]
- Mencin, A.; Kluwe, J.; Schwabe, R.F. Toll-like receptors as targets in chronic liver diseases. Gut 2009, 58, 704–720. [Google Scholar] [CrossRef]
- Jones, B.V.; Begley, M.; Hill, C.; Gahan, C.G.; Marchesi, J.R. Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc. Natl. Acad. Sci. USA 2008, 105, 13580–13585. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef] [PubMed]
- Kellogg, T.F.; Wostmann, B.S. Fecal neutral steroids and bile acids from germfree rats. J. Lipid Res. 1969, 10, 495–503. [Google Scholar] [CrossRef]
- Selwyn, F.P.; Csanaky, I.L.; Zhang, Y.; Klaassen, C.D. Importance of Large Intestine in Regulating Bile Acids and Glucagon-Like Peptide-1 in Germ-Free Mice. Drug Metab. Dispos. 2015, 43, 1544–1556. [Google Scholar] [CrossRef]
- Wahlstrom, A.; Sayin, S.I.; Marschall, H.U.; Backhed, F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016, 24, 41–50. [Google Scholar] [CrossRef]
- Devlin, A.S.; Fischbach, M.A. A biosynthetic pathway for a prominent class of microbiota-derived bile acids. Nat. Chem. Biol. 2015, 11, 685–690. [Google Scholar] [CrossRef]
- Hirano, S.; Masuda, N. Transformation of bile acids by Eubacterium lentum. Appl. Environ. Microbiol. 1981, 42, 912–915. [Google Scholar] [CrossRef]
- Hirano, S.; Masuda, N.; Oda, H.; Mukai, H. Transformation of bile acids by Clostridium perfringens. Appl. Environ. Microbiol. 1981, 42, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Kuipers, F.; Bloks, V.W.; Groen, A.K. Beyond intestinal soap--bile acids in metabolic control. Nat. Rev. Endocrinol. 2014, 10, 488–498. [Google Scholar] [CrossRef]
- Xie, G.; Wang, X.; Huang, F.; Zhao, A.; Chen, W.; Yan, J.; Zhang, Y.; Lei, S.; Ge, K.; Zheng, X.; et al. Dysregulated hepatic bile acids collaboratively promote liver carcinogenesis. Int. J. Cancer 2016, 139, 1764–1775. [Google Scholar] [CrossRef]
- Dermadi, D.; Valo, S.; Ollila, S.; Soliymani, R.; Sipari, N.; Pussila, M.; Sarantaus, L.; Linden, J.; Baumann, M.; Nystrom, M. Western Diet Deregulates Bile Acid Homeostasis, Cell Proliferation, and Tumorigenesis in Colon. Cancer Res. 2017, 77, 3352–3363. [Google Scholar] [CrossRef] [PubMed]
- Pai, R.; Tarnawski, A.S.; Tran, T. Deoxycholic acid activates beta-catenin signaling pathway and increases colon cell cancer growth and invasiveness. Mol. Biol. Cell 2004, 15, 2156–2163. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Ung, T.T.; Kim, N.H.; Jung, Y.D. Role of bile acids in colon carcinogenesis. World J. Clin. Cases 2018, 6, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, R.F.; Cole, C.E.; Hawk, E.T.; Lubet, R.A. Ursodeoxycholate/Sulindac combination treatment effectively prevents intestinal adenomas in a mouse model of polyposis. Gastroenterology 2004, 127, 838–844. [Google Scholar] [CrossRef]
- Clements, W.D.; Parks, R.; Erwin, P.; Halliday, M.I.; Barr, J.; Rowlands, B.J. Role of the gut in the pathophysiology of extrahepatic biliary obstruction. Gut 1996, 39, 587–593. [Google Scholar] [CrossRef]
- Kakiyama, G.; Pandak, W.M.; Gillevet, P.M.; Hylemon, P.B.; Heuman, D.M.; Daita, K.; Takei, H.; Muto, A.; Nittono, H.; Ridlon, J.M.; et al. Modulation of the fecal bile acid profile by gut microbiota in cirrhosis. J. Hepatol. 2013, 58, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Carrio, J.; Salazar, N.; Margolles, A.; Gonzalez, S.; Gueimonde, M.; de Los Reyes-Gavilan, C.G.; Suarez, A. Free Fatty Acids Profiles Are Related to Gut Microbiota Signatures and Short-Chain Fatty Acids. Front. Immunol. 2017, 8, 823. [Google Scholar] [CrossRef]
- Sharon, G.; Garg, N.; Debelius, J.; Knight, R.; Dorrestein, P.C.; Mazmanian, S.K. Specialized metabolites from the microbiome in health and disease. Cell Metab. 2014, 20, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Wachtershauser, A.; Stein, J. Rationale for the luminal provision of butyrate in intestinal diseases. Eur. J. Nutr. 2000, 39, 164–171. [Google Scholar] [CrossRef]
