
DNA Polymerase B1 Binding Protein 1 Is Important for DNA Repair by Holoenzyme PolB1 in the Extremely Thermophilic Crenarchaeon Sulfolobus acidocaldarius


Abstract
1. Introduction
2. Materials and Methods
2.1. Strains and Growth Conditions
2.2. General DNA Manipulation
2.3. Construction of the PolB1-binding Protein Gene-Deleted Strains
2.4. Construction of the pyrE-Proficient Strains
2.5. Growth Temperature Range
2.6. DNA Damage Sensitivity Tests
2.7. Spontaneous Mutation Analysis
3. Results
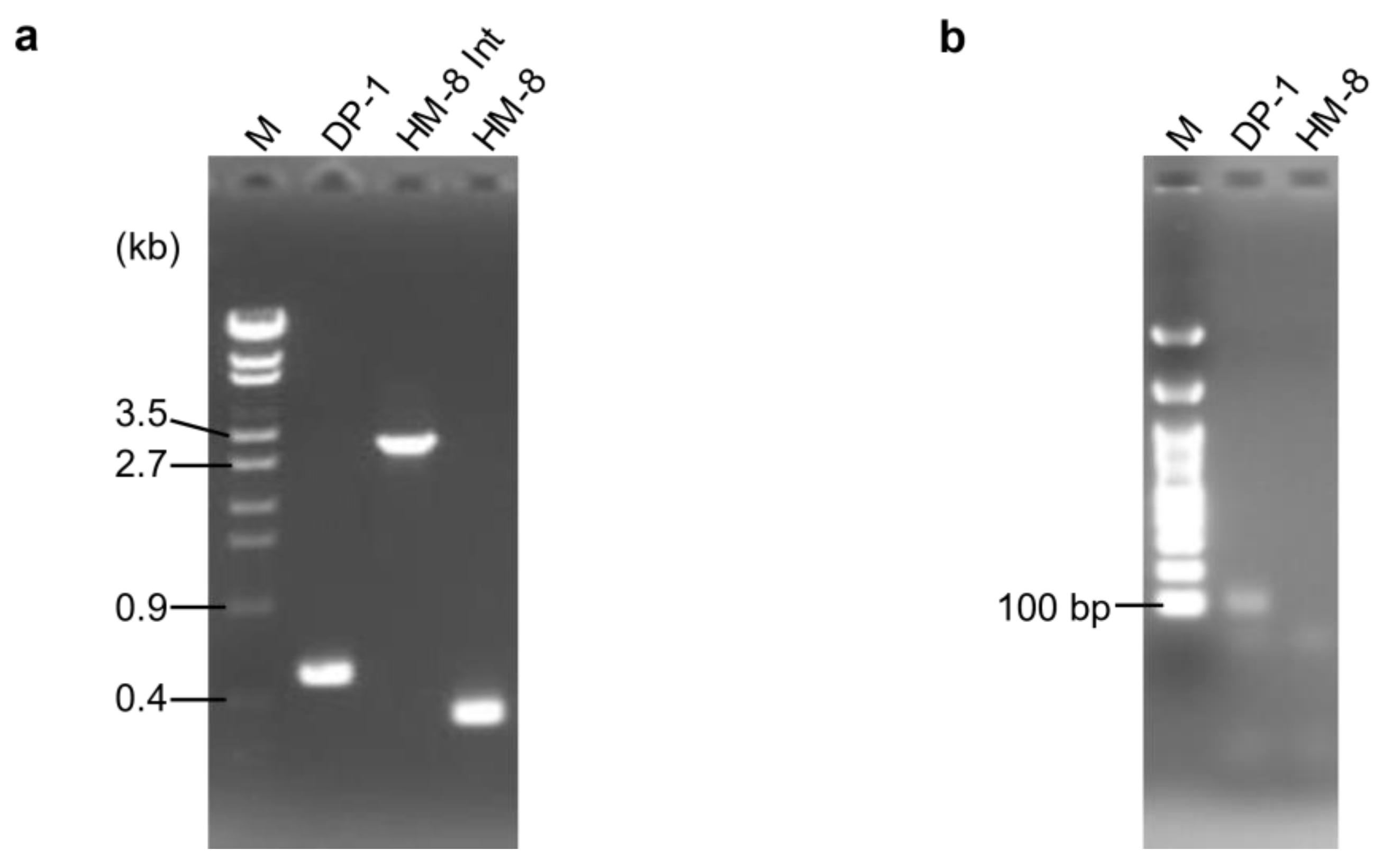
3.1. Deletion of PolB1-Binding Protein Genes
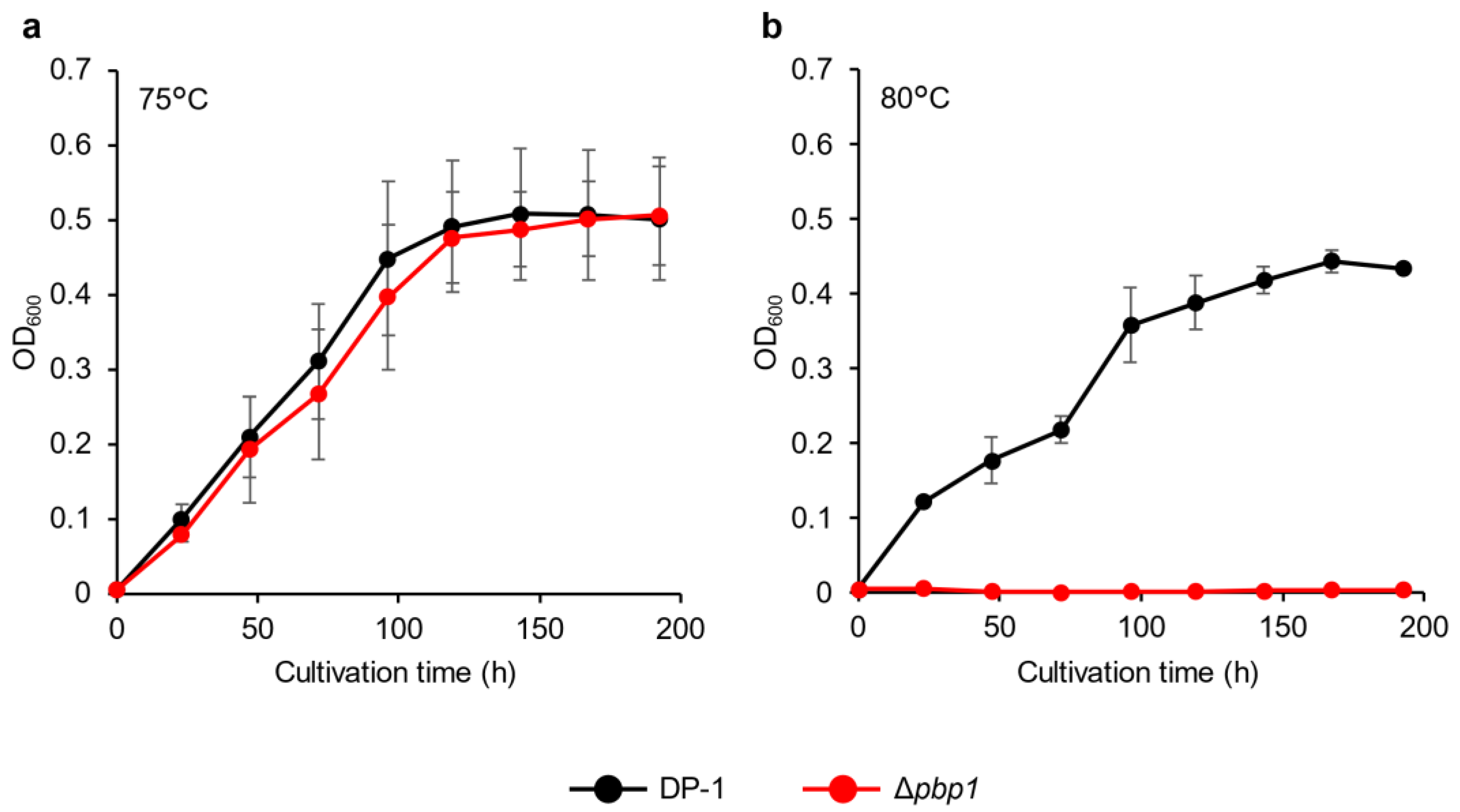
3.2. Growth Properties at Various Temperatures
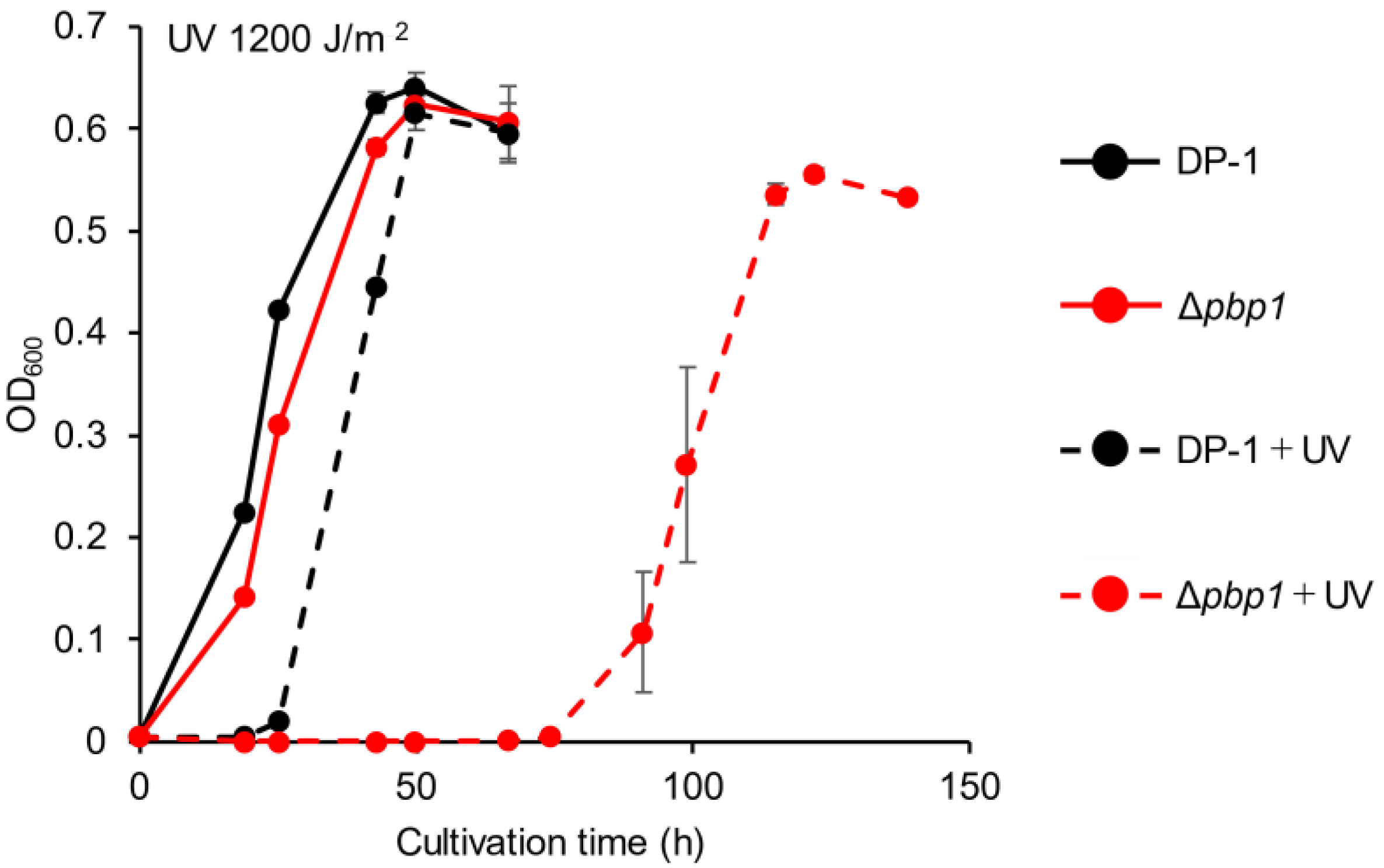
3.3. Sensitivity to UV Irradiation
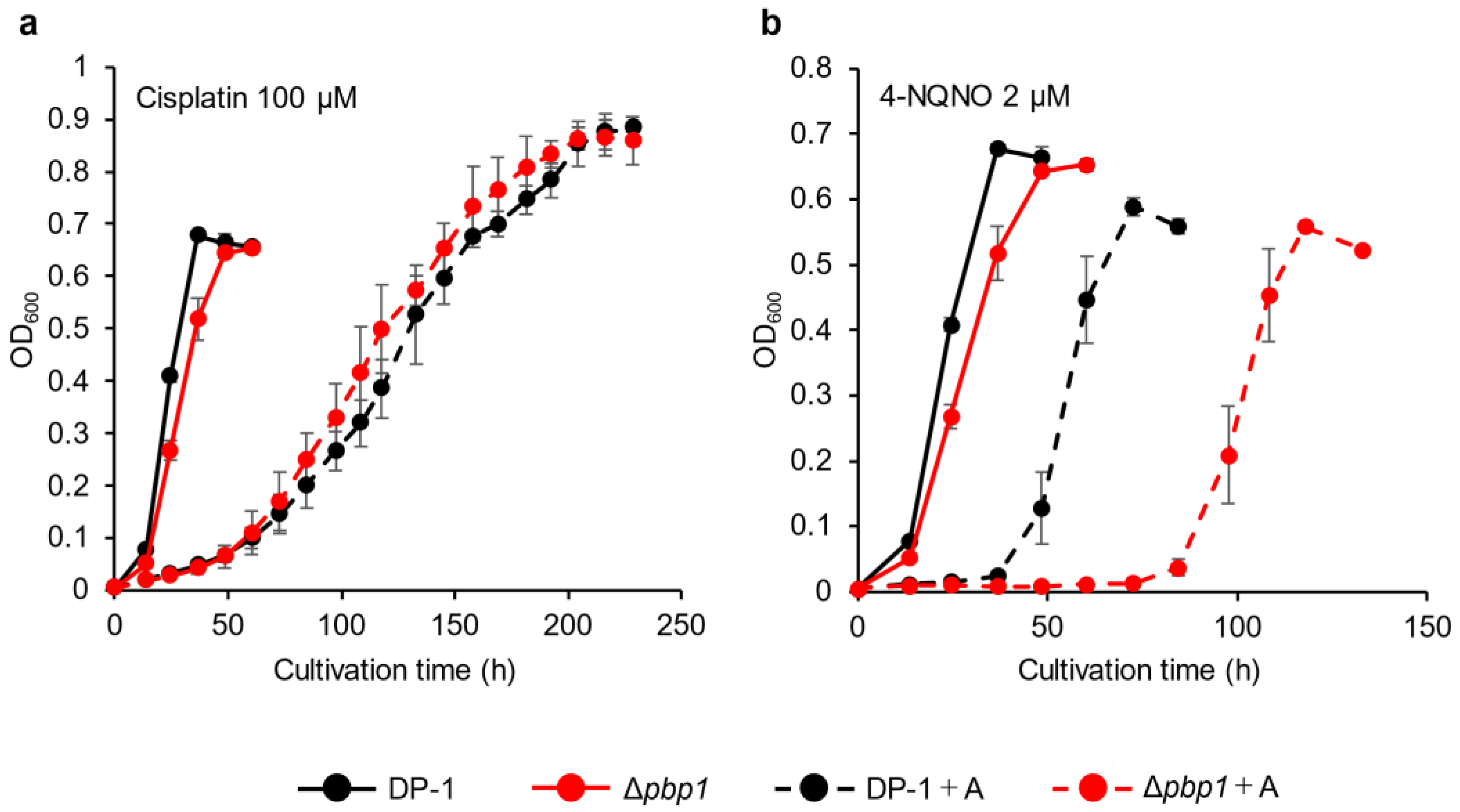
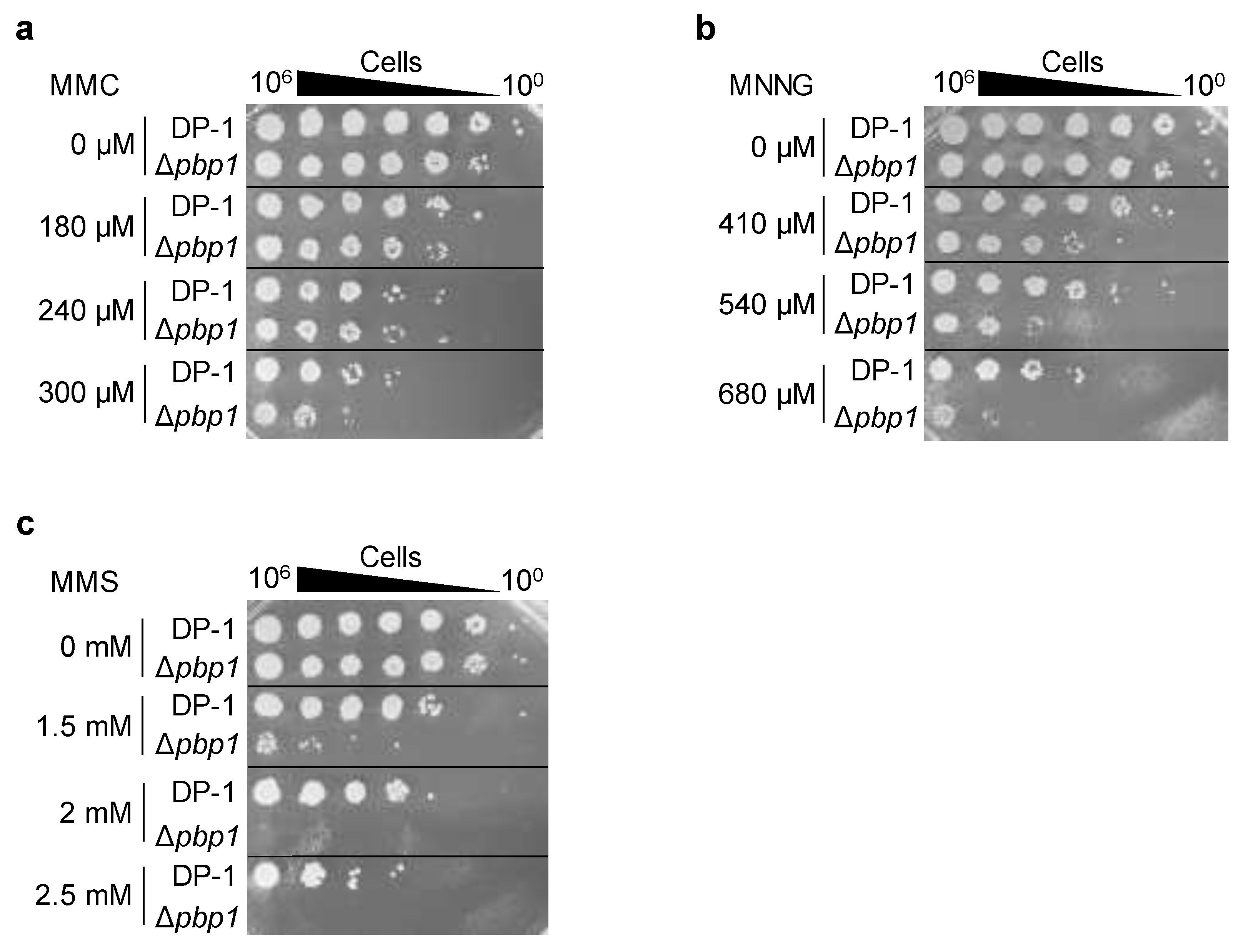
3.4. Sensitivity to Chemical Mutagens
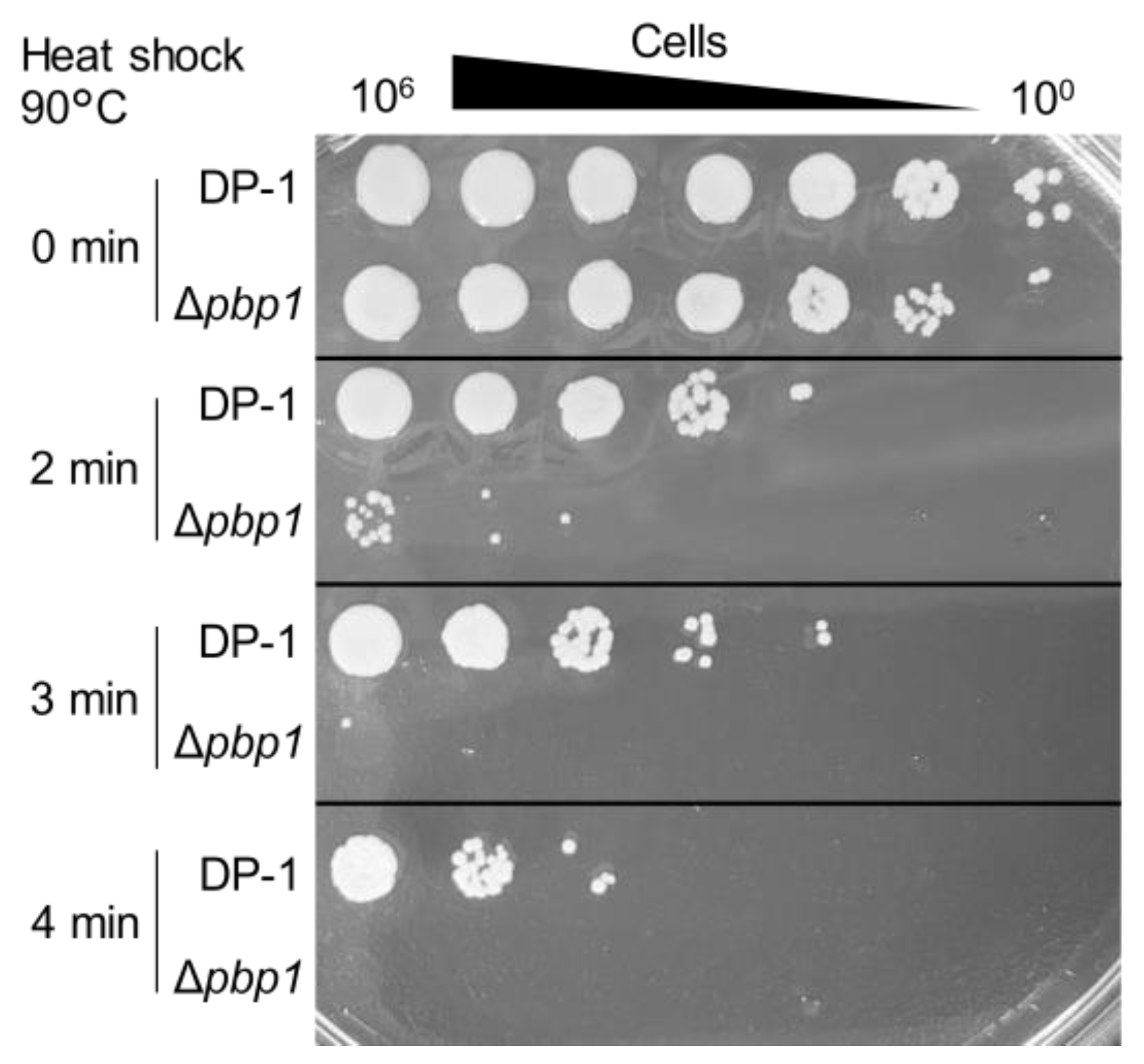
3.5. Sensitivity to Heat-Shock Treatment
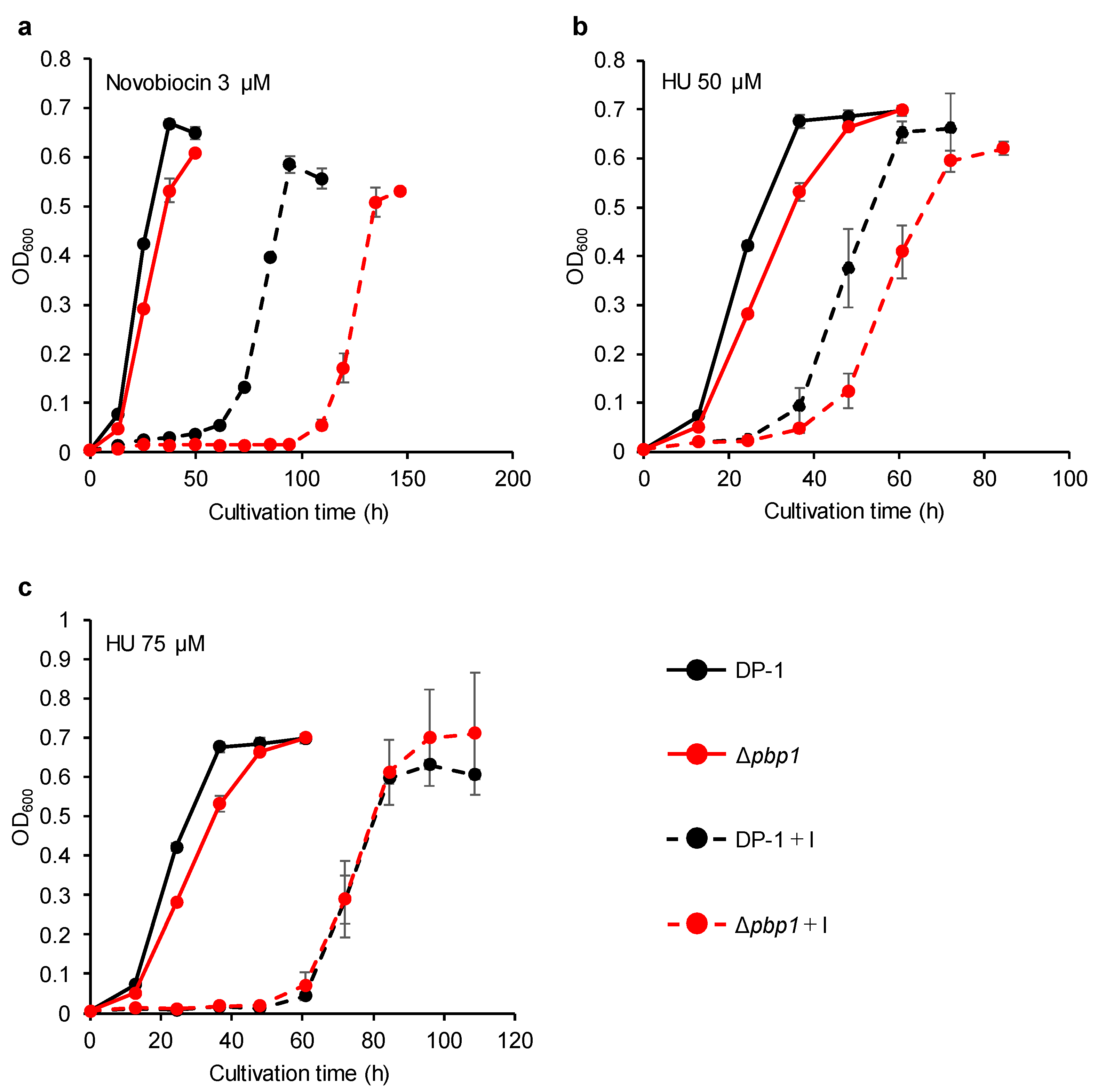
3.6. Sensitivity to DNA Replication Inhibitors
3.7. Estimation of Mutation Rates
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Johansson, E.; Dixon, N. Replicative DNA polymerases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012799. [Google Scholar] [CrossRef] [PubMed]
- White, M.F. DNA repair. In Archaea: Evolution, Physiology and Molecular Biology; Garrett, R.A., Klenk, H.P., Eds.; Blackwell Publishing Ltd.: Hoboken, NJ, USA, 2007; pp. 171–183. [Google Scholar]
- White, M.F.; Allers, T. DNA repair in the archaea–an emerging picture. FEMS Microbiol. Rev. 2018, 42, 514–526. [Google Scholar] [CrossRef]
- McHenry, C.S. Bacterial replicases and related polymerases. Curr. Opin. Chem. Biol. 2011, 15, 587–594. [Google Scholar] [CrossRef]
