
Metagenomic Snapshots of Viral Components in Guinean Bats


 ,
,  ,
,  , and
, and 
Abstract
1. Introduction
2. Materials and Methods
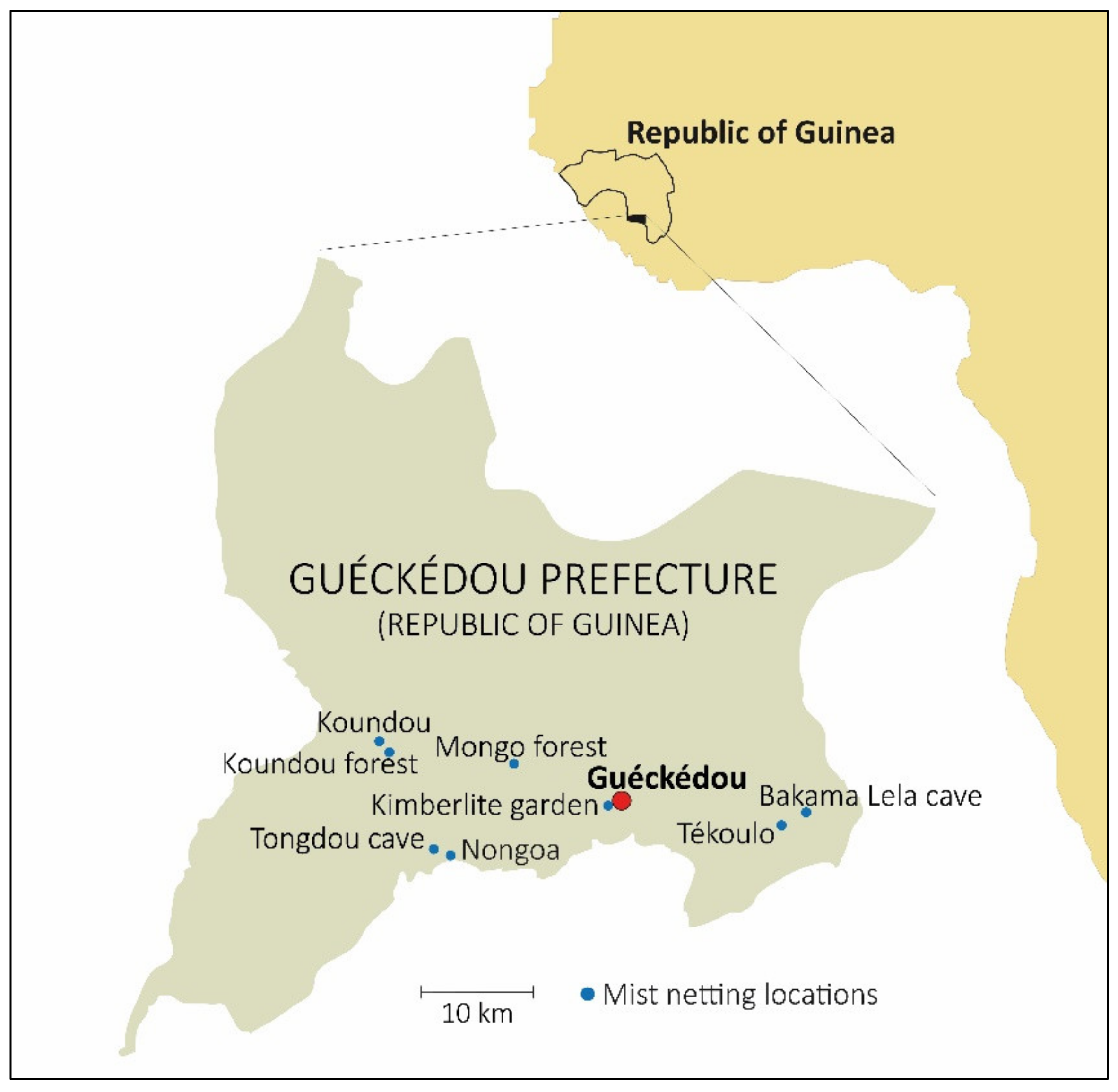
2.1. Bat Capturing and Sampling
2.2. Molecular Identification of the Bat Species
2.3. Sample Processing and Characterization of the Bat Virome
2.4. Sequence Data and Phylogenetic Analysis
2.5. Identification of Nonretroviral Integrated RNA Virus (NIRV) Elements
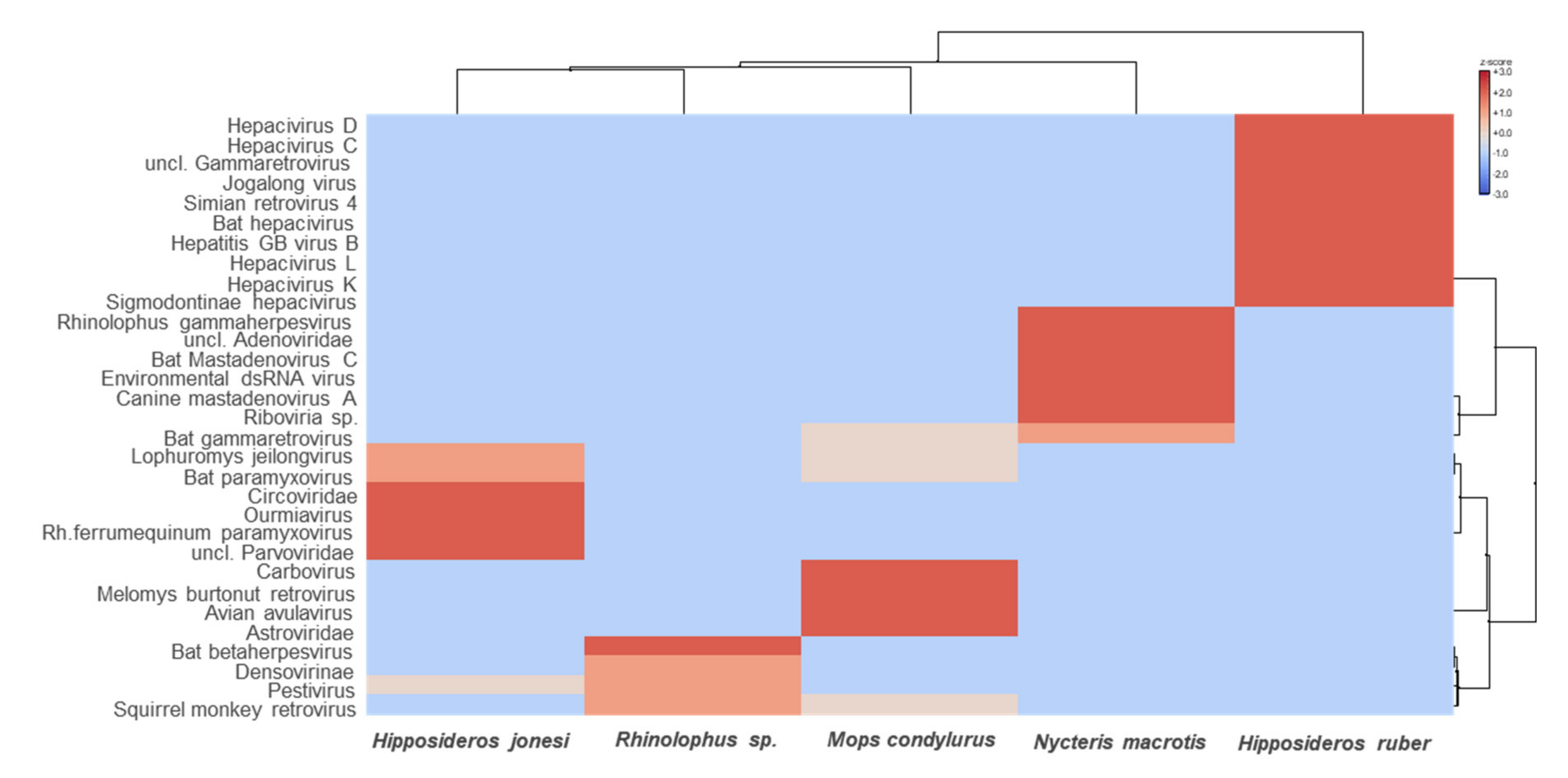
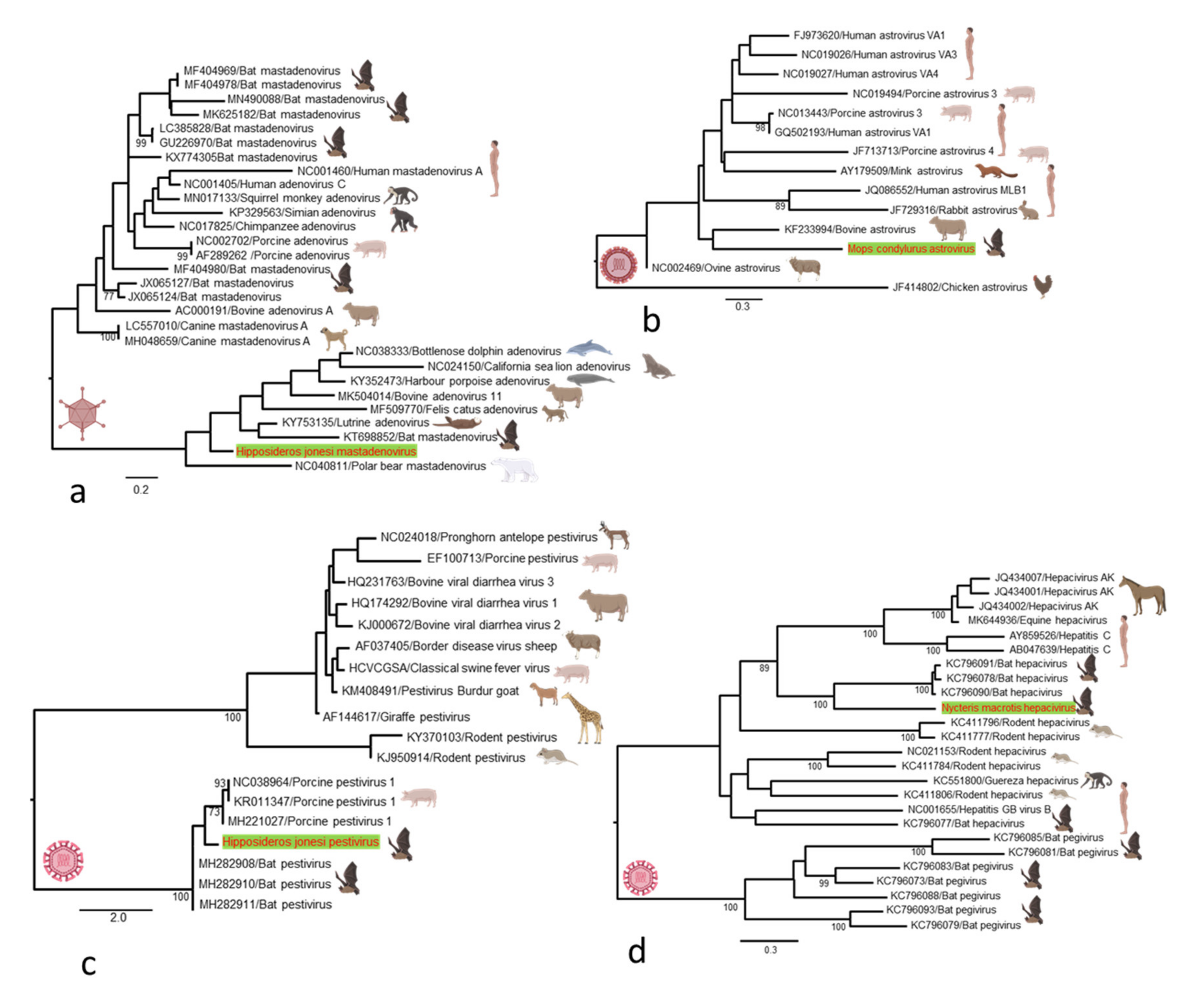
3. Results
3.1. Bat Species Identification
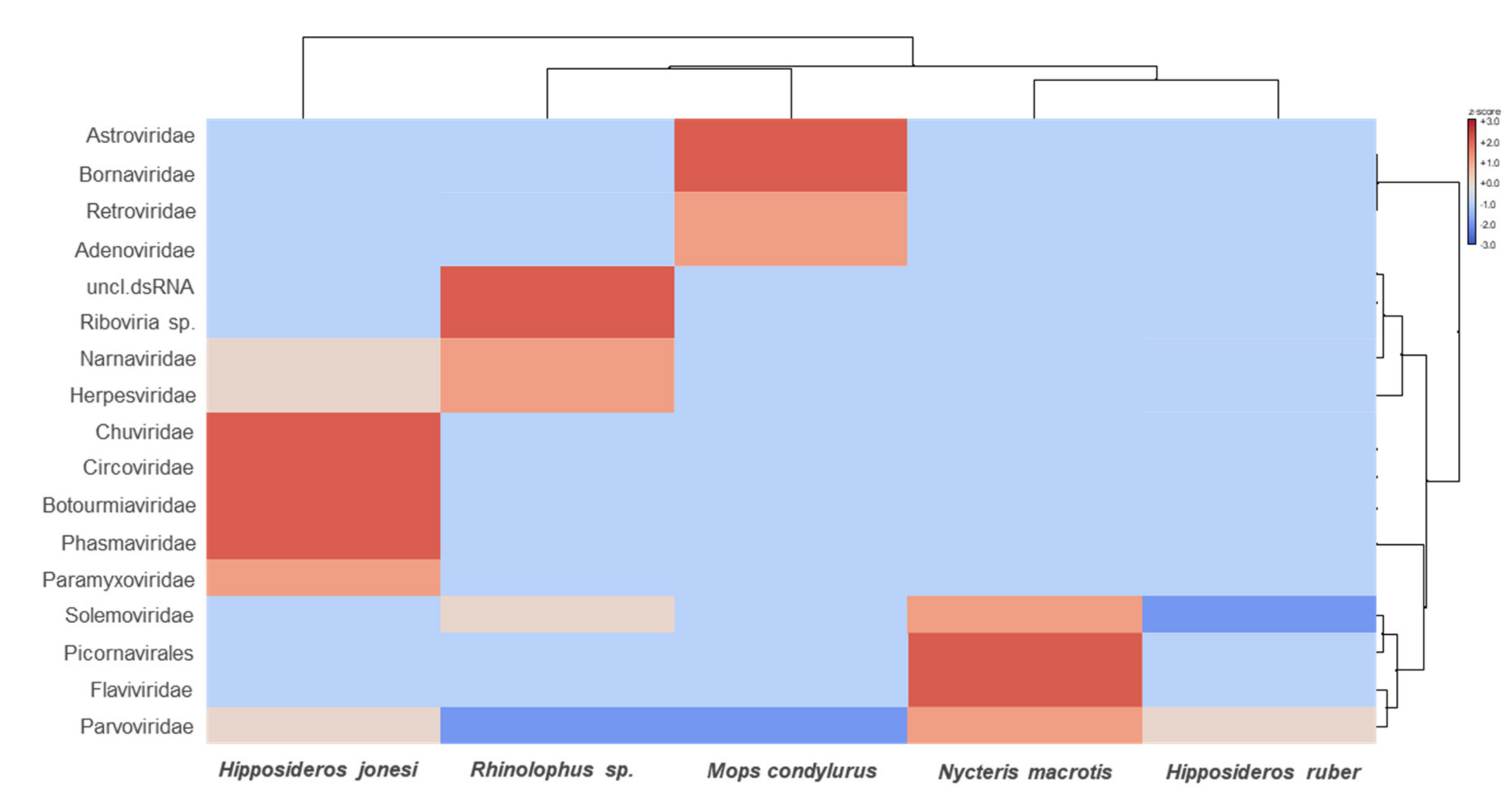
3.2. Characterization of the Bat Virome
3.3. Filovirus Paleoviral Sequences
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kemunto, N.; Mogoa, E.; Osoro, E.; Bitek, A.; Njenga, M.K.; Thumbi, S.M. Zoonotic disease research in East Africa. BMC Infect. Dis. 2018, 18, 545. [Google Scholar] [CrossRef]
- Taylor, L.H.; Latham, S.M.; Woolhouse, M.E. Risk factors for human disease emergence. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2001, 356, 983–989. [Google Scholar] [CrossRef]
- Jones, K.E.; Patel, N.G.; Levy, M.A.; Storeygard, A.; Balk, D.; Gittleman, J.L.; Daszak, P. Global trends in emerging infectious diseases. Nature 2008, 451, 990–993. [Google Scholar] [CrossRef] [PubMed]
- Bean, A.G.; Baker, M.L.; Stewart, C.R.; Cowled, C.; Deffrasnes, C.; Wang, L.F.; Lowenthal, J.W. Studying immunity to zoonotic diseases in the natural host—Keeping it real. Nat. Rev. Immunol. 2013, 13, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.F.; Anderson, D.E. Viruses in bats and potential spillover to animals and humans. Curr. Opin. Virol. 