
Characterization of Erysipelothrix rhusiopathiae Isolates from Diseased Pigs in 15 Chinese Provinces from 2012 to 2018

 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Isolates
2.2. MLST Analysis
2.3. Mouse Infection Experiment
2.4. Antimicrobial Susceptibility Assay
2.5. Genome Sequencing and Comparative Genomic Analysis
2.6. Detection of the Distribution of Drug Resistance Genes and Mutants
2.7. Analysis of the Contribution of Suspected Drug Resistance Genes and Mutants
2.8. Accession Numbers
2.9. Statistical Analysis
3. Results
3.1. MLST and Virulence o thef Isolates
3.2. Antimicrobial Susceptibility
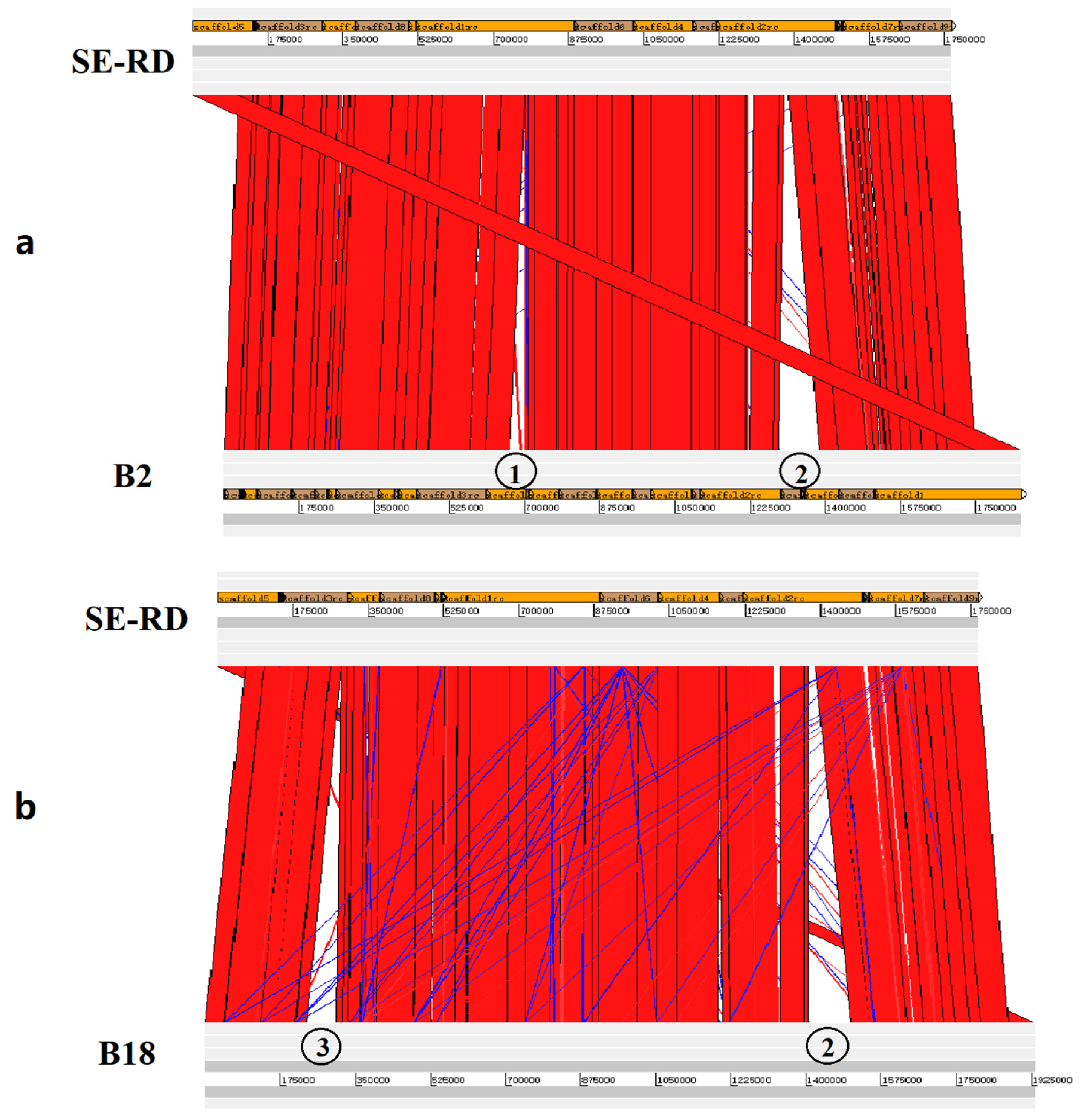
3.3. Genome Sequencing and Comparative Genomic Analysis
3.4. Distribution of Drug Resistance Genes and Resistance-Conferring Mutants
3.5. Contribution of the Newly Identified Drug Resistance Genes and Resistance-Conferring Mutants
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pal, N.; Bender, J.S.; Opriessnig, T. Rapid detection and differentiation of Erysipelothrix spp. by a novel multiplex real-time PCR assay. J. Appl. Microbiol. 2010, 108, 1083–1093. [Google Scholar] [CrossRef]
- Janssen, T.; Voss, M.; Kuhl, M.; Semmler, T.; Philipp, H.C.; Ewers, C. A combinational approach of multilocus sequence typing and other molecular typing methods in unravelling the epidemiology of Erysipelothrix rhusiopathiae strains from poultry and mammals. Vet. Res. 2015, 46, 84. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, H.; Brannstrom, S.; Skarin, H.; Chirico, J. Characterization of Erysipelothrix rhusiopathiae isolates from laying hens and poultry red mites (Dermanyssus gallinae) from an outbreak of erysipelas. Avian Pathol. 2010, 39, 505–509. [Google Scholar] [CrossRef]
- Ersdal, C.; Jorgensen, H.J.; Lie, K.I. Acute and Chronic Erysipelothrix rhusiopathiae Infection in Lambs. Vet. Pathol. 2015, 52, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, A.; Lebarbenchon, C.; Bourret, V.; Bastien, M.; Lagadec, E.; Thiebot, J.B.; Boulinier, T.; Delord, K.; Barbraud, C.; Marteau, C.; et al. Avian cholera outbreaks threaten seabird species on Amsterdam Island. PLoS ONE 2018, 13, e0197291. [Google Scholar] [CrossRef]
- Lee, J.J.; Kim, D.H.; Lim, J.J.; Kim, D.G.; Chang, H.H.; Lee, H.J.; Kim, S.H.; Rhee, M.H.; Endale, M.; Imada, Y.; et al. Characterization and identification of Erysipelothrix rhusiopathiae isolated from an unnatural host, a cat, with a clinical manifestation of depression. J. Vet. Med. Sci. 2011, 73, 149–154. [Google Scholar] [CrossRef]
