
Vector-Borne Blood Parasites of the Great-Tailed Grackle (Quiscalus mexicanus) in East-Central Texas, USA

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Subjects
2.2. Study Area
2.3. Bird Collection
2.4. Trypanosome and Filarioid Nematode Infection Prevalence
2.5. Haemosporida Infection Prevalence and Lineage Determination
2.6. Trypanosome and Filarioid Nematode Phylogenetic Relatedness
2.7. Polyparasitism Assembly Analysis
3. Results
3.1. Bird Processing
3.2. Trypanosome and Filarioid Nematode Infection Prevalence
3.3. Haemosporida Infection Prevalence and Lineage Determination
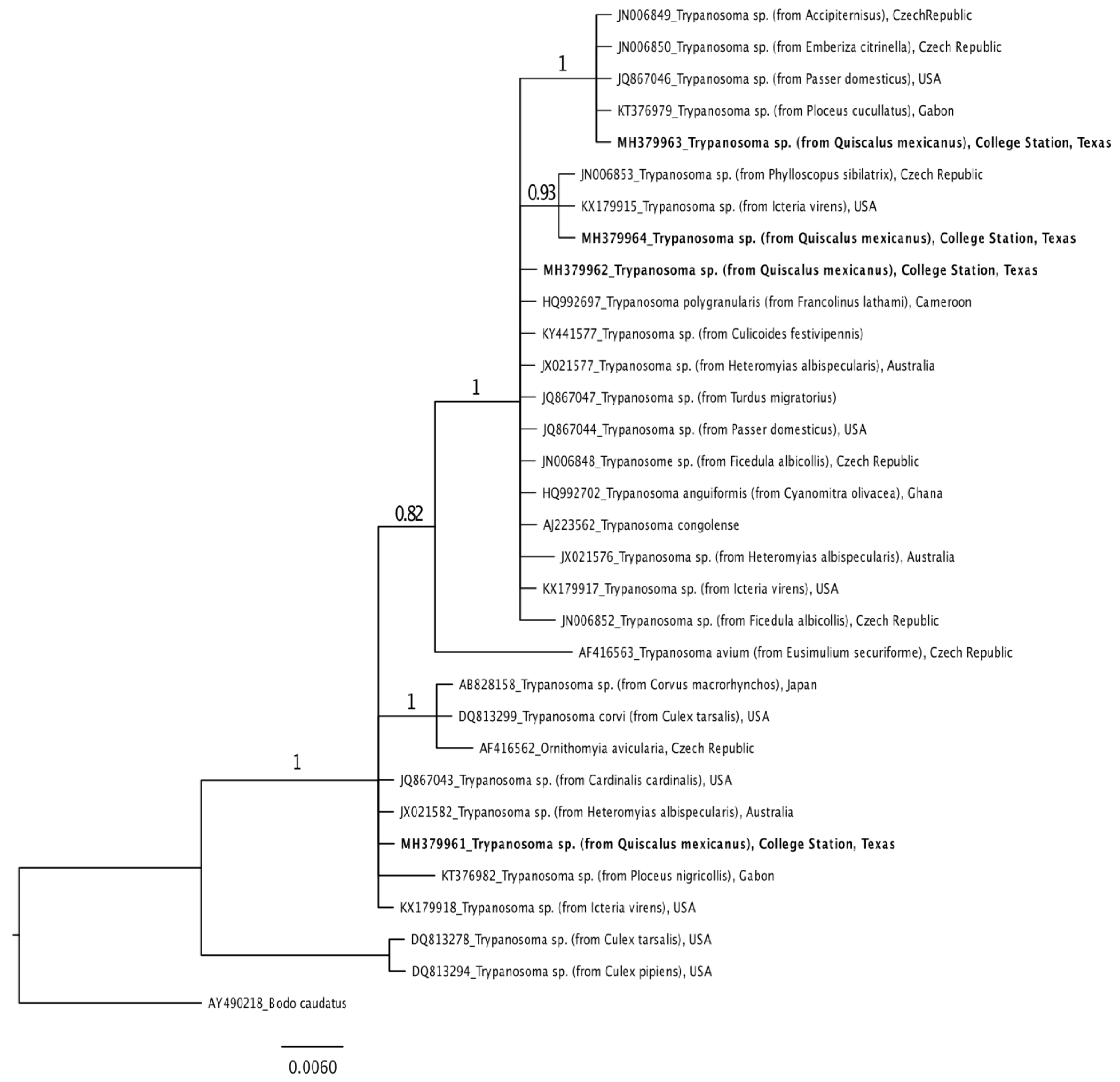
3.4. Trypanosome and Filarioid Nematode Phylogenetic Relatedness
3.5. Polyparasitism Assembly Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- DaCosta, J.M.; Wehtje, W.; Klicka, J. Historic genetic structuring and paraphyly within the great-tailed grackle. Condor 2018, 110, 170–177. [Google Scholar] [CrossRef] [Green Version]
- Wehtje, W. The range expansion of the great-tailed grackle (Quiscalus mexicanus Gmelin) in North America since 1880. J. Biogeogr. 2003, 30, 1593–1607. [Google Scholar] [CrossRef]
- Grabrucker, S.; Grabrucker, A.M. Rare Feeding Behavior of Great-Tailed Grackles (Quiscalus mexicanus) in the Extreme Habitat of Death Valley. Open Ornithol. J. 2010, 3, 101–104. [Google Scholar] [CrossRef] [Green Version]
- Rappole, J.H.; Kane, A.H.; Flores, R.H.; Tipton, A.R.; Koerth, N. Seasonal variation in habitat use by great-tailed grackles in the lower Rio Grande Valley. In Proceedings of the Great Plains Wild Damage Control Workshop, Fort Collins, CO, USA, 17–20 April 1989; p. 407. [Google Scholar]
- Greiner, E.C.; Bennett, G.F.; White, E.M.; Coombs, R.F. Distribution of the avian hematozoa of North America. Can. J. Zool. 1975, 53, 1762–1787. [Google Scholar] [CrossRef]
- Manwell, R. Leucocytozoa and other blood parasites of the Purple Grackle, Quiscalus quiscala quiscala. J. Parasitol. 1951, 37, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Herman, C.M. The blood protozoa of North American birds. Bird-Banding 1944, 1, 89–112. [Google Scholar] [CrossRef]
- Robinson, E.J., Jr. Culicoides crepuscularis (Malloch) (Diptera: Ceratopogonidae) as a host for Chandlerella quiscali (von Linstow, 1904) comb. N. (Filarioidea: Onchoceridae). J. Parasitol. 1971, 57, 772–776. [Google Scholar] [CrossRef]
- Merino, S.; Moreno, J.; Sanz, J.J.; Arriero, E. Are avian blood parasites pathogenic in the wild? A medication experiment in blue tits (Parus caeruleus). Proc. Royal Soc. B. 2000, 267, 2507–2510. [Google Scholar] [CrossRef] [Green Version]
- Valkiunas, G. Avian Malaria Parasites and Other Haemosporidia; CRC Press: Boca Raton, FL, USA, 2004. [Google Scholar]
- Atkinson, C.T.; Thomas, N.J.; Hunter, D.B. Parasitic Diseases of Wild Birds; John Wiley & Sons: Hoboken, NJ, USA, 2009. [Google Scholar]