- Ziegler, K.; Kerimi, A.; Poquet, L.; Williamson, G. Butyric acid increases transepithelial transport of ferulic acid through upregulation of the monocarboxylate transporters SLC16A1 (MCT1) and SLC16A3 (MCT4). Arch. Biochem. Biophys. 2016, 599, 3–12. [Google Scholar] [CrossRef]
- Cresci, G.A.; Glueck, B.; McMullen, M.R.; Xin, W.; Allende, D.; Nagy, L.E. Prophylactic tributyrin treatment mitigates chronic-binge ethanol-induced intestinal barrier and liver injury. J. Gastroenterol. Hepatol. 2017, 32, 1587–1597. [Google Scholar] [CrossRef]
- Yamashita, H.; Maruta, H.; Jozuka, M.; Kimura, R.; Iwabuchi, H.; Yamato, M.; Saito, T.; Fujisawa, K.; Takahashi, Y.; Kimoto, M.; et al. Effects of acetate on lipid metabolism in muscles and adipose tissues of type 2 diabetic Otsuka Long-Evans Tokushima Fatty (OLETF) rats. Biosci. Biotechnol. Biochem. 2009, 73, 570–576. [Google Scholar] [CrossRef] [PubMed]
- Al-Lahham, S.; Roelofsen, H.; Rezaee, F.; Weening, D.; Hoek, A.; Vonk, R.; Venema, K. Propionic acid affects immune status and metabolism in adipose tissue from overweight subjects. Eur. J. Clin. Investig. 2012, 42, 357–364. [Google Scholar] [CrossRef]
- Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Cheng, J.; Duncan, A.E.; Kau, A.L.; Griffin, N.W.; Lombard, V.; Henrissat, B.; Bain, J.R.; et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 2013, 341, 1241214. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Yin, J.; Zhang, J.; Ward, R.E.; Martin, R.J.; Lefevre, M.; Cefalu, W.T.; Ye, J. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 2009, 58, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Vrieze, A.; Van Nood, E.; Holleman, F.; Salojarvi, J.; Kootte, R.S.; Bartelsman, J.F.; Dallinga-Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 2012, 143, 913–916.e917. [Google Scholar] [CrossRef]
- Chambers, E.S.; Viardot, A.; Psichas, A.; Morrison, D.J.; Murphy, K.G.; Zac-Varghese, S.E.; MacDougall, K.; Preston, T.; Tedford, C.; Finlayson, G.S.; et al. Effects of targeted delivery of propionate to the human colon on appetite regulation, body weight maintenance and adiposity in overweight adults. Gut 2015, 64, 1744–1754. [Google Scholar] [CrossRef] [PubMed]
- Szabo, G.; Mandrekar, P. Focus on: Alcohol and the liver. Alcohol Res. Health 2010, 33, 87–96. [Google Scholar]
- Chen, P.; Miyamoto, Y.; Mazagova, M.; Lee, K.C.; Eckmann, L.; Schnabl, B. Microbiota Protects Mice Against Acute Alcohol-Induced Liver Injury. Alcohol Clin. Exp. Res. 2015, 39, 2313–2323. [Google Scholar] [CrossRef]
- Hamarneh, S.R.; Kim, B.M.; Kaliannan, K.; Morrison, S.A.; Tantillo, T.J.; Tao, Q.; Mohamed, M.M.R.; Ramirez, J.M.; Karas, A.; Liu, W.; et al. Intestinal Alkaline Phosphatase Attenuates Alcohol-Induced Hepatosteatosis in Mice. Dig. Dis. Sci. 2017, 62, 2021–2034. [Google Scholar] [CrossRef]
- Rao, R.K. Acetaldehyde-induced barrier disruption and paracellular permeability in Caco-2 cell monolayer. Methods Mol. Biol. 2008, 447, 171–183. [Google Scholar] [CrossRef]
- Uesugi, T.; Froh, M.; Arteel, G.E.; Bradford, B.U.; Thurman, R.G. Toll-like receptor 4 is involved in the mechanism of early alcohol-induced liver injury in mice. Hepatology 2001, 34, 101–108. [Google Scholar] [CrossRef]
- Adachi, Y.; Moore, L.E.; Bradford, B.U.; Gao, W.; Thurman, R.G. Antibiotics prevent liver injury in rats following long-term exposure to ethanol. Gastroenterology 1995, 108, 218–224. [Google Scholar] [CrossRef]
- Elamin, E.E.; Masclee, A.A.; Dekker, J.; Jonkers, D.M. Ethanol metabolism and its effects on the intestinal epithelial barrier. Nutr. Rev. 2013, 71, 483–499. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Cope, K.; Risby, T.H.; Diehl, A.M. Obesity and female gender increase breath ethanol concentration: Potential implications for the pathogenesis of nonalcoholic steatohepatitis. Am. J. Gastroenterol. 2001, 96, 1200–1204. [Google Scholar] [CrossRef]
- Cope, K.; Risby, T.; Diehl, A.M. Increased gastrointestinal ethanol production in obese mice: Implications for fatty liver disease pathogenesis. Gastroenterology 2000, 119, 1340–1347. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, S.H.; da Costa, K.A. Choline: An essential nutrient for public health. Nutr. Rev. 2009, 67, 615–623. [Google Scholar] [CrossRef]
