
rt269I Type of Hepatitis B Virus (HBV) Polymerase versus rt269L Is More Prone to Mutations within HBV Genome in Chronic Patients Infected with Genotype C2: Evidence from Analysis of Full HBV Genotype C2 Genome

 , , and
, , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. HBV Sequence Acquisition and Processing
2.2. HBV Phylogenetic Analysis and Intergenotypic Recombination Analysis
2.3. Analysis of Mutation Rates Based on the HBV Full Genome and 5 HBV Regions (preS1, S, preC/C, X and Pol)
2.4. Statistical Analysis
3. Results
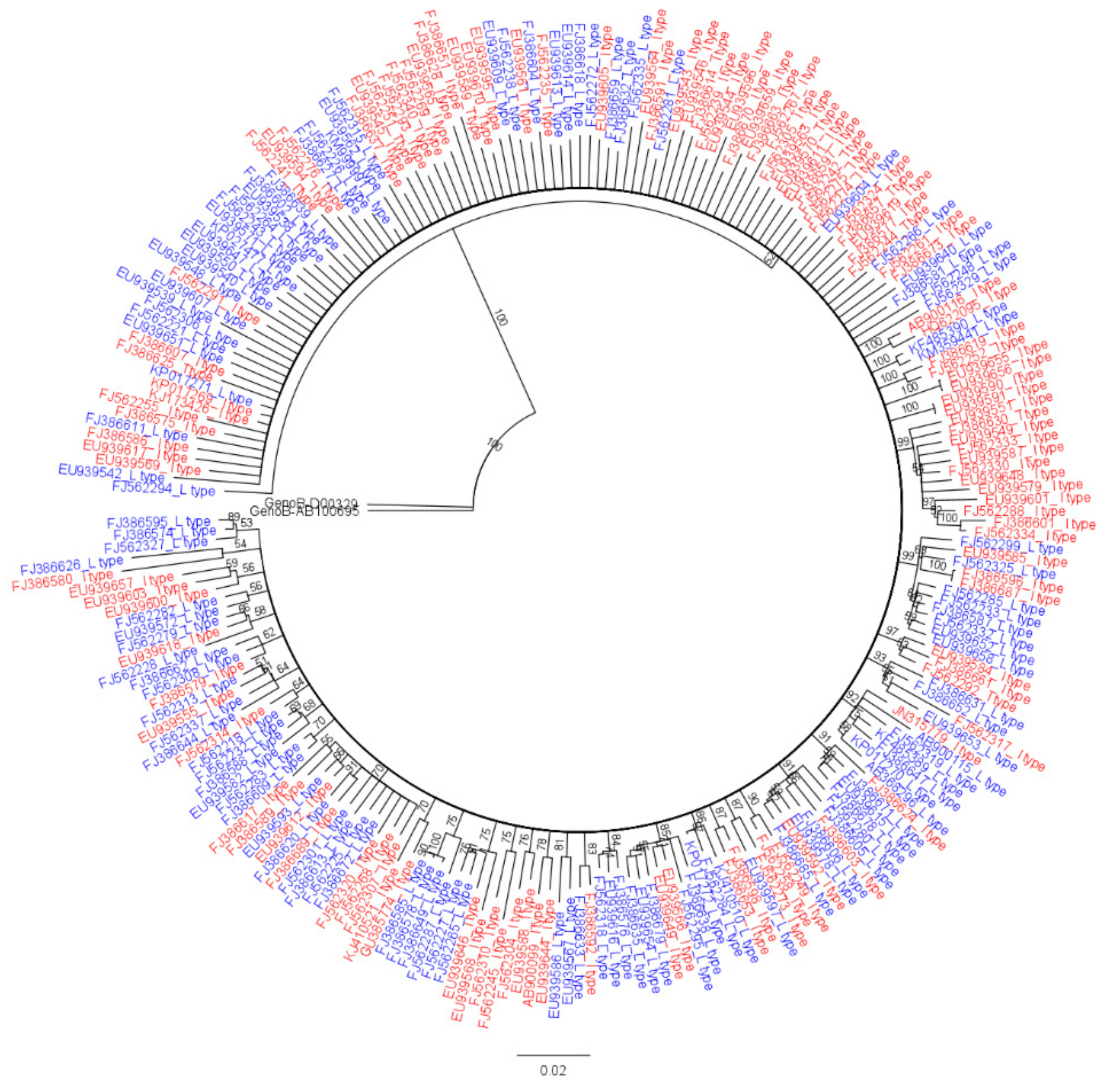
3.1. Genotype Determination of HBV Full Genome Sequences via Phylogenetic Analysis
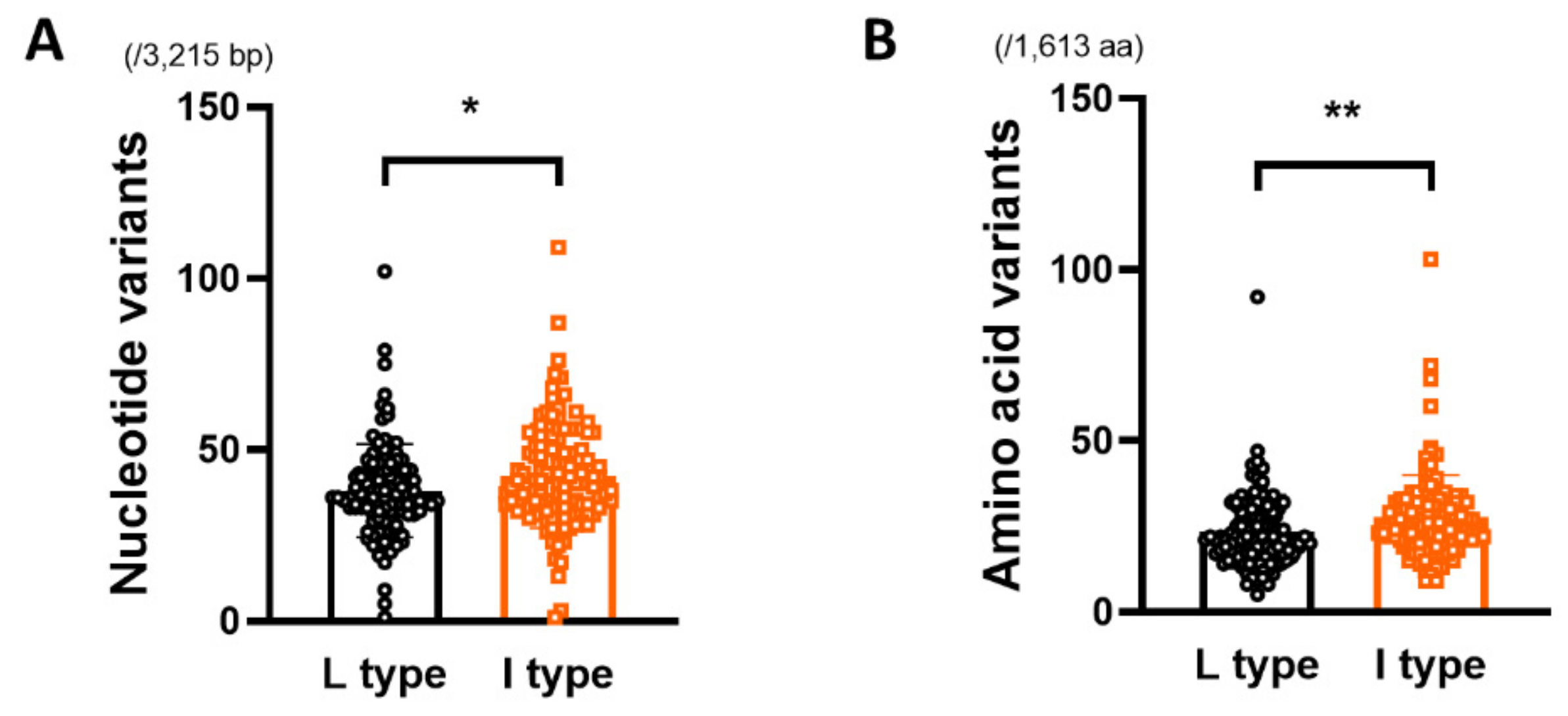
3.2. Comparison of Genetic Diversity of the HBV Full Genome between rt269L and rt269I
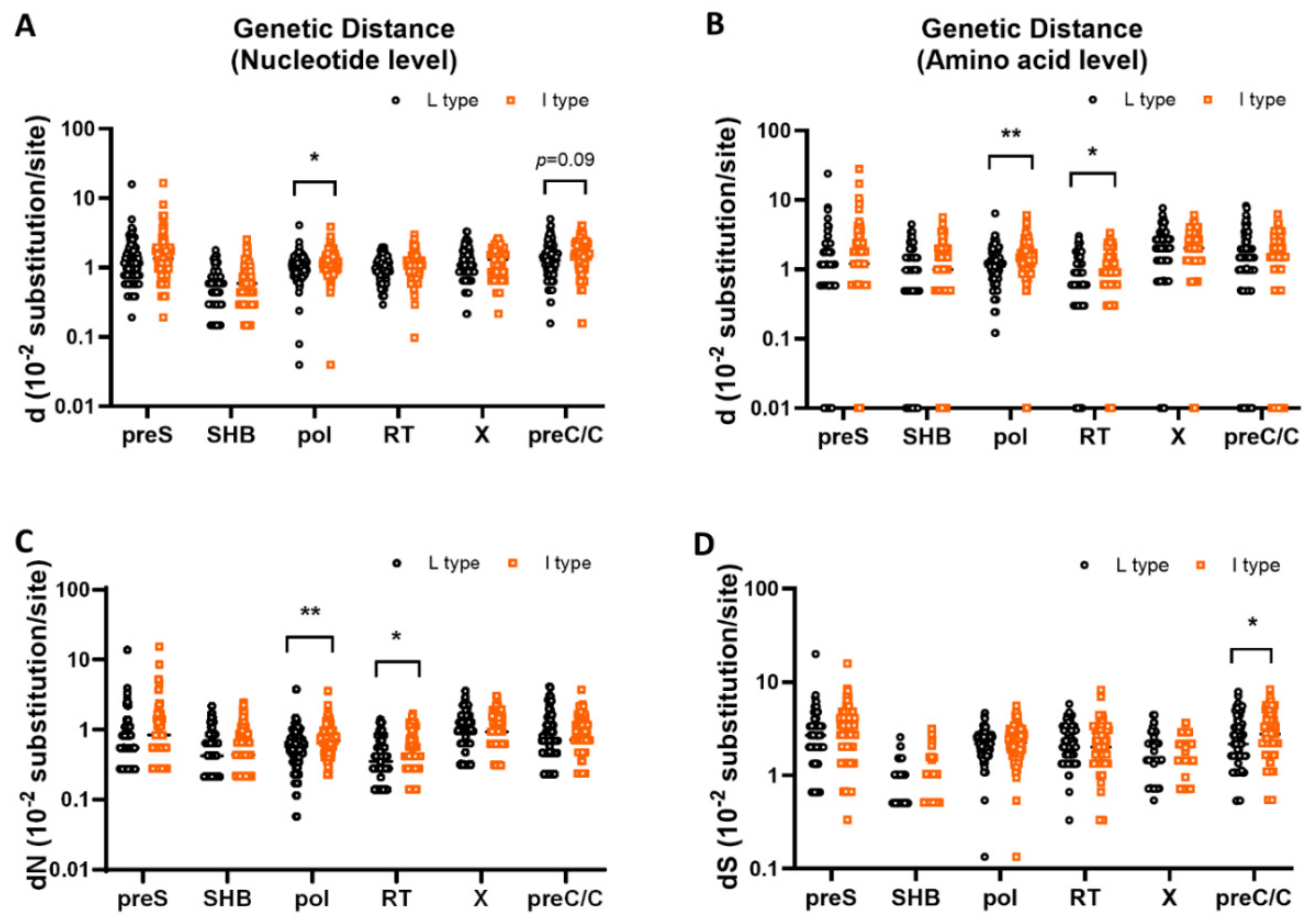
3.3. Comparison of Genetic Diversity in the PreS, S, X, PreC/C and Pol Regions between rt269L and rt269I
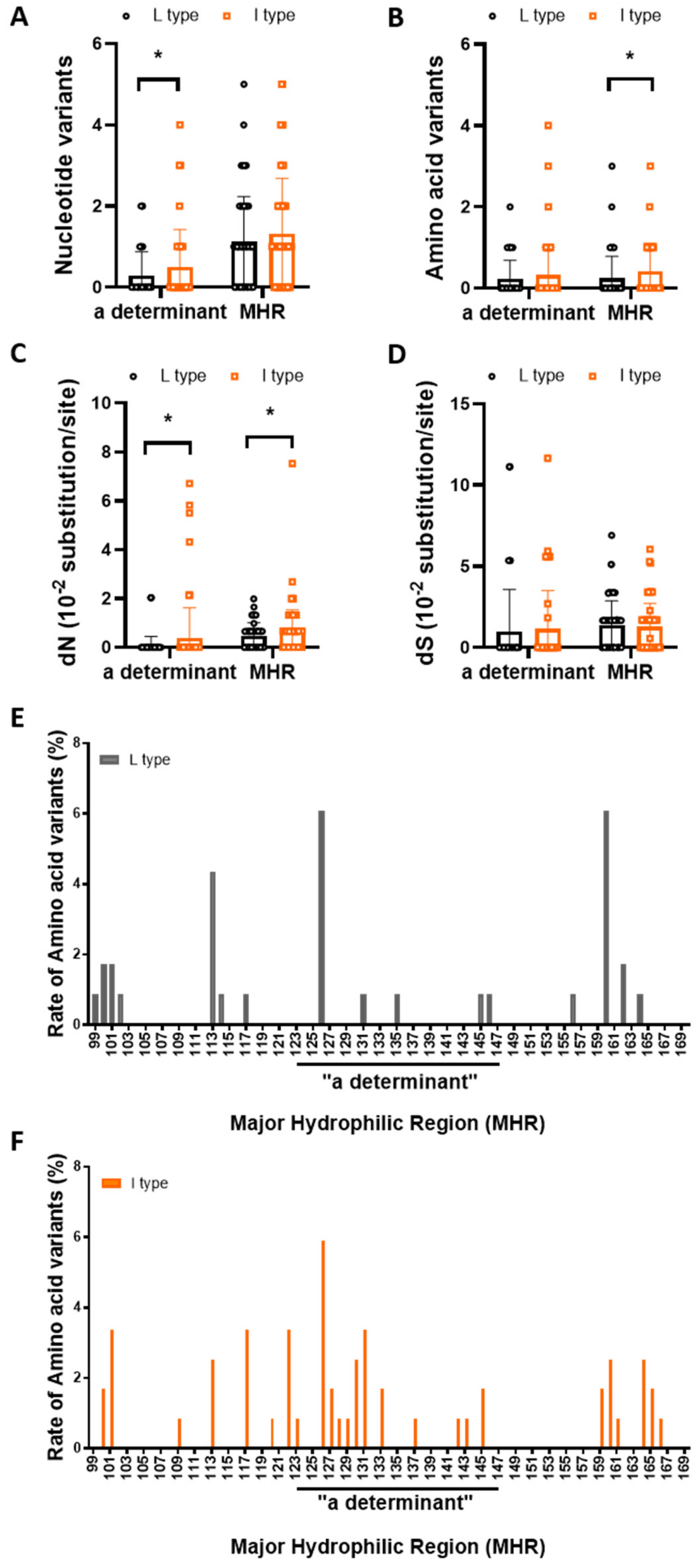
3.4. rt269I versus rt269L Is More Prone to NS Mutations in the MHR or “a” Determinant Region
3.5. Identification of Signature Nonsynonymous Mutations Specific to rt269L or rt269I
3.5.1. PreS and S Region
3.5.2. X Region
3.5.3. PreC/C region

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation Region | Amino Acid | Reference Strains | FDR Corrected p-Value (q-Value) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| L Type (n = 115) | I Type (n = 119) | Function | Ref | ||||||
| preS1 | I84T/M/L/V | Ile (I) Thr (T) Met (M) Leu (L) Val (V) | 99 (86.09%) 9 (7.83%) 1 (0.87%) 1 (0.87%) 2 (1.74%) | 82 (68.91%) 18 (15.13%) 3 (2.52%) 14 (11.76%) 2 (1.68%) | <0.001 c | Transactivator domain, Occult infection | [17] | ||
| A90T/V | Ala (A) Th r(T) Val (V) | 84 (73%) 31(27%) | 57(47.9%) 1 (0.8%) 61 (51.3%) | <0.001 c | |||||
| preS2 | F141L | Phe (F) Leu (L) | 100 (86.96%) 12 (10.43%) | 95 (79.83%) 24 (20.17%) | 0.046 a | HCC | [27] | ||
| S | sI68T | Ile (I) Thr (T) | 81 (70.43%) 34 (29.57%) | 103 (86.55%) 16 (13.45%) | 0.004 b | HCC | [57] | ||
| sL213I/S | Leu (L) Ile (I) Ser (S) | 110 (96.65%) 5 (4.35%) 0 (0.00%) | 105 (88.24%) 13 (10.92%) 1 (0.84%) | 0.054 | Disease progression | [58] | |||
| Terminal protein | D16A/E/N/V | Asp (D) Ala (A) Glu (E) Asn (N) Val (V) | 110 (95.65%) 1 (0.87%) 2 (1.74%) 1 (0.87%) 1 (0.87%) | 105 (88.24%) 3 (2.52%) 4 (3.36%) 3 (2.52%) 4 (3.36%) | 0.054 | N-terminal helix1 mutation | [59] | ||
| Spacer | S314P | Ser (S) Pro (P) | 78 (67.83%) 37 (32.17%) | 50 (42.01%) 69 (57.98%) | <0.001 c | ||||
| F321L | Phe (F) Leu (L) | 79 (68.70%) 36 (31.30%) | 51 (42.86%) 68 (57.14%) | <0.001 c | |||||
| Reverse transcriptase | rtN139K/H | Asn (N) Lys (K) His (H) | 112 (98.26%) 0(0.00%) 1 (1.74%) | 112 (94.96%) 2 (1.68%) 5 (4.20%) | 0.066 | Pretreatment mutation | [60] | ||
| rtM204I/V | Met(M) Ile (I) Val (V) | 111(96.52%) 4 (3.48%) 0 (0.00%) | 103 (86.55%) 14 (11.76%) 2 (1.68%) | 0.009 b | resistance to LMV, LdT, ADV, TNF | [61,62] | |||
| rtI224V | Ile (I) Val (V) | 110 (95.65%) 4 (3.48%) | 106 (89.08%) 13 (10.92%) | 0.042 a | Pretreatment mutation | [60] | |||
| RNaseH | D828A/V | Asp (D) Ala (A) Val (V) | 105(91.30%) 6(5.22%) 4 (3.48%) | 88 (73.95%) 5 (4.20%) 26 (21.85%) | 0.001 b | putative catalytic site mutants | [63] | ||
| HBx | V/L5M | Val (V) Leu (L) Met (M) | 98 (85.22%) 6 (5.22%) 10 (8.7%) | 92 (77.31%) 6 (5.04%) 21 (17.65%) | 0.054 | Independent predictors of HCC survival, miRNA binding site | [11] | ||