- Raia, P.; Delarue, M.; Sauguet, L. An updated structural classification of replicative DNA polymerases. Biochem. Soc. Trans. 2019, 47, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, T.A.; Burgers, P.M. Dividing the workload at a eukaryotic replication fork. Trends Cell Biol. 2008, 18, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Sarmiento, F.; Long, F.; Cann, I.; Whitman, W.B. Diversity of the DNA replication system in the archaea domain. Archaea 2014, 2014, 675946. [Google Scholar] [CrossRef]
- Doublie, S.; Zahn, K.E. Structural insights into eukaryotic DNA replication. Front. Microbiol. 2014, 5, 444. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Aggarwal, A.K.; Rechkoblit, O. Eukaryotic DNA polymerases. Curr. Opin. Struct. Biol. 2018, 53, 77–87. [Google Scholar] [CrossRef]
- Makarova, K.S.; Krupovic, M.; Koonin, E.V. Evolution of replicative DNA polymerases in archaea and their contributions to the eukaryotic replication machinery. Front. Microbiol. 2014, 5, 354. [Google Scholar] [CrossRef]
- Cooper, C.D.O. Archaeal DNA polymerases: New frontiers in DNA replication and repair. Emerg. Top. Life Sci. 2018, 2, 503–516. [Google Scholar] [CrossRef]
- Cubonová, L.; Richardson, T.; Burkhart, B.W.; Kelman, Z.; Connolly, B.A.; Reeve, J.N.; Santangelo, T.J. Archaeal DNA polymerase D but not DNA polymerase B is required for genome replication in Thermococcus kodakarensis. J. Bacteriol. 2013, 195, 2322–2328. [Google Scholar] [CrossRef] [PubMed]
- Sarmiento, F.; Mrázek, J.; Whitman, W.B. Genome-scale analysis of gene function in the hydrogenotrophic methanogenic archaeon Methanococcus maripaludis. Proc. Natl. Acad. Sci. USA 2013, 110, 4726–4731. [Google Scholar] [CrossRef] [PubMed]