2019, 34, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Letko, M.; Seifert, S.N.; Olival, K.J.; Plowright, R.K.; Munster, V.J. Bat-borne virus diversity, spillover and emergence. Nat. Rev. Microbiol. 2020, 18, 461–471. [Google Scholar] [CrossRef]
- Boni, M.F.; Lemey, P.; Jiang, X.; Lam, T.T.; Perry, B.W.; Castoe, T.A.; Rambaut, A.; Robertson, D.L. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat. Microbiol. 2020, 5, 1408–1417. [Google Scholar] [CrossRef]
- Berto, A.; Anh, P.H.; Carrique-Mas, J.J.; Simmonds, P.; Van Cuong, N.; Tue, N.T.; Van Dung, N.; Woolhouse, M.E.; Smith, I.; Marsh, G.A.; et al. Detection of potentially novel paramyxovirus and coronavirus viral RNA in bats and rats in the Mekong Delta region of southern Viet Nam. Zoonoses Public Health 2018, 65, 30–42. [Google Scholar] [CrossRef]
- Forbes, K.M.; Webala, P.W.; Jääskeläinen, A.J.; Abdurahman, S.; Ogola, J.; Masika, M.M.; Kivistö, I.; Alburkat, H.; Plyusnin, I.; Levanov, L.; et al. Bombali Virus in Mops condylurus Bat, Kenya. Emerg. Infect. Dis. 2019, 25, 955–957. [Google Scholar] [CrossRef]
- Towner, J.S.; Amman, B.R.; Sealy, T.K.; Carroll, S.A.; Comer, J.A.; Kemp, A.; Swanepoel, R.; Paddock, C.D.; Balinandi, S.; Khristova, M.L.; et al. Isolation of genetically diverse Marburg viruses from Egyptian fruit bats. PLoS Pathog. 2009, 5, e1000536. [Google Scholar] [CrossRef]
- Jones, M.E.; Schuh, A.J.; Amman, B.R.; Sealy, T.K.; Zaki, S.R.; Nichol, S.T.; Towner, J.S. Experimental Inoculation of Egyptian Rousette Bats (Rousettus aegyptiacus) with Viruses of the Ebolavirus and Marburgvirus Genera. Viruses 2015, 7, 3420–3442. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.L.; Tan, C.W.; Anderson, D.E.; Jiang, R.D.; Li, B.; Zhang, W.; Zhu, Y.; Lim, X.F.; Zhou, P.; Liu, X.L.; et al. Characterization of a filovirus (Mengla virus) from Rousettus bats in China. Nat. Microbiol. 2019, 4, 390–395. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, Z.; Cai, J.; Yan, X.; Zhang, F.; Wu, J.; Xu, L.; Zhao, Z.; Hu, T.; Tu, C.; et al. Seroreactive Profiling of Filoviruses in Chinese Bats Reveals Extensive Infection of Diverse Viruses. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Leroy, E.M.; Kumulungui, B.; Pourrut, X.; Rouquet, P.; Hassanin, A.; Yaba, P.; Délicat, A.; Paweska, J.T.; Gonzalez, J.P.; Swanepoel, R. Fruit bats as reservoirs of Ebola virus. Nature 2005, 438, 575–576. [Google Scholar] [CrossRef] [PubMed]
- Olival, K.J.; Hayman, D.T. Filoviruses in bats: Current knowledge and future directions. Viruses 2014, 6, 1759–1788. [Google Scholar] [CrossRef]
- Horie, M.; Honda, T.; Suzuki, Y.; Kobayashi, Y.; Daito, T.; Oshida, T.; Ikuta, K.; Jern, P.; Gojobori, T.; Coffin, J.M.; et al. Endogenous non-retroviral RNA virus elements in mammalian genomes. Nature 2010, 463, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Honda, T.; Tomonaga, K. Endogenous non-retroviral RNA virus elements evidence a novel type of antiviral immunity. Mob. Genet. Elem. 2016, 6, e1165785. [Google Scholar] [CrossRef]
- Taylor, D.J.; Dittmar, K.; Ballinger, M.J.; Bruenn, J.A. Evolutionary maintenance of filovirus-like genes in bat genomes. BMC Evol. Biol. 2011, 11, 336. [Google Scholar] [CrossRef] [PubMed]