- Marshall, K.R.; Walton, S.A.; Boyd, M.; Bishop, B.; Wellehan, J.; Craft, W.; Santoro, D. Erysipeloid lesions caused by Erysipelothrix rhusiopathiae in a dog: Clinical and histopathological findings, molecular diagnosis and treatment. Vet. Dermatol. 2019, 30, 434-e134. [Google Scholar] [CrossRef] [PubMed]
- Shimoji, Y.; Osaki, M.; Ogawa, Y.; Shiraiwa, K.; Nishikawa, S.; Eguchi, M.; Yamamoto, T.; Tsutsui, T. Wild boars: A potential source of Erysipelothrix rhusiopathiae infection in Japan. Microbiol. Immunol. 2019, 63, 465–468. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Garcia, A.I.; Muller, F.; Rodler, F.S.; Traub, F.; Heilmann, R.M. Infective endocarditis due to Erysipelothrix rhusiopathiae in a dog—A case report. BMC Vet. Res. 2020, 16, 328. [Google Scholar] [CrossRef] [PubMed]
- Cano-Manuel, F.J.; Lopez-Olvera, J.; Fandos, P.; Soriguer, R.C.; Perez, J.M.; Granados, J.E. Long-term monitoring of 10 selected pathogens in wild boar (Sus scrofa) in Sierra Nevada National Park, southern Spain. Vet. Microbiol. 2014, 174, 148–154. [Google Scholar] [CrossRef]
- McNeil, M.; Gerber, P.F.; Thomson, J.; Williamson, S.; Opriessnig, T. Serotypes and Spa types of Erysipelothrix rhusiopathiae isolates from British pigs (1987 to 2015). Vet. J. 2017, 225, 13–15. [Google Scholar] [CrossRef]
- To, H.; Sato, H.; Tazumi, A.; Tsutsumi, N.; Nagai, S.; Iwata, A.; Nagano, T. Characterization of Erysipelothrix rhusiopathiae strains isolated from recent swine erysipelas outbreaks in Japan. J. Vet. Med. Sci. 2012, 74, 949–953. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ding, Y.; Zhu, D.; Zhang, J.; Yang, L.; Wang, X.; Chen, H.; Tan, C. Virulence determinants, antimicrobial susceptibility, and molecular profiles of Erysipelothrix rhusiopathiae strains isolated from China. Emerg. Microbes Infect. 2015, 4, e69. [Google Scholar] [CrossRef] [PubMed]
- Kwok, A.H.; Li, Y.; Jiang, J.; Jiang, P.; Leung, F.C. Complete genome assembly and characterization of an outbreak strain of the causative agent of swine erysipelas—Erysipelothrix rhusiopathiae SY1027. BMC Microbiol. 2014, 14, 176. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhu, Y.; Peng, Z.; Ding, Y.; Jie, K.; Wang, Z.; Peng, Y.; Tang, X.; Wang, X.; Chen, H.; et al. Comparative Genome Analysis of a Pathogenic Erysipelothrix rhusiopathiae Isolate WH13013 from Pig Reveals Potential Genes Involve in Bacterial Adaptions and Pathogenesis. Vet. Sci. 2020, 7, 74. [Google Scholar] [CrossRef] [PubMed]