- Zídková, L.; Cepicka, I.; Szabová, J.; Svobodová, M. Biodiversity of avian trypanosomes. Infect. Genet. Evol. 2012, 12, 102–112. [Google Scholar] [CrossRef]
- Grigar, M.K.; Cummings, K.J.; Rodriguez-Rivera, L.D.; Rankin, S.C.; Johns, K.; Hamer, G.L.; Hamer, S.A. Salmonella Surveillance Among great-tailed grackles (Quiscalus mexicanus) and Other Urban Bird Species in Eastern Texas. Vector-Borne Zoonotic Dis. 2016, 16, 752–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamer, G.L.; Anderson, T.K.; Berry, G.E.; Makohon-Moore, A.P.; Crafton, J.C.; Brawn, J.D.; Dolinski, A.C.; Krebs, B.L.; Ruiz, M.O.; Muzzall, P.M. Prevalence of filarioid nematodes and trypanosomes in American robins and house sparrows, Chicago USA. Int. J. Parasitol. 2013, 2, 42–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallon, S.M.; Ricklefs, R.E.; Swanson, B.; Bermingham, E. Detecting avian malaria: An improved polymerase chain reaction diagnostic. J. Parasitol. 2003, 89, 1044–1047. [Google Scholar] [CrossRef]
- Fecchio, A.; Lima, M.R.; Svensson-Coelho, M.; Marini, M.Â.; Ricklefs, R.E. Structure and organization of an avian haemosporidian assemblage in a Neotropical savanna in Brazil. Parasitology 2013, 140, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Bensch, S.; Hellgren, O.; Pérez-tris, J. MalAvi: A public database of malaria parasites and related haemosporidians in avian hosts based on mitochondrial cytochrome b lineages. Mol. Ecol. Resour. 2009, 9, 1353–1358. [Google Scholar] [CrossRef]
- Benson, D.A.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Wheeler, D.L. GenBank. Nucleic Acids Res. 2008, 36, D25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, S. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2008, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Ricklefs, R.E.; Swanson, B.L.; Fallon, S.M.; MartÍnez-AbraÍn, A.; Scheuerlein, A.; Gray, J.; Latta, S.C. Community relationships of avian malaria parasites in southern Missouri. Ecol. Monogr. 2005, 75, 543–559. [Google Scholar] [CrossRef]
- Sehgal, R.N.; Jones, H.I.; Smith, T.B. Host specificity and incidence of Trypanosoma in some African rainforest birds: A molecular approach. Mol. Ecol. 2001, 10, 2319–2327. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fungiflora, O.; Gascuel, O. A simple, fast and accurate method to estimate large phylogenies by maximum-likelihood. Syst. Biol. 2003, 52, 696704. [Google Scholar]
- Janovy, J.; Clopton, R.; Clopton, D.; Snyder, S.D.; Efting, A.; Krebs, L. Species density distributions as null models for ecologically significant interactions of parasite species in an assemblage. Ecol. Model. 1995, 77, 189–196. [Google Scholar] [CrossRef]
- Poulin, R. Richness, nestedness, and randomness in parasite infracommunity structure. Oecologia 1996, 105, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Toomey, M.B.; Butler, M.W.; Meadows, M.G.; Taylor, L.A.; Fokidis, H.B.; McGraw, K.J. A novel method for quantifying the glossiness of animals. Behav. Ecol. Sociobiol. 2010, 64, 1047–1055. [Google Scholar] [CrossRef]