| T36P/S/A | Thr (T) Pro (P) Ser (S) Ala (A) | 57 (50%) 22 (19.3%) 22 (19.3%) 13 (11.4%) | 89 (75.42%) 7 (5.93%) 9 (7.63%) 13 (11.02%) | <0.001 c | B cell epitope | [64] | |||
| HBx | S101P/A | Ser (S) Ala (A) Pro (P) | 103(89.6%) 10 (8.7%) 1 (0.90%) | 78 (65.5%) 18 (15.1%) 23 (19.3%) | <0.001 c | ||||
| V131I | Val (V) Ile (I) | 30 (26.79%) 82(73.21%) | 14 (11.76%) 105 (88.24%) | 0.004 b | HCC | [65] | |||
| Precore | G1896A | Trp (W) Stop | 84 (73.04%) 31 (26.96%) | 61 (51.26%) 58 (48.74%) | <0.001 c | HBeAg-negative serostatus | [14] | ||
| Core | A2189T/C, C2191T | Ile (I) Phe (F) Leu (L) | 75 (65.22%) 5 (4.35%) 35 (30.43%) | 62 (52.10%) 4 (3.36%) 53 (44.54%) | 0.047 a | HBeAg-negative serostatus, HCC-related HBcAg mutation | [14,66] | ||
| C2444A/T, A2445G | Gln (Q) Tyr (Y) His (H) Stop | 111 (96.52%) 1 (0.87%) 0 (0.00%) 3 (2.61%) | 107 (89.92%) 2 (1.68%) 4 (3.36%) 6 (5.04%) | 0.068 | HCC-related HBcAg mutation | [14] | |||
3.5.4. Pol Region
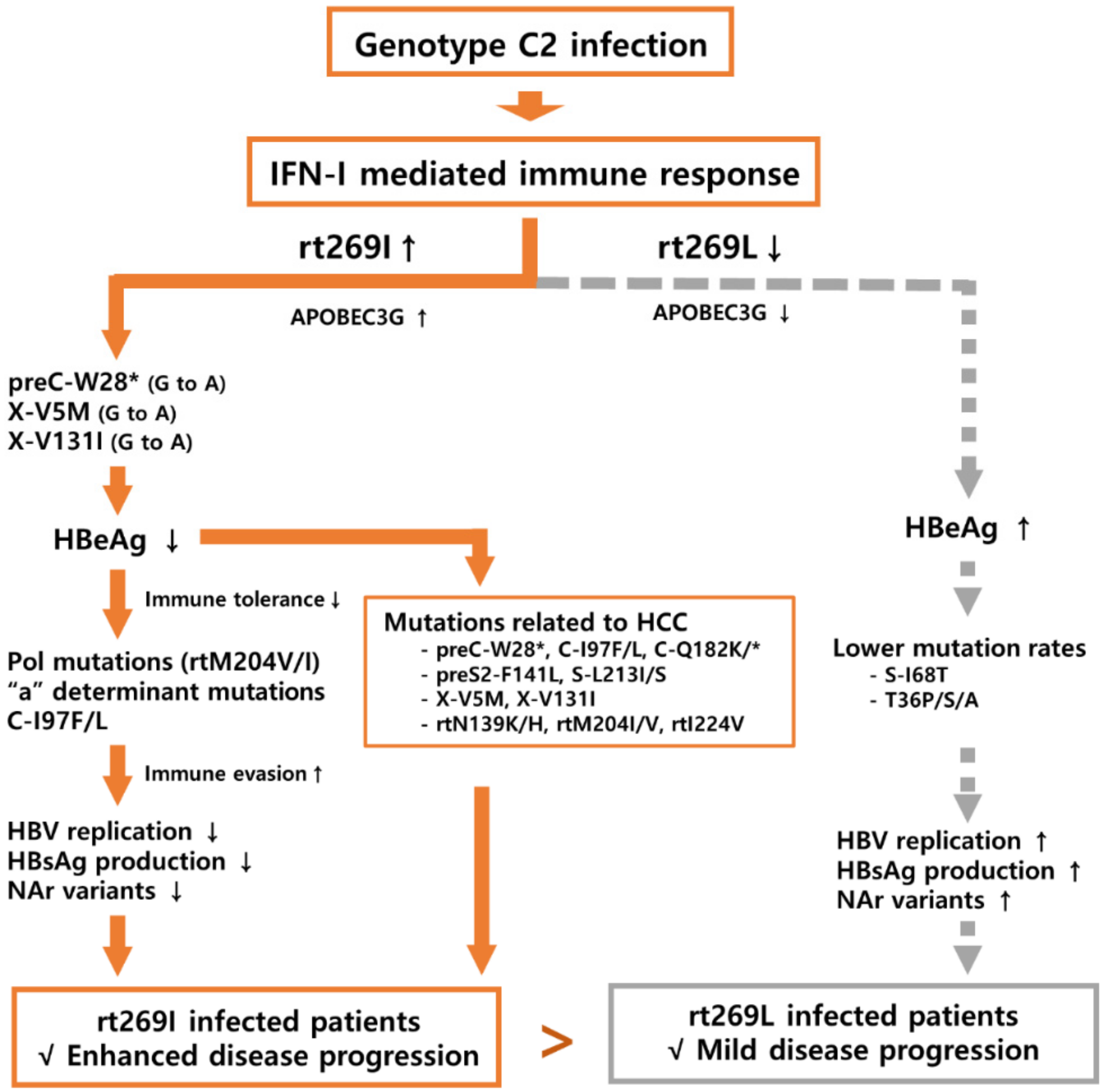
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schweitzer, A.; Horn, J.; Mikolajczyk, R.T.; Krause, G.; Ott, J. Estimations of worldwide prevalence of chronic hepatitis B virus infection: A systematic review of data published between 1965 and 2013. Lancet 2015, 386, 1546–1555. [Google Scholar] [CrossRef]
- Yim, S.Y.; Kim, J.H. The epidemiology of hepatitis B virus infection in Korea. Korean J. Intern. Med. 2019, 34, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.J.; Hepatitis, B. The virus and disease. Hepatology 2009, 49, S13–S21. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.A.; Bonhoeffer, S.; Hill, A.M.; Boehme, R.; Thomas, H.C.; McDade, H. Viral dynamics in hepatitis B virus infection. Proc. Natl. Acad. Sci. USA 1996, 93, 4398–4402. [Google Scholar] [CrossRef] [PubMed]
- Kurbanov, F.; Tanaka, Y.; Mizokami, M. Geographical and genetic diversity of the human hepatitis B virus. Hepatol. Res. 2010, 40, 14–30. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.L.; Wong, M.L.; Hui, A.Y.; Hung, L.C.; Chan, F.K.; Sung, J.J. Hepatitis B virus genotype C takes a more aggressive disease course than hepatitis B virus genotype B in hepatitis B e antigen-positive patients. J. Clin. Microbiol. 2003, 41, 1277–1279. [Google Scholar] [CrossRef]
- Osiowy, C.; Giles, E.; Trubnikov, M.; Choudhri, Y.; Andonov, A. Characterization of acute and chronic hepatitis B virus genotype in Canada. PLoS ONE 2015, 10, e0136074. [Google Scholar] [CrossRef]
- Croagh, C.M.; Desmond, P.V.; Bell, S.J. Genotypes and viral variants in chronic hepatitis B: A review of epidemiology and clinical relevance. World J. Hepatol. 2015, 7, 289–303. [Google Scholar] [CrossRef]
- Chan, H.L.; Hui, A.Y.; Wong, M.L.; Tse, A.M.; Hung, L.C.; Wong, V.W.; Sung, J.J. Genotype C hepatitis B virus infection is associated with an increased risk of hepatocellular carcinoma. Gut 2004, 53, 1494–1498. [Google Scholar] [CrossRef]
- Kim, H.; Kim, B.J. Association of preS/S mutations with occult hepatitis B virus (HBV) infection in South Korea: Transmission potential of distinct occult HBV variants. Int. J. Mol. Sci. 2015, 16, 13595–13609. [Google Scholar] [CrossRef]
- Kim, H.; Jee, Y.M.; Song, B.C.; Shin, J.W.; Yang, S.H.; Mun, H.S.; Kim, H.J.; Oh, E.J.; Yoon, J.H.; Kim, Y.J.; et al. Molecular epidemiology of hepatitis B virus (HBV) genotypes and serotypes in patients with chronic HBV infection in Korea. Intervirology 2007, 50, 52–57. [Google Scholar] [CrossRef]