- Kushida, T.; Narumi, I.; Ishino, S.; Ishino, Y.; Fujiwara, S.; Imanaka, T.; Higashibata, H. Pol B, a family B DNA polymerase, in Thermococcus kodakarensis is important for DNA repair, but not DNA replication. Microbes Environ. 2019, 34, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Miyabayashi, H.; Jain, R.; Suzuki, S.; Grogan, D.W.; Kurosawa, N. PolB1 is sufficient for DNA replication and repair under normal growth conditions in the extremely thermophilic crenarchaeon Sulfolobus acidocaldarius. Front. Microbiol. 2020, 11, 613375. [Google Scholar] [CrossRef]
- Feng, X.; Liu, X.; Xu, R.; Zhao, R.; Feng, W.; Liao, J.; Han, W.; She, Q. A unique B-Family DNA polymerase facilitating error-prone DNA damage tolerance in Crenarchaeota. Front. Microbiol. 2020, 11, 1585. [Google Scholar] [CrossRef] [PubMed]
- Cann, I.K.O.; Komori, K.; Toh, H.; Kanai, S.; Ishino, Y. A heterodimeric DNA polymerase: Evidence that members of Euryarchaeota possess a distinct DNA polymerase. Proc. Natl. Acad. Sci. USA 1998, 95. [Google Scholar] [CrossRef] [PubMed]
- Sauguet, L.; Raia, P.; Henneke, G.; Delarue, M. Shared active site architecture between archaeal PolD and multi-subunit RNA polymerases revealed by X-ray crystallography. Nat. Commun. 2016, 7, 12227–12238. [Google Scholar] [CrossRef]
- Klimczak, L.J.; Grummt, F.; Burger, K.J. Purification and characterization of DNA polymerase from the archaebacterium Sulfolobus acidocaldarius. Nucleic Acids Res. 1985, 13, 5269–5282. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Beattie, T.R.; Rojas, A.L.; Schermerhorn, K.; Gristwood, T.; Trinidad, J.C.; Albers, S.V.; Roversi, P.; Gardner, A.F.; Abrescia, N.G.A.; et al. Identification and characterization of a heterotrimeric archaeal DNA polymerase holoenzyme. Nat. Commun. 2017, 8, 15075. [Google Scholar] [CrossRef]
- Sakai, H.D.; Kurosawa, N. Saccharolobus caldissimus gen. nov., sp. nov., a facultatively anaerobic iron-reducing hyperthermophilic archaeon isolated from an acidic terrestrial hot spring, and reclassification of Sulfolobus solfataricus as Saccharolobus solfataricus comb. nov. and Sulfolobus shibatae as Saccharolobus shibatae comb. nov. Int. J. Syst. Evol. Microbiol. 2018, 68, 1271–1278. [Google Scholar] [CrossRef]