- Selander, R.K.; Giller, D.R. Analysis of sympatry of great-tailed and boat-tailed grackles. Condor 1961, 63, 29–86. [Google Scholar] [CrossRef]
- Carlson, J.S.; Walther, E.; TroutFryxell, R.; Staley, S.; Tell, L.A.; Sehgal, R.N.; Barker, C.M.; Cornel, A.J. Identifying avian malaria vectors: Sampling methods influence outcomes. Parasit. Vectors 2015, 8, 365. [Google Scholar] [CrossRef] [Green Version]
- Medeiros, M.C.; Hamer, G.L.; Ricklefs, R.E. Host compatibility rather than vector–host-encounter rate determines the host range of avian Plasmodium parasites. Proc. Royal Soc. B. 2013, 280, 20122947. [Google Scholar] [CrossRef] [Green Version]
- Stone, W.; Weber, B.; Parks, F. Morbidity and mortality of birds due to avian malaria. NY Fish Game J. 1971, 18, 62–63. [Google Scholar]
- Medeiros, M.C.; Ellis, V.; Ricklefs, R. Specialized avian Haemosporida trade reduced host breadth for increased prevalence. J. Evol. Biol. 2014, 27, 2520–2528. [Google Scholar] [CrossRef]
- Medeiros, M.C.; Ricklefs, R.E.; Brawn, J.D.; Ruiz, M.O.; Goldberg, T.L.; Hamer, G.L. Overlap in the seasonal infection patterns of avian malaria parasites and West Nile virus in vectors and hosts. Am. J. Trop. Med. Hyg. 2016, 95, 1121–1129. [Google Scholar] [CrossRef] [PubMed]
- Votýpka, J.; Svobodova, M. Trypanosoma avium: Experimental transmission from black flies to canaries. Parasitol. Res. 2004, 92, 147–151. [Google Scholar] [CrossRef]
- Martin, E.; Chu, E.; Shults, P.; Golnar, A.; Swanson, D.A.; Benn, J.; Kim, D.; Schneider, P.; Pena, S.; Culver, C.; et al. Culicoides species community composition and infection status with parasites in an urban environment of east central Texas, USA. Parasites Vectors 2019, 12, 39. [Google Scholar] [CrossRef]
- Kelly, D.; Paterson, R.; Townsend, C.; Poulin, R.; Tompkins, D. Parasite spillback: A neglected concept in invasion ecology? Ecology 2009, 90, 2047–2056. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Kinsella, J.M.; Snow, R.W.; Hayes, M.M.; Falk, B.G.; Reed, R.N.; Mazzotti, F.J.; Guyer, C.; Romagosa, C.M. Parasite spillover: Indirect effects of invasive Burmese pythons. Ecol. Evol. 2018, 8, 830–840. [Google Scholar] [CrossRef]
- Nelson, F.B.; Brown, G.P.; Shilton, C.; Shine, R. Helpful invaders: Can cane toads reduce the parasite burdens of native frogs? Int. J. Parasitol. 2015, 4, 295–300. [Google Scholar] [CrossRef] [Green Version]
- Edwards, E.E.; Dangoudoubiyam, S.; Hoppes, S.M.; Porter, B.F. Granulomatous filarial encephalomyelitis caused by Chandlerella quiscali in a northern crested caracara (Caracara cheriway). J. Zoo Wildl. Med. 2017, 48, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Telfer, S.; Bown, K. The effects of invasion on parasite dynamics and communities. Funct. Ecol. 2012, 26, 1288–1299. [Google Scholar] [CrossRef]