- Hause, B.M.; Nelson, E.A.; Christopher-Hennings, J. North American Big Brown Bats (Eptesicus fuscus) Harbor an Exogenous Deltaretrovirus. Msphere 2020, 5. [Google Scholar] [CrossRef]
- Zhdanov, V.M. Integration of viral genomes. Nature 1975, 256, 471–473. [Google Scholar] [CrossRef]
- Naville, M.; Warren, I.A.; Haftek-Terreau, Z.; Chalopin, D.; Brunet, F.; Levin, P.; Galiana, D.; Volff, J.N. Not so bad after all: Retroviruses and long terminal repeat retrotransposons as a source of new genes in vertebrates. Clin. Microbiol. Infect. 2016, 22, 312–323. [Google Scholar] [CrossRef]
- Leray, M.; Yang, J.Y.; Meyer, C.P.; Mills, S.C.; Agudelo, N.; Ranwez, V.; Boehm, J.T.; Machida, R.J. A new versatile primer set targeting a short fragment of the mitochondrial COI region for metabarcoding metazoan diversity: Application for characterizing coral reef fish gut contents. Front. Zool. 2013, 10, 34. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez, C.; García-Mudarra, J.L.; Ruedi, M.; Stadelmann, B.; Juste, J. The Iberian contribution to cryptic diversity in European bats. Acta Chiropterol. 2006, 8, 277–297. [Google Scholar] [CrossRef]
- Smith, M.F.; Patton, J.L. The diversification of South. American murid rodents: Evidence from mitochondrial DNA sequence data for the akodontine tribe. Biol. J. Linn. Soc. 1993, 50, 149–177. [Google Scholar] [CrossRef]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.J.; Tappu, R. MEGAN Community Edition-Interactive Exploration and Analysis of Large-Scale Microbiome Sequencing Data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Hyndman, T.H.; Shilton, C.M.; Stenglein, M.D.; Wellehan, J.F., Jr. Divergent bornaviruses from Australian carpet pythons with neurological disease date the origin of extant Bornaviridae prior to the end-Cretaceous extinction. PLoS Pathog. 2018, 14, e1006881. [Google Scholar] [CrossRef] [PubMed]