- Cranford, M.T.; Kaszubowski, J.D.; Trakselis, M.A. A hand-off of DNA between archaeal polymerases allows high-fidelity replication to resume at a discrete intermediate three bases past 8-oxoguanine. Nucleic Acids Res. 2020, 48, 10986–10997. [Google Scholar] [CrossRef]
- Suzuki, S.; Kurosawa, N. Development of the multiple gene knockout system with one-step PCR in thermoacidophilic crenarchaeon Sulfolobus acidocaldarius. Archaea 2017, 2017, 7459310. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Kurosawa, N. Disruption of the gene encoding restriction endonuclease SuaI and development of a host-vector system for the thermoacidophilic archaeon Sulfolobus acidocaldarius. Extremophiles 2016, 20, 139–148. [Google Scholar] [CrossRef]
- Grogan, D.W. Isolation of Sulfolobus acidocaldarius mutants. In Archaea: A Laboratory Manual-Thermophiles; Robb, F.T., Place, A.R., Sowers, K.R., Schreier, H.J., DasSarma, S., Fleishmann, E.M., Eds.; CSH Press: Cold Spring Harbor, NY, USA, 1995; pp. 125–132. [Google Scholar]
- Suzuki, S.; Kurosawa, N. Endonucleases responsible for DNA repair of helix-distorting DNA lesions in the thermophilic crenarchaeon Sulfolobus acidocaldarius in vivo. Extremophiles 2019, 23, 613–624. [Google Scholar] [CrossRef]
- Reilly, M.S.; Grogan, D.W. Characterization of intragenic recombination in a hyperthermophilic archaeon via conjugational DNA exchange. J. Bacteriol. 2001, 183, 2943–2946. [Google Scholar] [CrossRef]
- Courcelle, J.; Crowley, D.J.; Hanawalt, P.C. Recovery of DNA replication in UV-irradiated Escherichia coli requires both excision repair and RecF protein function. J. Bacteriol. 1999, 181, 916–922. [Google Scholar] [CrossRef]
- Lopes, M.; Foiani, M.; Sogo, J.M. Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol. Cell 2006, 21, 15–27. [Google Scholar] [CrossRef]