- Kim, H.; Jee, Y.M.; Song, B.C.; Hyun, J.W.; Mun, H.S.; Kim, H.J.; Oh, E.J.; Yoon, J.H.; Kim, Y.J.; Lee, H.S.; et al. Analysis of hepatitis B virus quasispecies distribution in a Korean chronic patient based on the full genome sequences. J. Med. Virol. 2007, 79, 212–219. [Google Scholar] [CrossRef]
- Kim, B.J. Hepatitis B virus mutations related to liver disease progression of Korean patients. World J. Gastroenterol. 2014, 20, 460–467. [Google Scholar] [CrossRef]
- Kim, D.W.; Lee, S.A.; Hwang, E.S.; Kook, Y.H.; Kim, B.J. Naturally occurring precore/core region mutations of hepatitis B virus genotype C related to hepatocellular carcinoma. PLoS ONE 2012, 7, e47372. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, S.A.; Kim, D.W.; Lee, S.H.; Kim, B.J. Naturally occurring mutations in large surface genes related to occult infection of hepatitis B virus genotype C. PLoS ONE 2013, 8, e54486. [Google Scholar] [CrossRef]
- Lee, S.A.; Kim, K.J.; Kim, D.W.; Kim, B.J. Male-specific W4P/R mutation in the pre-S1 region of hepatitis B virus, increasing the risk of progression of liver diseases in chronic patients. J. Clin. Microbiol. 2013, 51, 3928–3936. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Gong, J.R.; Lee, S.A.; Kim, B.J. Discovery of a novel mutation (X8Del) resulting in an 8-bp deletion in the hepatitis B virus X gene associated with occult infection in Korean vaccinated individuals. PLoS ONE 2015, 10, e0139551. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.A.; Kim, H.; Won, Y.S.; Seok, S.H.; Na, Y.; Shin, H.B. Male-specific hepatitis B virus large surface protein variant W4P potentiates tumorigenicity and induces gender disparity. Mol. Cancer 2015, 14, 23. [Google Scholar] [CrossRef]
- Lee, S.A.; Kim, K.J.; Kim, H.; Choi, W.H.; Won, Y.S.; Kim, B.J. Hepatitis B virus preS1 deletion is related to viral replication increase and disease progression. World J. Gastroenterol. 2015, 21, 5039–5048. [Google Scholar] [CrossRef]
- Song, B.C.; Kim, S.H.; Kim, H.; Ying, Y.H.; Kim, H.J.; Kim, Y.J.; Yoon, J.H.; Lee, H.S.; Cha, C.Y.; Kook, Y.H.; et al. Prevalence of naturally occurring surface antigen variants of hepatitis B virus in Korean patients infected chronically. J. Med. Virol. 2005, 76, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Song, B.C.; Kim, H.; Kim, S.H.; Cha, C.Y.; Kook, Y.H.; Kim, B.J. Comparison of full length sequences of hepatitis B virus isolates in hepatocellular carcinoma patients and asymptomatic carriers of Korea. J. Med. Virol. 2005, 75, 13–19. [Google Scholar] [CrossRef]
- Kim, H.; Jee, Y.; Mun, H.S.; Park, J.H.; Yoon, J.H.; Kim, Y.J.; Lee, H.S.; Hyun, J.W.; Hwang, E.S.; Cha, C.Y.; et al. Characterization of two hepatitis B virus populations in a single Korean hepatocellular carcinoma patient with an HBeAg-negative serostatus: A novel X-Gene-deleted strain with inverted duplication sequences of upstream enhancer site II. Intervirology 2007, 50, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Jee, Y.; Mun, H.S.; Song, B.C.; Park, J.H.; Hyun, J.W.; Hwang, E.S.; Cha, C.Y.; Kook, Y.H.; Kim, B.J. Comparison of full genome sequences between two hepatitis B virus strains with or without preC mutation (A1896) from a single Korean hepatocellular carcinoma patient. Microbiol. Biotechnol. 2007, 17, 701–704. [Google Scholar]