- Baize, S.; Pannetier, D.; Oestereich, L.; Rieger, T.; Koivogui, L.; Magassouba, N.F.; Soropogui, B.; Sow, M.S.; Keïta, S.; De Clerck, H.; et al. Emergence of Zaire Ebola virus disease in Guinea. N. Engl. J. Med. 2014, 371, 1418–1425. [Google Scholar] [CrossRef]
- Carroll, M.W.; Matthews, D.A.; Hiscox, J.A.; Elmore, M.J.; Pollakis, G.; Rambaut, A.; Hewson, R.; García-Dorival, I.; Bore, J.A.; Koundouno, R.; et al. Temporal and spatial analysis of the 2014–2015 Ebola virus outbreak in West Africa. Nature 2015, 524, 97–101. [Google Scholar] [CrossRef]
- Marí Saéz, A.; Weiss, S.; Nowak, K.; Lapeyre, V.; Zimmermann, F.; Düx, A.; Kühl, H.S.; Kaba, M.; Regnaut, S.; Merkel, K.; et al. Investigating the zoonotic origin of the West African Ebola epidemic. EMBO Mol. Med. 2015, 7, 17–23. [Google Scholar] [CrossRef]
- Kareinen, L.; Ogola, J.; Kivistö, I.; Smura, T.; Aaltonen, K.; Jääskeläinen, A.J.; Kibiwot, S.; Masika, M.M.; Nyaga, P.; Mwaengo, D.; et al. Range Expansion of Bombali Virus in Mops condylurus Bats, Kenya, 2019. Emerg. Infect. Dis. 2020, 26, 3007–3010. [Google Scholar] [CrossRef] [PubMed]
- Karan, L.S.; Makenov, M.T.; Korneev, M.G.; Sacko, N.; Boumbaly, S.; Yakovlev, S.A.; Kourouma, K.; Bayandin, R.B.; Gladysheva, A.V.; Shipovalov, A.V.; et al. Bombali Virus in Mops condylurus Bats, Guinea. Emerg. Infect. Dis. 2019, 25, 1774–1775. [Google Scholar] [CrossRef] [PubMed]