- Chuma, T.; Kawamoto, T.; Shahada, F.; Fujimoto, H.; Okamoto, K. Antimicrobial susceptibility of Erysipelothrix rhusiopathiae isolated from pigs in Southern Japan with a modified agar dilution method. J. Vet. Med. Sci. 2010, 72, 643–645. [Google Scholar] [CrossRef]
- Yamamoto, K.; Kijima, M.; Yoshimura, H.; Takahashi, T. Antimicrobial susceptibilities of Erysipelothrix rhusiopathiae isolated from pigs with swine erysipelas in Japan, 1988–1998. J. Vet. Med. B Infect. Dis. Vet. Public Health 2001, 48, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Ku, X.; Yu, X.; Sun, Q.; Wu, H.; Chen, F.; Zhang, X.; Guo, L.; Tang, X.; He, Q. Prevalence and antimicrobial susceptibilities of bacterial pathogens in Chinese pig farms from 2013 to 2017. Sci. Rep. 2019, 9, 9908. [Google Scholar] [CrossRef]
- Yamamoto, K.; Sasaki, Y.; Ogikubo, Y.; Noguchi, N.; Sasatsu, M.; Takahashi, T. Identification of the tetracycline resistance gene, tet(M), in Erysipelothrix rhusiopathiae. J. Vet. Med. B Infect. Dis. Vet. Public Health 2001, 48, 293–301. [Google Scholar] [CrossRef]
- Zhang, A.; Xu, C.; Wang, H.; Lei, C.; Liu, B.; Guan, Z.; Yang, C.; Yang, Y.; Peng, L. Presence and new genetic environment of pleuromutilin-lincosamide-streptogramin A resistance gene lsa(E) in Erysipelothrix rhusiopathiae of swine origin. Vet. Microbiol. 2015, 177, 162–167. [Google Scholar] [CrossRef]
- Xu, C.W.; Zhang, A.Y.; Yang, C.M.; Pan, Y.; Guan, Z.B.; Lei, C.W.; Peng, L.Y.; Li, Q.Z.; Wang, H.N. First report of macrolide resistance gene erm(T) harbored by a novel small plasmid from Erysipelothrix rhusiopathiae. Antimicrob. Agents Chemother. 2015, 59, 2462–2465. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, Y.; Li, J.; Zhang, A.; Zhu, W.; Zhang, Q.; Xu, Z.; Yan, S.; Sun, X.; Chen, H.; Jin, M. iTRAQ-based quantitative proteomic analysis reveals potential virulence factors of Erysipelothrix rhusiopathiae. J. Proteom. 2017, 160, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Wang, Y.; Cai, C.; Li, J.; Wu, C.; Kang, C.; Jin, M. Erysipelothrix rhusiopathiae recruits host plasminogen via the major protective antigen SpaA. FEMS Microbiol. Lett. 2017, 364, 5. [Google Scholar] [CrossRef]
- Zhu, W.; Fan, Z.; Qiu, R.; Chen, L.; Wei, H.; Hu, B.; Chen, M.; Wang, F. Characterization of Pasteurella multocida isolates from rabbits in China. Vet. Microbiol. 2020, 244, 108649. [Google Scholar] [CrossRef] [PubMed]