- Han, J.; Dzierlenga, A.L.; Lu, Z.; Billheimer, D.D.; Torabzadeh, E.; Lake, A.D.; Li, H.; Novak, P.; Shipkova, P.; Aranibar, N.; et al. Metabolomic profiling distinction of human nonalcoholic fatty liver disease progression from a common rat model. Obesity 2017, 25, 1069–1076. [Google Scholar] [CrossRef]
- Muraki, Y.; Makita, Y.; Yamasaki, M.; Amano, Y.; Matsuo, T. Elevation of liver endoplasmic reticulum stress in a modified choline-deficient l-amino acid-defined diet-fed non-alcoholic steatohepatitis mouse model. Biochem. Biophys. Res. Commun. 2017, 486, 632–638. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, S.H.; Wishnok, J.S.; Blusztajn, J.K. Formation of methylamines from ingested choline and lecithin. J. Pharmacol. Exp. Ther. 1983, 225, 320–324. [Google Scholar]
- Al-Waiz, M.; Mikov, M.; Mitchell, S.C.; Smith, R.L. The exogenous origin of trimethylamine in the mouse. Metabolism 1992, 41, 135–136. [Google Scholar] [CrossRef]
- Lang, D.H.; Yeung, C.K.; Peter, R.M.; Ibarra, C.; Gasser, R.; Itagaki, K.; Philpot, R.M.; Rettie, A.E. Isoform specificity of trimethylamine N-oxygenation by human flavin-containing monooxygenase (FMO) and P450 enzymes: Selective catalysis by FMO3. Biochem. Pharmacol. 1998, 56, 1005–1012. [Google Scholar] [CrossRef]
- Tang, W.H.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N. Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef]
- Arslan, N. Obesity, fatty liver disease and intestinal microbiota. World J. Gastroenterol. 2014, 20, 16452–16463. [Google Scholar] [CrossRef]
- Dumas, M.E.; Barton, R.H.; Toye, A.; Cloarec, O.; Blancher, C.; Rothwell, A.; Fearnside, J.; Tatoud, R.; Blanc, V.; Lindon, J.C.; et al. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc. Natl. Acad. Sci. USA 2006, 103, 12511–12516. [Google Scholar] [CrossRef] [PubMed]
- Odamaki, T.; Kato, K.; Sugahara, H.; Hashikura, N.; Takahashi, S.; Xiao, J.Z.; Abe, F.; Osawa, R. Age-related changes in gut microbiota composition from newborn to centenarian: A cross-sectional study. BMC Microbiol. 2016, 16, 90. [Google Scholar] [CrossRef]
- Francino, M.P. Antibiotics and the Human Gut Microbiome: Dysbioses and Accumulation of Resistances. Front. Microbiol. 2015, 6, 1543. [Google Scholar] [CrossRef]
- Wu, H.; Esteve, E.; Tremaroli, V.; Khan, M.T.; Caesar, R.; Manneras-Holm, L.; Stahlman, M.; Olsson, L.M.; Serino, M.; Planas-Felix, M.; et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat. Med. 2017, 23, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.A.M.; Aronoff, D.M. The influence of non-steroidal anti-inflammatory drugs on the gut microbiome. Clin. Microbiol. Infect 2016, 22, 178.e1–e178.e9. [Google Scholar] [CrossRef]
- Hill, D.A.; Siracusa, M.C.; Abt, M.C.; Kim, B.S.; Kobuley, D.; Kubo, M.; Kambayashi, T.; Larosa, D.F.; Renner, E.D.; Orange, J.S.; et al. Commensal bacteria-derived signals regulate basophil hematopoiesis and allergic inflammation. Nat. Med. 2012, 18, 538–546. [Google Scholar] [CrossRef]
- Deplancke, B.; Gaskins, H.R. Microbial modulation of innate defense: Goblet cells and the intestinal mucus layer. Am. J. Clin. Nutr. 2001, 73, 1131S–1141S. [Google Scholar] [CrossRef] [PubMed]
- Gordon, H.A.; Pesti, L. The gnotobiotic animal as a tool in the study of host microbial relationships. Bacteriol. Rev. 1971, 35, 390–429. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.; McCoy, K.D.; Macpherson, A.J. Use of axenic animals in studying the adaptation of mammals to their commensal intestinal microbiota. Semin. Immunol. 2007, 19, 59–69. [Google Scholar] [CrossRef]
- Lecuyer, E.; Rakotobe, S.; Lengline-Garnier, H.; Lebreton, C.; Picard, M.; Juste, C.; Fritzen, R.; Eberl, G.; McCoy, K.D.; Macpherson, A.J.; et al. Segmented filamentous bacterium uses secondary and tertiary lymphoid tissues to induce gut IgA and specific T helper 17 cell responses. Immunity 2014, 40, 608–620. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, P.D.; Cebra, J.J. The preference for switching to IgA expression by Peyer’s patch germinal center B cells is likely due to the intrinsic influence of their microenvironment. J. Immunol. 1991, 147, 4126–4135. [Google Scholar] [PubMed]