- Dorazi, R.; Götz, D.; Munro, S.; Bernander, R.; White, M.F. Equal rates of repair of DNA photoproducts in transcribed and non-transcribed strands in Sulfolobus solfataricus. Mol. Microbiol. 2007, 63, 521–529. [Google Scholar] [CrossRef]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef]
- Hjort, K.; Bernander, R. Cell cycle regulation in the hyperthermophilic crenarchaeon Sulfolobus acidocaldarius. Mol. Microbiol. 2001, 40, 225–234. [Google Scholar] [CrossRef]
- Liew, L.P.; Lim, Z.Y.; Cohen, M.; Kong, Z.; Marjavaara, L.; Chabes, A.; Bell, S.D. Hydroxyurea-mediated cytotoxicity without inhibition of ribonucleotide reductase. Cell Rep. 2016, 17, 1657–1670. [Google Scholar] [CrossRef]
- Cariello, N.F.; Swenberg, J.A.; Skopek, T.R. Fidelity of Thermococcus litoralis DNA polymerase (VentTM) in PCR determined by denaturing gradient gel electrophoresis. Nucleic Acids Res. 1991, 19, 4193–4198. [Google Scholar] [CrossRef]
- Lundberg, K.S.; Shoemaker, D.D.; Adams, M.W.; Short, J.M.; Sorge, J.A.; Mathur, E.J. High-fidelity amplification using a thermostable DNA polymerase isolated from Pyrococcus furiosus. Gene 1991, 108, 1–6. [Google Scholar] [CrossRef]
- Takagi, M.; Nishioka, M.; Kakihara, H.; Kitabayashi, M.; Inoue, H.; Kawakami, B.; Oka, M.; Imanaka, T. Characterization of DNA polymerase from Pyrococcus sp. strain KOD1 and its application to PCR. Appl. Environ. Microbiol. 1997, 63, 4504–4510. [Google Scholar] [CrossRef]
- Kähler, M.; Antranikian, G. Cloning and characterization of a family B polymerase from the hyperthermophilic crenarchaeon Pyrobaculum islandicum. J. Bacteriol. 2000, 182, 655–663. [Google Scholar] [CrossRef]
- Seo, K.J.; Cho, S.S.; Ppyun, H.W.; Youn, M.H.; Kim, S.H.; Seo, B.S.; Kwon, S.T. Characterization of a family B DNA polymerase from the hyperthermophilic crenarchaeon Ignicoccus hospitalis KIN4/I and its application to PCR. Appl. Biochem. Biotechnol. 2014, 173, 1108–1120. [Google Scholar] [CrossRef]