- Kim, H.J.; Park, J.H.; Jee, Y.; Lee, S.A.; Kim, H.; Song, B.C.; Yang, S.; Lee, M.; Yoon, J.H.; Kim, Y.J.; et al. Hepatitis B virus X mutations occurring naturally associated with clinical severity of liver disease among Korean patients with chronic genotype C infection. J. Med. Virol. 2008, 80, 1337–1343. [Google Scholar] [CrossRef]
- Mun, H.S.; Lee, S.A.; Jee, Y.; Kim, H.; Park, J.H.; Song, B.C.; Yoon, J.H.; Kim, Y.J.; Lee, H.S.; Hyun, J.W.; et al. The prevalence of hepatitis B virus preS deletions occurring naturally in Korean patients infected chronically with genotype C. J. Med. Virol. 2008, 80, 1189–1194. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.A.; Cho, Y.K.; Lee, K.H.; Hwang, E.S.; Kook, Y.H.; Kim, B.J. Gender disparity in distribution of the major hydrophilic region variants of hepatitis B virus genotype C according to hepatitis B e antigen serostatus. J. Med. Virol. 2011, 83, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.A.; Mun, H.S.; Kim, H.; Lee, H.K.; Kim, B.J.; Hwang, E.S.; Kook, Y.H.; Kim, B.J. Naturally occurring hepatitis B virus X deletions and insertions among Korean chronic patients. J. Med. Virol. 2011, 83, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Mun, H.S.; Lee, S.A.; Kim, H.; Hwang, E.S.; Kook, Y.H.; Kim, B.J. Novel F141L pre-S2 mutation in hepatitis B virus increases the risk of hepatocellular carcinoma in patients with chronic genotype C infections. J. Virol. 2011, 85, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Lee, S.A.; Kim, H.; Won, Y.S.; Kim, B.J. Naturally occurring mutations in the nonstructural region 5B of hepatitis C virus (HCV) from treatment-naïve Korean patients chronically infected with HCV genotype 1b. PLoS ONE 2014, 9, e87773. [Google Scholar] [CrossRef] [PubMed]
- Hayer, J.; Jadeau, F.; Deléage, G.; Kay, A.; Zoulim, F.; Combet, C. HBVdb: A knowledge database for Hepatitis B Virus. Nucleic Acids Res. 2013, 41, D566–D570. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; He, K.; Wu, B.; Xu, M.; Du, L.; Liu, W.; Liao, P.; Liu, Y.; He, M. A systematic genotype and subgenotype re-ranking of hepatitis B virus under a novel classification standard. Heliyon 2019, 5, e02556. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar]
- Minin, V.N.; Dorman, K.S.; Fang, F.; Suchard, M.A. Dual multiple change-point model leads to more accurate recombination detection. Bioinformatics 2005, 21, 3034–3042. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.L.; Sabin, C.A.; Dong, B.Q.; Wei, S.C.; Chen, Q.Y.; Fang, K.X.; Yang, J.Y.; Wang, X.Y.; Harrison, T.J. The association of HBV core promoter double mutations (A1762T and G1764A) with viral load differs between HBeAg positive and anti-HBe positive individuals: A longitudinal analysis. J. Hepatol. 2009, 50, 273–280. [Google Scholar] [CrossRef][Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Kwong, K.S.; Holland, B.; Cheung, S.H. A modified Benjamini-Hochberg multiple comparisons procedure for controlling the false discovery rate. J. Stat. Plan. Inference 2002, 104, 351–362. [Google Scholar] [CrossRef]
- Ferreira, J.A.; Zwinderman, A.H. Approximate power and sample size calculations with the Benjamini-Hochberg method. Int. J. Biostat. 2006, 2, 8. [Google Scholar] [CrossRef]
- Khodadad, N.; Seyedian, S.S.; Moattari, A.; Haghighi, S.B.; Pirmoradi, R.; Abbasi, S.; Makvandi, M. In silico functional and structural characterization of hepatitis B virus PreS/S-gene in Iranian patients infected with chronic hepatitis B virus genotype D. Heliyon 2020, 6, e04332. [Google Scholar] [CrossRef]