- Edwards, M.R.; Liu, H.; Shabman, R.S.; Ginell, G.M.; Luthra, P.; Ramanan, P.; Keefe, L.J.; Köllner, B.; Amarasinghe, G.K.; Taylor, D.J.; et al. Conservation of Structure and Immune Antagonist Functions of Filoviral VP35 Homologs Present in Microbat Genomes. Cell Rep. 2018, 24, 861–872. [Google Scholar] [CrossRef]
- Taylor, D.J.; Leach, R.W.; Bruenn, J. Filoviruses are ancient and integrated into mammalian genomes. BMC Evol. Biol. 2010, 10, 193. [Google Scholar] [CrossRef]
- Watanabe, S.; Noda, T.; Kawaoka, Y. Functional mapping of the nucleoprotein of Ebola virus. J. Virol. 2006, 80, 3743–3751. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location | Habitat | Lat (N) | Long (W) | Alt | Species |
|---|---|---|---|---|---|
| Tongdou cave | Cave | 8°30.867 | 10°21.226 | 400 | Nycteris macrotis ** |
| Nongoa | Rural core | 8°30.389 | 10°19.580 | 399 | Nanonycteris veldkampii **, Micropteropus pusillus **, Myonycteris leptodon ** |
| Mongo forest | Secondary forest | 8°36.540 | 10°15.375 | 417 | Hipposideros ruber **, Hipposideros jonesi **, Doryrhina cyclops **, Rhinolophus sp. *, Myonycteris angolensis smithii ** |
| Bakama Lela cave | Cave | 8°33.105 | 9°55.697 | 594 | Hipposideros ruber **, Hipposideros jonesi** |
| Tékoulo | House | 8°32.550 | 9°57.232 | 550 | Mops condylurus **, Chaerephon nigeriae ** |
| Koundou | House | 8°38.516 | 10°24.980 | 426 | Rhinolophus fumigatus ** |
| Koundou forest | Secondary forest | 8°37.596 | 10°24.648 | 411 | Hipposideros ruber **, Hipposideros abae **, Pseudoromicia brunnea ** |
| Kimberlite garden | Rural core | 8°33.130 | 10°8.975 | 428 | Hipposideros ruber **, Mops condylurus ** |
| Species | Number | Organs |
|---|---|---|