- Daimon, K.; Ishino, S.; Imai, N.; Nagumo, S.; Yamagami, T.; Matsukawa, H.; Ishino, Y. Two family B DNA polymerases from Aeropyrum pernix, based on revised translational frames. Front. Mol. Biosci. 2018, 5, 37. [Google Scholar] [CrossRef] [PubMed]
- Iwai, T. Functional and phylogenetic analysis of DNA polymerase and cofactor PCNA in Crenarchaeota. Ph.D. Thesis, Soka University, Tokyo, Japan, 2001. [Google Scholar]
- Sun, F.; Huang, L. Sulfolobus chromatin proteins modulate strand displacement by DNA polymerase B1. Nucleic Acids Res. 2013, 41, 8182–8195. [Google Scholar] [CrossRef]
- Choli, T.; Henning, P.; Wittmann-Liebold, B.; Reinhardt, R. Isolation, characterization and microsequence analysis of a small basic methylated DNA-binding protein from the Archaebacterium, Sulfolobus solfataricus. Biochim. Biophys. Acta 1988, 950, 193–203. [Google Scholar] [CrossRef]
- Zhang, C.; Phillips, A.P.R.; Wipfler, R.L.; Olsen, G.J.; Whitaker, R.J. The essential genome of the crenarchaeal model Sulfolobus islandicus. Nat. Commun. 2018, 9, 4908. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Feng, Y.; Zhang, Z.; Yao, H.; Luo, Y.; Wang, J.; Huang, L. Biochemical and structural characterization of Cren7, a novel chromatin protein conserved among Crenarchaea. Nucleic Acids Res. 2007, 36, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.H.; Suzuki, M.; Adman, E.; Shinkai, A.; Loeb, L.A. Prokaryotic DNA polymerase I: Evolution, structure, and “base flipping” mechanism for nucleotide selection. J. Mol. Biol. 2001, 308, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Yasui, A. Alternative Excision Repair Pathways. Cold Spring Harb. Perspect. Biol. 2013, 5, a012617. [Google Scholar] [CrossRef]