- Littlejohn, M.; Davies, J.; Yuen, L.; Edwards, R.; Sozzi, T.; Jackson, K.; Cowie, B.; Tong, S.; Davis, J.; Locarnini, S. Molecular virology of hepatitis B virus, sub-genotype C4 in northern Australian Indigenous populations. J. Med. Virol. 2014, 86, 695–706. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Wei, F.; Hu, D.; Li, Q.; Smith, D.; Li, N.; Chen, D. Mutations in the major hydrophilic region (MHR) of hepatitis B virus genotype C in North China. J. Med. Virol. 2012, 84, 1901–1906. [Google Scholar] [CrossRef]
- Choi, Y.M.; Lee, S.Y.; Kim, B.J. Naturally Occurring Hepatitis B Virus Mutations Leading to Endoplasmic Reticulum Stress and Their Contribution to the Progression of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2019, 20, 597. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Ghosh, A.; Dasgupta, D.; Ghosh, A.; Roychoudhury, S.; Roy, G.; Das, S.; Das, K.; Gupta, S.; Basu, K.; et al. Novel point and combo-mutations in the genome of hepatitis B virus-genotype D: Characterization and impact on liver disease progression to hepatocellular carcinoma. PLoS ONE 2014, 9, e110012. [Google Scholar] [CrossRef]
- Chen, C.H.; Changchien, C.S.; Lee, C.M.; Hung, C.H.; Hu, T.H.; Wang, J.H.; Wang, J.C.; Lu, S.N. Combined mutations in pre-s/surface and core promoter/precore regions of hepatitis B virus increase the risk of hepatocellular carcinoma: A case-control study. J. Infect. Dis. 2008, 198, 1634–1642. [Google Scholar] [CrossRef] [PubMed]
- Benhenda, S.; Cougot, D.; Buendia, M.A.; Neuveut, C. Chapter 4 hepatitis B virus X protein: Molecular functions and its role in virus life cycle and pathogenesis. Adv. Cancer Res. 2009, 103, 75–109. [Google Scholar] [CrossRef]
- Quarleri, J. Core promoter: A critical region where the hepatitis B virus makes decisions. World J. Gastroenterol. 2014, 20, 425–435. [Google Scholar] [CrossRef]
- Kim, H.; Lee, S.A.; Kim, B.J. X region mutations of hepatitis B virus related to clinical severity. World J. Gastroenterol. 2016, 22, 5467–5478. [Google Scholar] [CrossRef]
- Schlicht, H.J.; Wasenauer, G. The quaternary structure, antigenicity, and aggregational behavior of the secretory core protein of human hepatitis B virus are determined by its signal sequence. J. Virol. 1991, 65, 6817–6825. [Google Scholar] [CrossRef]
- Matsumura, S.; Yamamoto, K.; Shimada, N.; Okano, N.; Okamoto, R.; Suzuki, T.; Hakoda, T.; Mizuno, M.; Higashi, T.; Tsuji, T. High frequency of circulating HBcAg-specific CD8 T cells in hepatitis B infection: A flow cytometric analysis. Clin. Exp. Immunol. 2001, 124, 435–444. [Google Scholar] [CrossRef]
- Jung, M.C.; Diepolder, H.M.; Spengler, U.; Wierenga, E.A.; Zachoval, R.; Hoffmann, R.M.; Eichenlaub, D.; Frösner, G.; Will, H.; Pape, G.R. Activation of a heterogeneous hepatitis B (HB) core and e antigenspecific CD4+ T-cell population during seroconversion to antiHBe and anti-HBs in hepatitis B virus infection. J. Virol. 1995, 69, 3358–3368. [Google Scholar] [CrossRef]
- Yim, S.Y.; Um, S.H.; Young Jung, J.; Kim, T.H.; Kim, J.D.; Keum, B.; Seo, Y.S.; Yim, H.J.; Jeen, Y.T.; Lee, H.S.; et al. Clinical significance of hepatitis B virus precore and core promoter variants in Korean patients with chronic hepatitis B. J. Clin. Gastroenterol. 2015, 49, 61–68. [Google Scholar] [CrossRef]