- Bouskra, D.; Brezillon, C.; Berard, M.; Werts, C.; Varona, R.; Boneca, I.G.; Eberl, G. Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature 2008, 456, 507–510. [Google Scholar] [CrossRef]
- Macpherson, A.J.; Martinic, M.M.; Harris, N. The functions of mucosal T cells in containing the indigenous commensal flora of the intestine. Cell Mol. Life Sci. 2002, 59, 2088–2096. [Google Scholar] [CrossRef]
- Macpherson, A.J.; Hunziker, L.; McCoy, K.; Lamarre, A. IgA responses in the intestinal mucosa against pathogenic and non-pathogenic microorganisms. Microbes Infect. 2001, 3, 1021–1035. [Google Scholar] [CrossRef]
- Hall, J.A.; Bouladoux, N.; Sun, C.M.; Wohlfert, E.A.; Blank, R.B.; Zhu, Q.; Grigg, M.E.; Berzofsky, J.A.; Belkaid, Y. Commensal DNA limits regulatory T cell conversion and is a natural adjuvant of intestinal immune responses. Immunity 2008, 29, 637–649. [Google Scholar] [CrossRef]
- Uzbay, T. Germ-free animal experiments in the gut microbiota studies. Curr. Opin. Pharmacol. 2019, 49, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Chirlaque, C.; Aranda, C.J.; Ocon, B.; Capitan-Canadas, F.; Ortega-Gonzalez, M.; Carrero, J.J.; Suarez, M.D.; Zarzuelo, A.; Sanchez de Medina, F.; Martinez-Augustin, O. Germ-free and Antibiotic-treated Mice are Highly Susceptible to Epithelial Injury in DSS Colitis. J. Crohns Colitis 2016, 10, 1324–1335. [Google Scholar] [CrossRef] [PubMed]
- Al-Asmakh, M.; Zadjali, F. Use of Germ-Free Animal Models in Microbiota-Related Research. J. Microbiol. Biotechnol. 2015, 25, 1583–1588. [Google Scholar] [CrossRef]
- Tada, T.; Yamamura, S.; Kuwano, Y.; Abo, T. Level of myelopoiesis in the bone marrow is influenced by intestinal flora. Cell Immunol. 1996, 173, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Staber, F.G.; Tarcsay, L.; Dukor, P. Modulations of myelopoiesis in vivo by chemically pure preparations of cell wall components from gram-negative bacteria: Effects at different stages. Infect Immun. 1978, 20, 40–49. [Google Scholar] [CrossRef]
- Khosravi, A.; Yanez, A.; Price, J.G.; Chow, A.; Merad, M.; Goodridge, H.S.; Mazmanian, S.K. Gut microbiota promote hematopoiesis to control bacterial infection. Cell Host Microbe 2014, 15, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Menezes, J.S.; Umesaki, Y.; Mazmanian, S.K. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4615–4622. [Google Scholar] [CrossRef] [PubMed]
- Thaiss, C.A.; Zmora, N.; Levy, M.; Elinav, E. The microbiome and innate immunity. Nature 2016, 535, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Vijay-Kumar, M.; Aitken, J.D.; Carvalho, F.A.; Cullender, T.C.; Mwangi, S.; Srinivasan, S.; Sitaraman, S.V.; Knight, R.; Ley, R.E.; Gewirtz, A.T. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science 2010, 328, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, D.; Tang, M.S.; Bowcutt, R.; Loke, P.; Cadwell, K. Bacterial sensor Nod2 prevents inflammation of the small intestine by restricting the expansion of the commensal Bacteroides vulgatus. Immunity 2014, 41, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Macia, L.; Tan, J.; Vieira, A.T.; Leach, K.; Stanley, D.; Luong, S.; Maruya, M.; Ian McKenzie, C.; Hijikata, A.; Wong, C.; et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat. Commun. 2015, 6, 6734. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Thaiss, C.A.; Zeevi, D.; Dohnalova, L.; Zilberman-Schapira, G.; Mahdi, J.A.; David, E.; Savidor, A.; Korem, T.; Herzig, Y.; et al. Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell 2015, 163, 1428–1443. [Google Scholar] [CrossRef] [PubMed]
- Chudnovskiy, A.; Mortha, A.; Kana, V.; Kennard, A.; Ramirez, J.D.; Rahman, A.; Remark, R.; Mogno, I.; Ng, R.; Gnjatic, S.; et al. Host-Protozoan Interactions Protect from Mucosal Infections through Activation of the Inflammasome. Cell 2016, 167, 444–456 e414. [Google Scholar] [CrossRef] [PubMed]