- Belden, L.K.; Harris, R.N. Infectious diseases in wildlife: The community ecology context. Front. Ecol. Environ. 2007, 5, 533–539. [Google Scholar] [CrossRef]
- Medeiros, M.C.I.; Anderson, T.K.; Higashiguchi, J.M.; Kitron, U.D.; Walker, E.D.; Brawn, J.D.; Krebs, B.L.; Ruiz, M.O.; Goldberg, T.L.; Ricklefs, R.E.; et al. An inverse association between West Nile virus serostatus and avian malaria infection status. Parasites Vector 2014, 7, 415. [Google Scholar] [CrossRef] [Green Version]
- Young, H.S.; Parker, I.M.; Gilbert, G.S.; Guerra, A.S.; Nunn, C.L. Introduced Species, Disease Ecology, and Biodiversity-Disease Relationships. Trends Ecol. Evol. 2017, 32, 41–54. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Haematoparasite | Genus | Lineage | Count | Sample Size | Est. Prevalence | 95% C.I. |
|---|---|---|---|---|---|---|
| Filarioid nematode | - | - | 24 | 46 | 0.52 | 0.37–0.66 |
| Avian Trypanosome | - | - | 11 | 46 | 0.24 | 0.12–0.36 |
| Haemosporida | - | - | 44 | 60 | 0.73 | 0.62–0.85 |
| * Haemoproteus | - | 38 | 60 | 0.63 | 0.51–0.76 | |
| * Haemoproteus | CHI18PA | 31 | 60 | 0.52 | 0.37–0.63 | |
| * Haemoproteus | CHI22PA | 6 | 60 | 0.1 | 0.03–0.18 | |
| * Haemoproteus | CHI18PA/CHI22PA | 1 | 60 | 0.02 | 0–0.05 | |
| Plasmodium | Unclassified | 1 | 60 | 0.02 | 0–0.05 | |
| Undetermined | - | 5 | 60 | 0.08 | - |
| Infection Status | Sample Size | Expected | Observed | Estimated Portion of Population | 95% Confidence Interval |
|---|---|---|---|---|---|
| No infection | 45 | 4 | 6 | 0.13 | 0.03–0.22 |
| Haemosporida sp. (H) | 45 | 12 | 9 | 0.20 | 0.08–0.32 |
| Filarial Nematode sp. (F) | 45 | 5 | 4 | 0.09 | 0.01–0.17 |
| Trypanosome sp. (T) | 45 | 1 | 2 | 0.04 | 0–0.10 |
| H:F co-infection | 45 | 13 | 16 | 0.36 | 0.22–0.50 |
| H:T co-infection | 45 | 4 | 4 | 0.09 | 0.01–0.17 |
| F:T co-infection | 45 | 1 | 0 | 0.00 | 0 |
| H:F:T co-infection | 45 | 4 | 4 | 0.09 | 0.01–0.17 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Golnar, A.J.; Medeiros, M.C.I.; Rosenbaum, K.; Bejcek, J.; Hamer, S.A.; Hamer, G.L. Vector-Borne Blood Parasites of the Great-Tailed Grackle (Quiscalus mexicanus) in East-Central Texas, USA. Microorganisms 2021, 9, 504. https://doi.org/10.3390/microorganisms9030504
Golnar AJ, Medeiros MCI, Rosenbaum K, Bejcek J, Hamer SA, Hamer GL. Vector-Borne Blood Parasites of the Great-Tailed Grackle (Quiscalus mexicanus) in East-Central Texas, USA. Microorganisms. 2021; 9(3):504. https://doi.org/10.3390/microorganisms9030504
Chicago/Turabian StyleGolnar, Andrew J., Matthew C. I. Medeiros, Katlyn Rosenbaum, Justin Bejcek, Sarah A. Hamer, and Gabriel L. Hamer. 2021. "Vector-Borne Blood Parasites of the Great-Tailed Grackle (Quiscalus mexicanus) in East-Central Texas, USA" Microorganisms 9, no. 3: 504. https://doi.org/10.3390/microorganisms9030504