- Grogan, D.W. Understanding DNA repair in hyperthermophilic archaea: Persistent gaps and other reactions to focus on the fork. Archaea 2015, 2015, 942605. [Google Scholar] [CrossRef]
- Fujikane, R.; Ishino, S.; Ishino, Y.; Forterre, P. Genetic analysis of DNA repair in the hyperthermophilic archaeon, Thermococcus kodakarensis. Genes Genet. Syst. 2010, 85, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Tian, B.; Li, S.; Ao, X.; Dalgaard, K.; Gökce, S.; Liang, Y.; She, Q. Genetic manipulation in Sulfolobus islandicus and functional analysis of DNA repair genes. Biochem. Soc. Trans. 2013, 41, 405–410. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains or DNAs | Relevant Characteristic(s) | Source or Reference |
|---|---|---|
| Strains | ||
| S. acidocaldarius | ||
| DP-1 | SK-1 with Δphr (ΔpyrE ΔsuaI Δphr) | [23,24] |
| HM-8 | DP-1 with Δpbp1 (ΔpyrE ΔsuaI Δphr Δpbp1) | This study |
| Plasmid DNA | ||
| placSpyrE | Plasmid DNA carrying 0.8 kb of the 5′ and 3′ homologous regions of the suaI locus at both ends of the pyrE-lacS dual marker | [23] |
| PCR products | ||
| MONSTER-pbp1 | Linear DNA containing the 38-bp 5′ and 30-bp 3′ sequences of the pbp1 flanking regions and a 38-bp region of pbp1 as the Tg-arm at both ends of the pyrE-lacS dual marker | This study |
| MONSTER-pbp2 | Linear DNA containing the 38-bp 5′ and 30-bp 3′ sequences of the pbp2 flanking regions and a 38-bp region of pbp2 as the Tg-arm at both ends of the pyrE-lacS dual marker | This study |
| MONSTER-pbp2n | Linear DNA containing the 38-bp 5′ and 30-bp 3′ sequences of the pbp2 flanking regions and a 38-bp region of pbp2 as the Tg-arm at both ends of the pyrE-lacS dual marker | This study |
| Primers | Sequence (5′-3′) 1 |
|---|---|
| MONSTER-pbp1-F | tatacgtttcaaaatgcaaatattaaaaatagttagaagagcacgtactctcacataatttctcatacTGTTTTTCTCTATATCAATCTC |