| Nycteris macrotis | 2 | spleen, kidney, liver |
| Rhinolophus sp. | 1 | spleen, kidney, liver, thymus |
| Hipposideros jonesi | 2 | spleen, kidney, liver, thymus |
| Mops condylurus | 6 | spleen, kidney, liver, thymus |
| Hipposideros ruber | 3 | kidney, liver |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hermida Lorenzo, R.J.; Cadar, D.; Koundouno, F.R.; Juste, J.; Bialonski, A.; Baum, H.; García-Mudarra, J.L.; Hakamaki, H.; Bencsik, A.; Nelson, E.V.; et al. Metagenomic Snapshots of Viral Components in Guinean Bats. Microorganisms 2021, 9, 599. https://doi.org/10.3390/microorganisms9030599
Hermida Lorenzo RJ, Cadar D, Koundouno FR, Juste J, Bialonski A, Baum H, García-Mudarra JL, Hakamaki H, Bencsik A, Nelson EV, et al. Metagenomic Snapshots of Viral Components in Guinean Bats. Microorganisms. 2021; 9(3):599. https://doi.org/10.3390/microorganisms9030599
Chicago/Turabian StyleHermida Lorenzo, Roberto J., Dániel Cadar, Fara Raymond Koundouno, Javier Juste, Alexandra Bialonski, Heike Baum, Juan Luis García-Mudarra, Henry Hakamaki, András Bencsik, Emily V. Nelson, and et al. 2021. "Metagenomic Snapshots of Viral Components in Guinean Bats" Microorganisms 9, no. 3: 599. https://doi.org/10.3390/microorganisms9030599
APA StyleHermida Lorenzo, R. J., Cadar, D., Koundouno, F. R., Juste, J., Bialonski, A., Baum, H., García-Mudarra, J. L., Hakamaki, H., Bencsik, A., Nelson, E. V., Carroll, M. W., Magassouba, N., Günther, S., Schmidt-Chanasit, J., Muñoz Fontela, C., & Escudero-Pérez, B. (2021). Metagenomic Snapshots of Viral Components in Guinean Bats. Microorganisms, 9(3), 599. https://doi.org/10.3390/microorganisms9030599