- Kim, H.; Lee, S.A.; Do, S.Y.; Kim, B.J. Precore/core region mutation of hepatitis B virus related to clinical severity. World J. Gastroenterol. 2016, 22, 4287–4296. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Liu, S.; Zhao, Y.; Zhang, L.; Zhao, Y.; Liu, B. Precore/core region mutations in hepatitis B virus DNA predict postoperative survival in hepatocellular carcinoma. PLoS ONE 2015, 10, e0133393. [Google Scholar] [CrossRef]
- Hosono, S.; Tai, P.C.; Wang, W.; Ambroso, M.; Hwang, D.G.; Yuan, T.T. Core antigen mutations of human hepatitis B virus in hepatomas accumulate in MHC class II-restricted T cell epitopes. Virology 1995, 212, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.T.T.; Sahu, G.K.; Whitehead, W.E.; Greenberg, R.; Shih, C. The mechanism of an immature secretion phenotype of a highly frequent naturally occurring missense mutation at codon 97 of human hepatitis B virus core antigen. J. Virol. 1999, 73, 5731–5740. [Google Scholar] [CrossRef]
- Shirvani-Dastgerdi, E.; Winer, B.Y.; Celia-Terrassa, T.; Kang, Y.; Tabernero, D.; Yagmur, E.; Rodríguez-Frías, F.; Gregori, J.; Luedde, T.; Trautwein, C.; et al. Selection of the highly replicative and partially multidrug resistant rtS78T HBV polymerase mutation during TDF-ETV combination therapy. J. Hepatol. 2017, 67, 246–254. [Google Scholar] [CrossRef]
- Thedja, M.D.; Muljono, D.H.; Ie, S.I.; Sidarta, E.; Turyadi; Verhoef, J.; Marzuki, S. Genogeography and immune epitope characteristics of hepatitis B virus genotype C reveals two distinct types: Asian and Papua-pacific. PLoS ONE 2015, 10, e0132533. [Google Scholar] [CrossRef]
- Clark, D.N.; Flanagan, J.M.; Hu, J. Mapping of functional subdomain in the terminal protein domain of hepatitis B virus polymerase. J. Virol. 2016, 91, e01785-16. [Google Scholar] [CrossRef]
- Li, X.G.; Liu, B.M.; Xu, J.; Liu, X.E.; Ding, H.; Li, T. Discrepancy of potential antiviral resistance mutation profiles within the HBV reverse transcriptase between nucleos(t)ide analogue-untreated and –treated patients with chronic hepatitis B in a hospital in China. J. Med. Virol. 2011, 84, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Lok, A.S.; Zoulim, F.; Locarnini, S.; Bartholomeusz, A.; Ghany, M.G.; Pawlotsky, J.M.; Liaw, Y.F.; Mizokami, M.; Kuiken, C. Hepatitis B virus drug resistance working group. Antiviral drug-resistant HBV: Standardization of nomeclature and assays and recommendations for management. Hepatology 2007, 46, 254–265. [Google Scholar] [CrossRef]
- Locarnini, S. Primary resistance, multidrug resistance, and cross-resistance pathways in HBV as a consequence of treatment failure. Hepatol. Int. 2008, 2, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.; Shin, Y.C.; Park, W.J.; Kim, S.; Kim, J.; Ryu, W.S. Residues Arg703, Asp777, and Arg781 of the RNase H domain of hepatitis B virus polymerase are critical for viral DNA synthesis. J. Virol. 2014, 88, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Liu, S.; Zhao, Y.; Guo, Z.; Xu, J. X protein mutations in hepatitis B virus DNA predict postoperative survival in hepatocellular carcinoma. Tumor. Biol. 2014, 35, 10325–10331. [Google Scholar] [CrossRef] [PubMed]
- Asim, M.; Malik, A.; Sarma, M.P.; Polipalli, S.K.; Begum, N.; Ahmad, I.; Khan, L.A.; Husain, S.A.; Akhtar, N.; Husain, S. Hepatitis B virus BCP, Precore/core, X gene mutations/genotypes and the risk of hepatocellular carcinoma in India. J. Med. Virol. 2010, 82, 1115–1125. [Google Scholar] [CrossRef] [PubMed]