- Cen, X.; Huang, Y.; Lu, Z.; Shao, W.; Zhuo, C.; Bao, C.; Feng, S.; Wei, C.; Tang, X.; Cen, L.; et al. LncRNA IGFL2-AS1 Promotes the Proliferation, Migration, and Invasion of Colon Cancer Cells and is Associated with Patient Prognosis. Cancer Manag. Res. 2021, 13, 5957–5968. [Google Scholar] [CrossRef]
- Chin, C.S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E.; et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Chaisson, M.J.; Tesler, G. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): Application and theory. BMC Bioinform. 2012, 13, 238. [Google Scholar] [CrossRef]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef]
- Carver, T.J.; Rutherford, K.M.; Berriman, M.; Rajandream, M.A.; Barrell, B.G.; Parkhill, J. ACT: The Artemis Comparison Tool. Bioinformatics 2005, 21, 3422–3423. [Google Scholar] [CrossRef] [PubMed]
- Makino, S.; Okada, Y.; Maruyama, T.; Ishikawa, K.; Takahashi, T.; Nakamura, M.; Ezaki, T.; Morita, H. Direct and rapid detection of Erysipelothrix rhusiopathiae DNA in animals by PCR. J. Clin. Microbiol. 1994, 32, 1526–1531. [Google Scholar] [CrossRef]
- Zhu, W.; Wu, C.; Kang, C.; Cai, C.; Wang, Y.; Li, J.; Zhang, Q.; Sun, X.; Jin, M. Evaluation of the protective efficacy of four newly identified surface proteins of Erysipelothrix rhusiopathiae. Vaccine 2018, 36, 8079–8083. [Google Scholar] [CrossRef] [PubMed]
- Estrada, A.A.; Gottschalk, M.; Rossow, S.; Rendahl, A.; Gebhart, C.; Marthaler, D.G. Serotype and Genotype (Multilocus Sequence Type) of Streptococcus suis Isolates from the United States Serve as Predictors of Pathotype. J. Clin. Microbiol. 2019, 57, e00377-19. [Google Scholar] [CrossRef]
- Zhu, W.; Wu, C.; Sun, X.; Zhang, A.; Zhu, J.; Hua, Y.; Chen, H.; Jin, M. Characterization of Streptococcus suis serotype 2 isolates from China. Vet. Microbiol. 2013, 166, 527–534. [Google Scholar] [CrossRef]
- Jani, K.; Srivastava, V.; Sharma, P.; Vir, A.; Sharma, A. Easy Access to Antibiotics; Spread of Antimicrobial Resistance and Implementation of One Health Approach in India. J. Epidemiol. Glob. Health 2021, 11, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.; Wang, Y.; Zhao, X. Research on the drug resistance mechanism of foodborne pathogens. Microb. Pathog. 2022, 162, 105306. [Google Scholar] [CrossRef]
- Putman, M.; van Veen, H.W.; Konings, W.N. Molecular properties of bacterial multidrug transporters. Microbiol. Mol. Biol. Rev. MMBR 2000, 64, 672–693. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Antibiotics | Number of Strains with MIC (μg/mL) | MIC(μg/mL) on the Breakpoint a of Resistance | Number of Resistant Strains (%) | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ≤0.015 | ≤0.03 | ≤0.06 | ≤0.125 | 0.25 | 0.5 | 1 | 2 | 4 | 8 | 16 | 32 | ≥64 | ≥128 | ≥256 | |||