- Gury-BenAri, M.; Thaiss, C.A.; Serafini, N.; Winter, D.R.; Giladi, A.; Lara-Astiaso, D.; Levy, M.; Salame, T.M.; Weiner, A.; David, E.; et al. The Spectrum and Regulatory Landscape of Intestinal Innate Lymphoid Cells Are Shaped by the Microbiome. Cell 2016, 166, 1231–1246.e1213. [Google Scholar] [CrossRef]
- Li, F.; Hao, X.; Chen, Y.; Bai, L.; Gao, X.; Lian, Z.; Wei, H.; Sun, R.; Tian, Z. The microbiota maintain homeostasis of liver-resident gammadeltaT-17 cells in a lipid antigen/CD1d-dependent manner. Nat. Commun. 2017, 7, 13839. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Littman, D.R. The microbiota in adaptive immune homeostasis and disease. Nature 2016, 535, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Peterson, D.A.; McNulty, N.P.; Guruge, J.L.; Gordon, J.I. IgA response to symbiotic bacteria as a mediator of gut homeostasis. Cell Host Microbe 2007, 2, 328–339. [Google Scholar] [CrossRef]
- Kunisawa, J.; Kiyono, H. Alcaligenes is Commensal Bacteria Habituating in the Gut-Associated Lymphoid Tissue for the Regulation of Intestinal IgA Responses. Front Immunol. 2012, 3, 65. [Google Scholar] [CrossRef]
- Bunker, J.J.; Flynn, T.M.; Koval, J.C.; Shaw, D.G.; Meisel, M.; McDonald, B.D.; Ishizuka, I.E.; Dent, A.L.; Wilson, P.C.; Jabri, B.; et al. Innate and Adaptive Humoral Responses Coat Distinct Commensal Bacteria with Immunoglobulin A. Immunity 2015, 43, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Pabst, O. New concepts in the generation and functions of IgA. Nat. Rev. Immunol. 2012, 12, 821–832. [Google Scholar] [CrossRef]
- Bemark, M.; Boysen, P.; Lycke, N.Y. Induction of gut IgA production through T cell-dependent and T cell-independent pathways. Ann. N. Y. Acad. Sci. 2012, 1247, 97–116. [Google Scholar] [CrossRef] [PubMed]
- Macpherson, A.J.; Gatto, D.; Sainsbury, E.; Harriman, G.R.; Hengartner, H.; Zinkernagel, R.M. A primitive T cell-independent mechanism of intestinal mucosal IgA responses to commensal bacteria. Science 2000, 288, 2222–2226. [Google Scholar] [CrossRef] [PubMed]
- Moon, C.; Baldridge, M.T.; Wallace, M.A.; Burnham, C.A.D.; Virgin, H.W.; Stappenbeck, T.S. Vertically transmitted faecal IgA levels determine extra-chromosomal phenotypic variation. Nature 2015, 521, 90–93. [Google Scholar] [CrossRef]
- Fransen, F.; Zagato, E.; Mazzini, E.; Fosso, B.; Manzari, C.; El Aidy, S.; Chiavelli, A.; D’Erchia, A.M.; Sethi, M.K.; Pabst, O.; et al. BALB/c and C57BL/6 Mice Differ in Polyreactive IgA Abundance, which Impacts the Generation of Antigen-Specific IgA and Microbiota Diversity. Immunity 2015, 43, 527–540. [Google Scholar] [CrossRef]
- Viladomiu, M.; Kivolowitz, C.; Abdulhamid, A.; Dogan, B.; Victorio, D.; Castellanos, J.G.; Woo, V.; Teng, F.; Tran, N.L.; Sczesnak, A.; et al. IgA-coated E. coli enriched in Crohn’s disease spondyloarthritis promote TH17-dependent inflammation. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Ando, M.; Kamada, N.; Nagano, Y.; Narushima, S.; Suda, W.; Imaoka, A.; Setoyama, H.; Nagamori, T.; et al. Th17 Cell Induction by Adhesion of Microbes to Intestinal Epithelial Cells. Cell 2015, 163, 367–380. [Google Scholar] [CrossRef]
- Britton, G.J.; Contijoch, E.J.; Mogno, I.; Vennaro, O.H.; Llewellyn, S.R.; Ng, R.; Li, Z.; Mortha, A.; Merad, M.; Das, A.; et al. Microbiotas from Humans with Inflammatory Bowel Disease Alter the Balance of Gut Th17 and RORgammat(+) Regulatory T Cells and Exacerbate Colitis in Mice. Immunity 2019, 50, 212–224.e214. [Google Scholar] [CrossRef] [PubMed]