- Preikschat, P.; Gunther, S.; Reinhold, S.; Will, H.; Budde, K.; Neumayer, H.H.; Kruger, D.H.; Meisel, H. Complex HBV populations with mutations in core promoter, C gene, and pre-S region are associated with development of cirrhosis in long-term renal transplant recipients. Hepatology 2002, 35, 466–477. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, J.; Sun, L.; Liu, G.; Li, B.; Zheng, Y.; Li, X.; Tao, J. Prevalence of mutations in HBV DNA polymerase gene associated with nucleos(t)ide resistance in treatment-naïve patients with chronic hepatitis B in central China. Braz. J. Infect. Dis. 2016, 20, 173–178. [Google Scholar] [CrossRef]
- Kim, J.E.; Lee, S.Y.; Kim, H.; Kim, K.J.; Choe, W.H.; Kim, B.J. Naturally occurring mutations in the reverse transcriptase region of hepatitis B virus polymerase from treatment-naïve Korean patients infected with genotype C2. World J. Gastroenterol. 2017, 23, 4222–4232. [Google Scholar] [CrossRef]
- Yuen, L.K.; Locarnini, S. Genetic variability of hepatitis B virus and response to antiviral treatments: Searching for a bigger picture. J. Hepatol. 2009, 50, 445–448. [Google Scholar] [CrossRef][Green Version]
- Zheng, J.; Zeng, Z.; Zhang, D.; Yu, Y.; Wang, F.; Pan, C.Q. Prevalence and significance of hepatitis B reverse transcriptase mutants in different disease stages of untreated patients. Liver Int. 2012, 32, 1535–1542. [Google Scholar] [CrossRef]
- Liu, B.M.; Li, T.; Xu, J.; Li, X.G.; Dong, J.P.; Yan, P.; Yang, J.X.; Yan, L.; Gao, Z.Y.; Li, W.P.; et al. Characterization of potential antiviral resistance mutations in hepatitis B virus reverse transcriptase sequences in treatmentnaïve Chinese patients. Antivir. Res. 2010, 85, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.P.; Han, F.Z.; Duan, H.L.; Ji, F.; Yan, X.B.; Fan, Y.C.; Wang, K. Hepatitis B virus pre-existing drug resistant mutation is related to the genotype and disease progression. J. Infect. Dev. Ctries 2017, 11, 727–732. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fan, J.; Zhang, Y.; Xiong, H.; Wang, Y.; Guo, X. Nucleotide analogueresistant mutations in hepatitis B viral genomes found in hepatitis B patients. J. Gen. Virol. 2015, 96, 663–670. [Google Scholar] [CrossRef]
- Lin, C.L.; Kao, J.H. The clinical implications of hepatitis B virus genotype: Recent advances. J. Gastroenterol. Hepatol. 2011, 26, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.S.Y.; Covert, E.; Wilson, E.; Kottilil, S. Chronic hepatitis B infection: A review. JAMA 2018, 319, 1802–1813. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Choi, Y.M.; Oh, S.J.; Yang, S.B.; Lee, J.H.; Choe, W.H.; Kook, Y.H.; Kim, B.J. rt269I type of hepatitis B virus (HBV) leads to HBV e antigen negative infections and liver disease progression via mitochondrial stress mediated type I interferon production in chronic patients with genotype C infections. Front. Immunol. 2019, 10, 1735. [Google Scholar] [CrossRef]
- Noguchi, C.; Hiraga, N.; Mori, N.; Tsuge, M.; Imamura, M.; Takahashi, S.; Fujimoto, Y.; Ochi, H.; Abe, H.; Maekawa, T.; et al. Dual effect of APOBEC3G on hepatitis B virus. J. Gen. Virol. 2007, 88 Pt 2, 432–440. [Google Scholar] [CrossRef]





| L Type (n = 115) | I Type (n = 119) | p-Value from t-Test | |
|---|---|---|---|
| Number of nucleotide variants | |||