| TE | 1 | 2 | 6 | 29 | 17 | 1 | 3 | 46 | 13 | 2 | 16 | 61 (50.8%) | |||||
| ERY | 53 | 2 | 1 | 1 | 19 | 20 | 4 | 4 | 6 | 6 | 4 | 2 | 64 (53.3%) | ||||
| CLI | 31 | 5 | 7 | 9 | 3 | 1 | 13 | 51 | 0.5 | 84 (70.0%) | |||||||
| CIP | 5 | 5 | 8 | 96 | 6 | 2 | 110 (91.7%) | ||||||||||
| EFX | 3 | 2 | 2 | 4 | 10 | 92 | 4 | 3 | 4 | 99 (82.5%) | |||||||
| MEM | 111 | 9 | 1 | 0 (0%) | |||||||||||||
| CTX | 18 | 77 | 25 | 2 | 0 (0%) | ||||||||||||
| Antibiotic a | Gene or Mutants | MIC (μg/mL) | Times Increase in MIC e | ||
|---|---|---|---|---|---|
| C600 b | C600(pSET2) c | C600 (Recombinant pSET2) d | |||
| ERY | erm(A)-like | 64 | 64 | >256 | >4 |
| CIP | wild-type gyrA | <0.06 | <0.06 | <0.06 | —f |
| wild-type parC | <0.06 | — | |||
| gyrA(90D-N) | <0.06 | — | |||
| gyrA(86T-I) | <0.06 | — | |||
| parC(81S-I) | <0.06 | — | |||
| EFX | wild-type gyrA | <0.03 | <0.03 | <0.03 | — |
| wild-type parC | <0.03 | — | |||
| gyrA(90D-N) | <0.03 | — | |||
| gyrA(86T-I) | 0.06 | >2 | |||
| parC(81S-I) | 0.06 | >2 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, C.; Lv, C.; Zhao, Y.; Zhu, W.; Liu, L.; Wang, T.; Kang, C.; Yang, Y.; Sun, X.; Zhang, Q.; et al. Characterization of Erysipelothrix rhusiopathiae Isolates from Diseased Pigs in 15 Chinese Provinces from 2012 to 2018. Microorganisms 2021, 9, 2615. https://doi.org/10.3390/microorganisms9122615
Wu C, Lv C, Zhao Y, Zhu W, Liu L, Wang T, Kang C, Yang Y, Sun X, Zhang Q, et al. Characterization of Erysipelothrix rhusiopathiae Isolates from Diseased Pigs in 15 Chinese Provinces from 2012 to 2018. Microorganisms. 2021; 9(12):2615. https://doi.org/10.3390/microorganisms9122615
Chicago/Turabian StyleWu, Chao, Changjie Lv, Ya Zhao, Weifeng Zhu, Liang Liu, Ting Wang, Chao Kang, Ying Yang, Xiaomei Sun, Qiang Zhang, and et al. 2021. "Characterization of Erysipelothrix rhusiopathiae Isolates from Diseased Pigs in 15 Chinese Provinces from 2012 to 2018" Microorganisms 9, no. 12: 2615. https://doi.org/10.3390/microorganisms9122615
APA StyleWu, C., Lv, C., Zhao, Y., Zhu, W., Liu, L., Wang, T., Kang, C., Yang, Y., Sun, X., Zhang, Q., & Jin, M. (2021). Characterization of Erysipelothrix rhusiopathiae Isolates from Diseased Pigs in 15 Chinese Provinces from 2012 to 2018. Microorganisms, 9(12), 2615. https://doi.org/10.3390/microorganisms9122615