- Tanoue, T.; Atarashi, K.; Honda, K. Development and maintenance of intestinal regulatory T cells. Nat. Rev. Immunol. 2016, 16, 295–309. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 2011, 331, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Round, J.L.; Mazmanian, S.K. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc. Natl. Acad. Sci. USA 2010, 107, 12204–12209. [Google Scholar] [CrossRef]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly, Y.M.; Glickman, J.N.; Garrett, W.S. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kim, M.; Kang, S.G.; Jannasch, A.H.; Cooper, B.; Patterson, J.; Kim, C.H. Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR-S6K pathway. Mucosal Immunol. 2015, 8, 80–93. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- Weiss, D.S. The great escape: Microbiotal LPS takes a toll on the liver. Cancer Prev. Res. 2012, 5, 1078–1080. [Google Scholar] [CrossRef]
- Carpino, G.; Del Ben, M.; Pastori, D.; Carnevale, R.; Baratta, F.; Overi, D.; Francis, H.; Cardinale, V.; Onori, P.; Safarikia, S.; et al. Increased Liver Localization of Lipopolysaccharides in Human and Experimental NAFLD. Hepatology 2019. [Google Scholar] [CrossRef]
- Szabo, G.; Velayudham, A.; Romics, L., Jr.; Mandrekar, P. Modulation of non-alcoholic steatohepatitis by pattern recognition receptors in mice: The role of toll-like receptors 2 and 4. Alcohol Clin. Exp. Res. 2005, 29, 140S–145S. [Google Scholar] [CrossRef]
- Ramos, H.C.; Rumbo, M.; Sirard, J.C. Bacterial flagellins: Mediators of pathogenicity and host immune responses in mucosa. Trends Microbiol. 2004, 12, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Liu, F.; Yang, J.; Zhong, M.; Zhang, E.; Li, Y.; Zhou, D.; Cao, Y.; Li, W.; Yu, J.; et al. Over-activation of TLR5 signaling by high-dose flagellin induces liver injury in mice. Cell Mol. Immunol. 2015, 12, 729–742. [Google Scholar] [CrossRef]
- Weiss, E.; Kretschmer, D. Formyl-Peptide Receptors in Infection, Inflammation, and Cancer. Trends Immunol. 2018, 39, 815–829. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Chen, K.; Yoshimura, T.; Liu, Y.; Gong, W.; Wang, A.; Gao, J.L.; Murphy, P.M.; Wang, J.M. Formylpeptide receptors are critical for rapid neutrophil mobilization in host defense against Listeria monocytogenes. Sci. Rep. 2012, 2, 786. [Google Scholar] [CrossRef]
- Pasare, C.; Medzhitov, R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science 2003, 299, 1033–1036. [Google Scholar] [CrossRef]
- Roh, Y.S.; Zhang, B.; Loomba, R.; Seki, E. TLR2 and TLR9 contribute to alcohol-mediated liver injury through induction of CXCL1 and neutrophil infiltration. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G30–G41. [Google Scholar] [CrossRef]
- Bergheim, I.; Weber, S.; Vos, M.; Kramer, S.; Volynets, V.; Kaserouni, S.; McClain, C.J.; Bischoff, S.C. Antibiotics protect against fructose-induced hepatic lipid accumulation in mice: Role of endotoxin. J. Hepatol. 2008, 48, 983–992. [Google Scholar] [CrossRef]
- Li, Z.; Yang, S.; Lin, H.; Huang, J.; Watkins, P.A.; Moser, A.B.; Desimone, C.; Song, X.Y.; Diehl, A.M. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology 2003, 37, 343–350. [Google Scholar] [CrossRef]
- Cani, P.D.; Neyrinck, A.M.; Fava, F.; Knauf, C.; Burcelin, R.G.; Tuohy, K.M.; Gibson, G.R.; Delzenne, N.M. Selective increases of bifidobacteria in gut microflora improve high-fat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia 2007, 50, 2374–2383. [Google Scholar] [CrossRef]
- Wagnerberger, S.; Spruss, A.; Kanuri, G.; Stahl, C.; Schroder, M.; Vetter, W.; Bischoff, S.C.; Bergheim, I. Lactobacillus casei Shirota protects from fructose-induced liver steatosis: A mouse model. J. Nutr. Biochem. 2013, 24, 531–538. [Google Scholar] [CrossRef]
- Esposito, E.; Iacono, A.; Bianco, G.; Autore, G.; Cuzzocrea, S.; Vajro, P.; Canani, R.B.; Calignano, A.; Raso, G.M.; Meli, R. Probiotics reduce the inflammatory response induced by a high-fat diet in the liver of young rats. J. Nutr. 2009, 139, 905–911. [Google Scholar] [CrossRef]
- Daubioul, C.A.; Horsmans, Y.; Lambert, P.; Danse, E.; Delzenne, N.M. Effects of oligofructose on glucose and lipid metabolism in patients with nonalcoholic steatohepatitis: Results of a pilot study. Eur. J. Clin. Nutr. 2005, 59, 723–726. [Google Scholar] [CrossRef]