| HBV full genome (/3,215 bp) | 38.03 ± 13.65 | 42.34 ± 15.17 | 0.0234 * |
| Number of amino acid variants | |||
| HBV full genome (/1,613 aa) | 23.43 ± 10.75 | 27.68 ± 12.39 | 0.0056 ** |
| d (10−2 substitution/site) (nucleotide level) | |||
| preS SHB pol RT X preC/C | 1.39 ± 1.59 0.58 ± 0.37 1.09 ± 0.46 0.97 ± 0.40 1.24 ± 0.61 1.38 ± 0.86 | 1.71 ± 1.78 0.64 ± 0.45 1.24 ± 0.50 1.03 ± 0.48 1.28 ± 0.60 1.56 ± 0.80 | 0.1559 0.2522 0.0154 * 0.3216 0.6752 0.0989 |
| d (10−2 substitution/site) (amino acid level) | |||
| preS SHB pol RT X preC/C | 1.60 ± 2.50 1.10 ± 0.95 1.26 ± 0.79 0.88 ± 0.67 2.19 ± 1.43 1.64 ± 1.56 | 2.23 ± 3.29 1.29 ± 1.01 1.60 ± 0.86 1.06 ± 0.72 2.34 ± 1.30 1.66 ± 1.29 | 0.0989 0.1494 0.0019 **a 0.0465 * 0.3796 0.9128 |
| dN (10−2 substitution/site) | |||
| preS SHB pol RT X preC/C | 0.81 ± 1.40 0.52 ± 0.45 0.61 ± 0.42 0.42 ± 0.32 1.04 ± 0.70 0.82 ± 0.81 | 1.11 ± 1.74 0.62 ± 0.49 0.78 ± 0.45 0.51 ± 0.35 1.10 ± 0.62 0.82 ± 0.65 | 0.1449 0.1142 0.0044 **a 0.0327 * 0.4405 0.9903 |
| dS (10−2 substitution/site) | |||
| preS SHB pol RT X preC/C | 2.78 ± 2.23 0.68 ± 0.55 2.21 ± 0.74 2.30 ± 1.05 1.72 ± 1.05 2.45 ± 1.62 | 3.22 ± 2.07 0.69 ± 0.65 2.31 ± 0.86 2.26 ± 1.23 1.68 ± 0.90 2.94 ± 1.62 | 0.1188 0.9058 0.2855 0.8048 0.7902 0.0212 * |
| L Type (n = 115) | I Type (n = 119) | p-Value from t-Test | |
|---|---|---|---|
| Number of nucleotide variants | |||
| a determinant (/72 bp) MHR (/213 bp) | 0.29 ± 0.59 1.14 ± 1.10 | 0.50 ± 0.92 1.31 ± 1.38 | 0.0311 * 0.2935 |
| Number of amino acid variants | |||
| a determinant (/24 aa) MHR (/71 aa) | 0.23 ± 0.45 0.24 ± 0.54 | 0.33 ± 0.68 0.41 ± 0.69 | 0.2182 0.0399 * |
| dN (10−2 substitution/site) | |||
| a determinant MHR | 0.07 ± 0.38 0.48 ± 0.53 | 0.38 ± 1.24 0.67 ± 0.87 | 0.0109 * 0.0497 * |
| dS (10−2 substitution/site) | |||
| a determinant MHR | 0.99 ± 2.58 1.39 ± 1.48 | 1.14 ± 2.36 1.26 ± 1.45 | 0.6425 0.5065 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, H.; Kim, D.H.; Choi, Y.-M.; Choi, H.; Kim, D.; Kim, B.-J. rt269I Type of Hepatitis B Virus (HBV) Polymerase versus rt269L Is More Prone to Mutations within HBV Genome in Chronic Patients Infected with Genotype C2: Evidence from Analysis of Full HBV Genotype C2 Genome. Microorganisms 2021, 9, 601. https://doi.org/10.3390/microorganisms9030601
Jeong H, Kim DH, Choi Y-M, Choi H, Kim D, Kim B-J. rt269I Type of Hepatitis B Virus (HBV) Polymerase versus rt269L Is More Prone to Mutations within HBV Genome in Chronic Patients Infected with Genotype C2: Evidence from Analysis of Full HBV Genotype C2 Genome. Microorganisms. 2021; 9(3):601. https://doi.org/10.3390/microorganisms9030601
Chicago/Turabian StyleJeong, Hyein, Dong Hyun Kim, Yu-Min Choi, HyeLim Choi, Donghyun Kim, and Bum-Joon Kim. 2021. "rt269I Type of Hepatitis B Virus (HBV) Polymerase versus rt269L Is More Prone to Mutations within HBV Genome in Chronic Patients Infected with Genotype C2: Evidence from Analysis of Full HBV Genotype C2 Genome" Microorganisms 9, no. 3: 601. https://doi.org/10.3390/microorganisms9030601
APA StyleJeong, H., Kim, D. H., Choi, Y.-M., Choi, H., Kim, D., & Kim, B.-J. (2021). rt269I Type of Hepatitis B Virus (HBV) Polymerase versus rt269L Is More Prone to Mutations within HBV Genome in Chronic Patients Infected with Genotype C2: Evidence from Analysis of Full HBV Genotype C2 Genome. Microorganisms, 9(3), 601. https://doi.org/10.3390/microorganisms9030601