| MONSTER-pbp1-R | gttttccattttggcgtccaacgtgtagttgatgacatACTCCTAGATCTAAAACTAAAG |
| MONSTER-pbp2-F | attattatatagtaatggaatttataaggtgaagcttaaaggctcttggaataagtgatccagagaaaTGTTTTTCTCTATATCAATCTC |
| MONSTER-pbp2-R | aaatatttcttcgccttctctaattcgtcctctggcaaACTCCTAGATCTAAAACTAAAG |
| MONSTER-pbp2n-F | aggacgaattagagaaggcgaagaaatatttccagaacaaggctcttggaataagtgatccagagaaaTGTTTTTCTCTATATCAATCTC |
| MONSTER-pbp2n-R | aattccctgaccgctaaaatctcgcctacggaaactacACTCCTAGATCTAAAACTAAAG |
| pbp1-out-F | tgatgatgacaatttgaatctc |
| pbp1-out-R | aattcctcctagcatgtatac |
| pbp1-in-F | aagatatagatatctgttttgac |
| pbp1-in-R | tttggcgtattaccttttttac |
| SAMR31-F | gatttcgtgaaagctctacttg |
| SAMR31-R | tttttctcagctctgatgtatc |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miyabayashi, H.; Sakai, H.D.; Kurosawa, N. DNA Polymerase B1 Binding Protein 1 Is Important for DNA Repair by Holoenzyme PolB1 in the Extremely Thermophilic Crenarchaeon Sulfolobus acidocaldarius. Microorganisms 2021, 9, 439. https://doi.org/10.3390/microorganisms9020439
Miyabayashi H, Sakai HD, Kurosawa N. DNA Polymerase B1 Binding Protein 1 Is Important for DNA Repair by Holoenzyme PolB1 in the Extremely Thermophilic Crenarchaeon Sulfolobus acidocaldarius. Microorganisms. 2021; 9(2):439. https://doi.org/10.3390/microorganisms9020439
Chicago/Turabian StyleMiyabayashi, Hiroka, Hiroyuki D. Sakai, and Norio Kurosawa. 2021. "DNA Polymerase B1 Binding Protein 1 Is Important for DNA Repair by Holoenzyme PolB1 in the Extremely Thermophilic Crenarchaeon Sulfolobus acidocaldarius" Microorganisms 9, no. 2: 439. https://doi.org/10.3390/microorganisms9020439
APA StyleMiyabayashi, H., Sakai, H. D., & Kurosawa, N. (2021). DNA Polymerase B1 Binding Protein 1 Is Important for DNA Repair by Holoenzyme PolB1 in the Extremely Thermophilic Crenarchaeon Sulfolobus acidocaldarius. Microorganisms, 9(2), 439. https://doi.org/10.3390/microorganisms9020439