- Jena, P.K.; Sheng, L.; Nagar, N.; Wu, C.; Barile, D.; Mills, D.A.; Wan, Y.Y. Synbiotics Bifidobacterium infantis and milk oligosaccharides are effective in reversing cancer-prone nonalcoholic steatohepatitis using western diet-fed FXR knockout mouse models. J. Nutr. Biochem. 2018, 57, 246–254. [Google Scholar] [CrossRef]
- Dhiman, R.K.; Rana, B.; Agrawal, S.; Garg, A.; Chopra, M.; Thumburu, K.K.; Khattri, A.; Malhotra, S.; Duseja, A.; Chawla, Y.K. Probiotic VSL#3 reduces liver disease severity and hospitalization in patients with cirrhosis: A randomized, controlled trial. Gastroenterology 2014, 147, 1327–1337.e1323. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Kassam, Z.; Fagan, A.; Gavis, E.A.; Liu, E.; Cox, I.J.; Kheradman, R.; Heuman, D.; Wang, J.; Gurry, T.; et al. Fecal microbiota transplant from a rational stool donor improves hepatic encephalopathy: A randomized clinical trial. Hepatology 2017, 66, 1727–1738. [Google Scholar] [CrossRef]
- Wang, W.W.; Zhang, Y.; Huang, X.B.; You, N.; Zheng, L.; Li, J. Fecal microbiota transplantation prevents hepatic encephalopathy in rats with carbon tetrachloride-induced acute hepatic dysfunction. World J. Gastroenterol. 2017, 23, 6983–6994. [Google Scholar] [CrossRef] [PubMed]
- Vetizou, M.; Pitt, J.M.; Daillere, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.; et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [PubMed]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef]
- Berlin, D.A.; Gulick, R.M.; Martinez, F.J. Severe Covid-19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Fu, Y.; Zhu, R.; Bai, T.; Han, P.; He, Q.; Jing, M.; Xiong, X.; Zhao, X.; Quan, R.; Chen, C.; et al. Clinical Features of COVID-19-Infected Patients With Elevated Liver Biochemistries: A Multicenter, Retrospective Study. Hepatology 2020. [Google Scholar] [CrossRef]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk Factors Associated With Acute Respiratory Distress Syndrome and Death in Patients With Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern Med. 2020. [Google Scholar] [CrossRef]
- Sun, J.; Aghemo, A.; Forner, A.; Valenti, L. COVID-19 and liver disease. Liver Int. 2020, 40, 1278–1281. [Google Scholar] [CrossRef]
- Geva-Zatorsky, N.; Sefik, E.; Kua, L.; Pasman, L.; Tan, T.G.; Ortiz-Lopez, A.; Yanortsang, T.B.; Yang, L.; Jupp, R.; Mathis, D.; et al. Mining the Human Gut Microbiota for Immunomodulatory Organisms. Cell 2017, 168, 928–943.e911. [Google Scholar] [CrossRef] [PubMed]
- Chai, X.; Hu, L.; Zhang, Y.; Han, W.; Lu, Z.; Ke, A.; Zhou, J.; Shi, G.; Fang, N.; Fan, J. Specific ACE2 expression in cholangiocytes may cause liver damage after 2019-nCoV infection. biorxiv 2020. [Google Scholar] [CrossRef]
- Aly, A.M.; Adel, A.; El-Gendy, A.O.; Essam, T.M.; Aziz, R.K. Gut microbiome alterations in patients with stage 4 hepatitis C. Gut Pathog. 2016, 8, 42. [Google Scholar] [CrossRef]
- Zuo, T.; Liu, Q.; Zhang, F.; Lui, G.C.; Tso, E.Y.; Yeoh, Y.K.; Chen, Z.; Boon, S.S.; Chan, F.K.; Chan, P.K.; et al. Depicting SARS-CoV-2 faecal viral activity in association with gut microbiota composition in patients with COVID-19. Gut 2020. [Google Scholar] [CrossRef] [PubMed]
- Zuo, T.; Zhan, H.; Zhang, F.; Liu, Q.; Tso, E.Y.K.; Lui, G.C.Y.; Chen, N.; Li, A.; Lu, W.; Chan, F.K.L.; et al. Alterations in Fecal Fungal Microbiome of Patients With COVID-19 During Time of Hospitalization until Discharge. Gastroenterology 2020. [Google Scholar] [CrossRef]
- Oh, T.G.; Kim, S.M.; Caussy, C.; Fu, T.; Guo, J.; Bassirian, S.; Singh, S.; Madamba, E.V.; Bettencourt, R.; Richards, L.; et al. A Universal Gut-Microbiome-Derived Signature Predicts Cirrhosis. Cell Metab. 2020. [Google Scholar] [CrossRef] [PubMed]
- Walsh, N.C.; Kenney, L.L.; Jangalwe, S.; Aryee, K.E.; Greiner, D.L.; Brehm, M.A.; Shultz, L.D. Humanized Mouse Models of Clinical Disease. Annu. Rev. Pathol. 2017, 12, 187–215. [Google Scholar] [CrossRef]
- Guo, J.; Li, Y.; Shan, Y.; Shu, C.; Wang, F.; Wang, X.; Zheng, G.; He, J.; Hu, Z.; Yang, Y.G. Humanized mice reveal an essential role for human hepatocytes in the development of the liver immune system. Cell Death Dis. 2018, 9, 667. [Google Scholar] [CrossRef]



| Microbial Metabolites or Components | Function of Microbiota | Effects | Mechanisms | References |
|---|---|---|---|---|
| Bile acids | Microbiota deconjugates primary bile acids and changes the primary/secondary ratio of bile acids. | Effects in the gut | Tauro-conjugated beta-muricholic acids (TβMCA) inhibit FXR-dependent Fgf15 expression in the ileum, which then increase bile acid synthesis in the liver. | [99] |
| Effects In the liver | Bile acids activate FXR, vitamin D receptor, and TGR5, and regulate the metabolism of glucose, fatty acid, triglyceride and VLDL. | [100] | ||
| Effects on immune system | Primary bile acids regulate CXCL16 level on liver sinusoidal endothelial cells, which controls the NKT cells accumulation and inhibit liver tumor growth. | [11] | ||
| Short chain fatty acids | Microbiota produce short chain fatty acids through fermentation of polysaccharides | Effects in the gut | Butyrates are energy sources for enterocytes and help maintain the integrity of the intestinal barrier. | [101] |
| SCFAs act on G-protein receptors (GPCRs) GPR41 and GPR43 on gut enteroendocrine L cells and enhance nutrient absorption. | [102] | |||
| Effects In the liver | SCFAs could increase hepatic lipid oxidation and lower hepatic lipid synthesis. | [103] | ||
| Effects on immune system | Butyrates suppress inflammation through inducing the differentiation of colonic Treg cells. | [104] | ||
| Ethanol | Microbiota contribute to the metabolism of ethanol into acetaldehyde and acetate. Microbiota also produce ethanol through fermentation of carbohydrates. | Effects in the gut | Acetaldehyde could significantly increase intestine permeability. | [55] |
| Effects In the liver | Alcohol and its metabolites cause damage to hepatocytes through generation of free radicals, which cause oxidative stress. | [105] | ||
| Effects on immune system | Ethanol-induced gut barrier dysfunction and translocation leads to activation of Kupffer cells, infiltrating neutrophils and macrophages, which release proinflammatory cytokines and cause parenchymal cell death. | [106] | ||
| Choline | Choline can be processed to trimethylamine (TMA) by intestinal bacteria, which can lead to reduced availability of dietary choline. | Effects In the liver | Choline deficiency inhibits VLDL excretion from the liver and leads to the hepatic accumulation of triglycerides. | [107] |
| Trimethylamine N-oxide (TMAO), derived from TMA, is associated with liver damage. However, a causal relationship between the two still needs to be clarified. | [108] | |||
| Pathogen-associated molecular patterns, including cell wall components and DNA | LPS is the cell component of gram-negative bacteria. Flagellin, the primary structural component of flagella. | Effects in the gut immune system | Activation of TLRs on intestinal epithelial cells promotes epithelial cell proliferation, secretion of IgA and antimicrobial peptides. | [109] |
| Effects In the liver immune system | Activation of TLRs on hepatic Kupffer cells and HSCs leads to inflammation and fibrosis, through inducing a range of cytokines, including IL-1, IL-6 and TNF. | [110] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, R.; Mao, Z.; Ye, X.; Zuo, T. Human Gut Microbiome and Liver Diseases: From Correlation to Causation. Microorganisms 2021, 9, 1017. https://doi.org/10.3390/microorganisms9051017
Li R, Mao Z, Ye X, Zuo T. Human Gut Microbiome and Liver Diseases: From Correlation to Causation. Microorganisms. 2021; 9(5):1017. https://doi.org/10.3390/microorganisms9051017
Chicago/Turabian StyleLi, Rui, Zhengsheng Mao, Xujun Ye, and Tao Zuo. 2021. "Human Gut Microbiome and Liver Diseases: From Correlation to Causation" Microorganisms 9, no. 5: 1017. https://doi.org/10.3390/microorganisms9051017
APA StyleLi, R., Mao, Z., Ye, X., & Zuo, T. (2021). Human Gut Microbiome and Liver Diseases: From Correlation to Causation. Microorganisms, 9(5), 1017. https://doi.org/10.3390/microorganisms9051017