
Metagenome-Scale Metabolic Network Suggests Folate Produced by Bifidobacterium longum Might Contribute to High-Fiber-Diet-Induced Weight Loss in a Prader–Willi Syndrome Child


Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Investigation
2.2. Metagenomic Sequencing and Data Preprocessing
2.3. Quantitative Calculation of Diet Component Intakes
2.4. Construction of Metagenome-Scale Metabolic Network
2.5. Statistical Analysis
2.6. Data Visualization
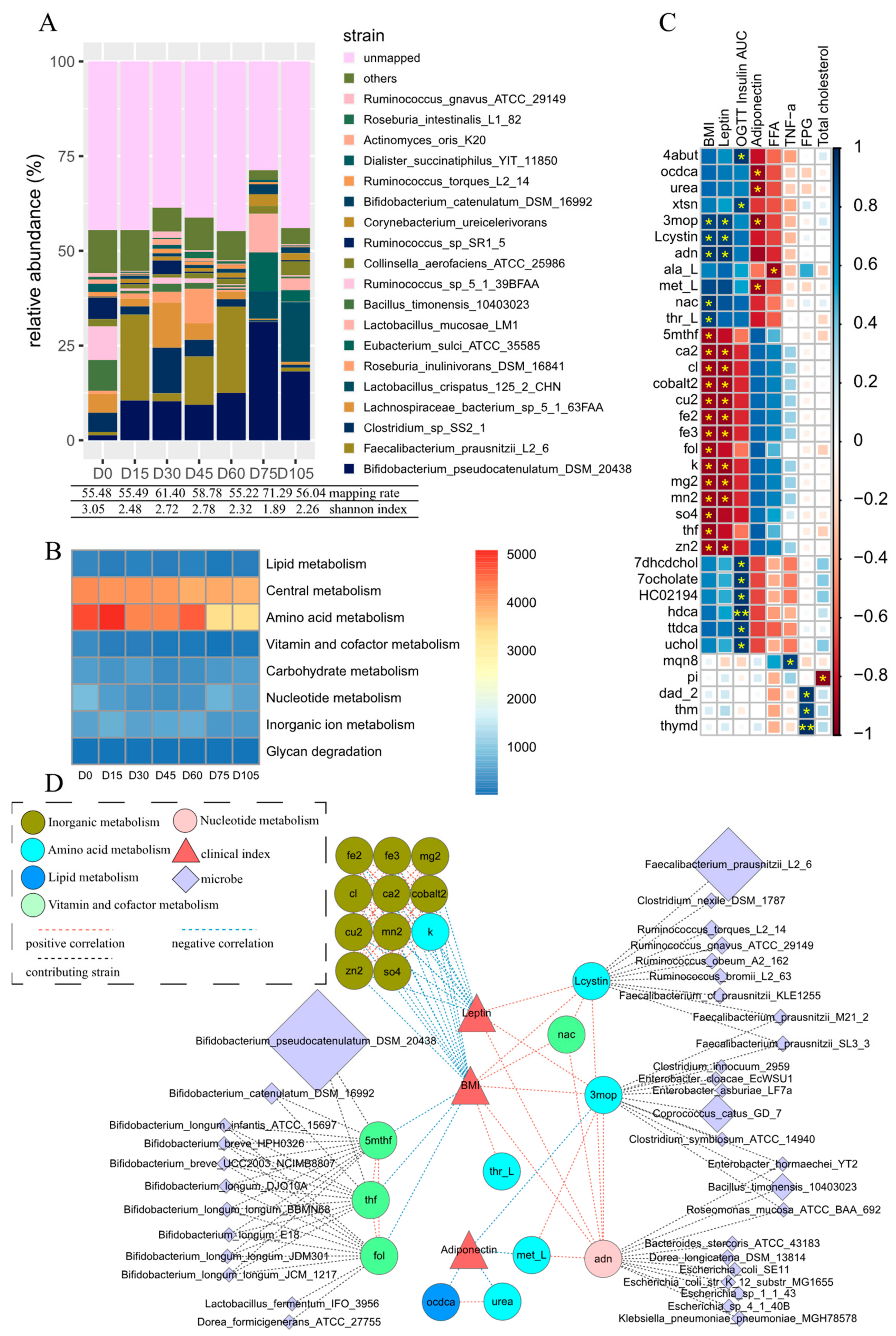
3. Results
3.1. Persistent Variation in Gut Microbiota during Dietary Intervention
3.2. Metabolites Simulated from the Reconstructed Microbial Metabolic Network
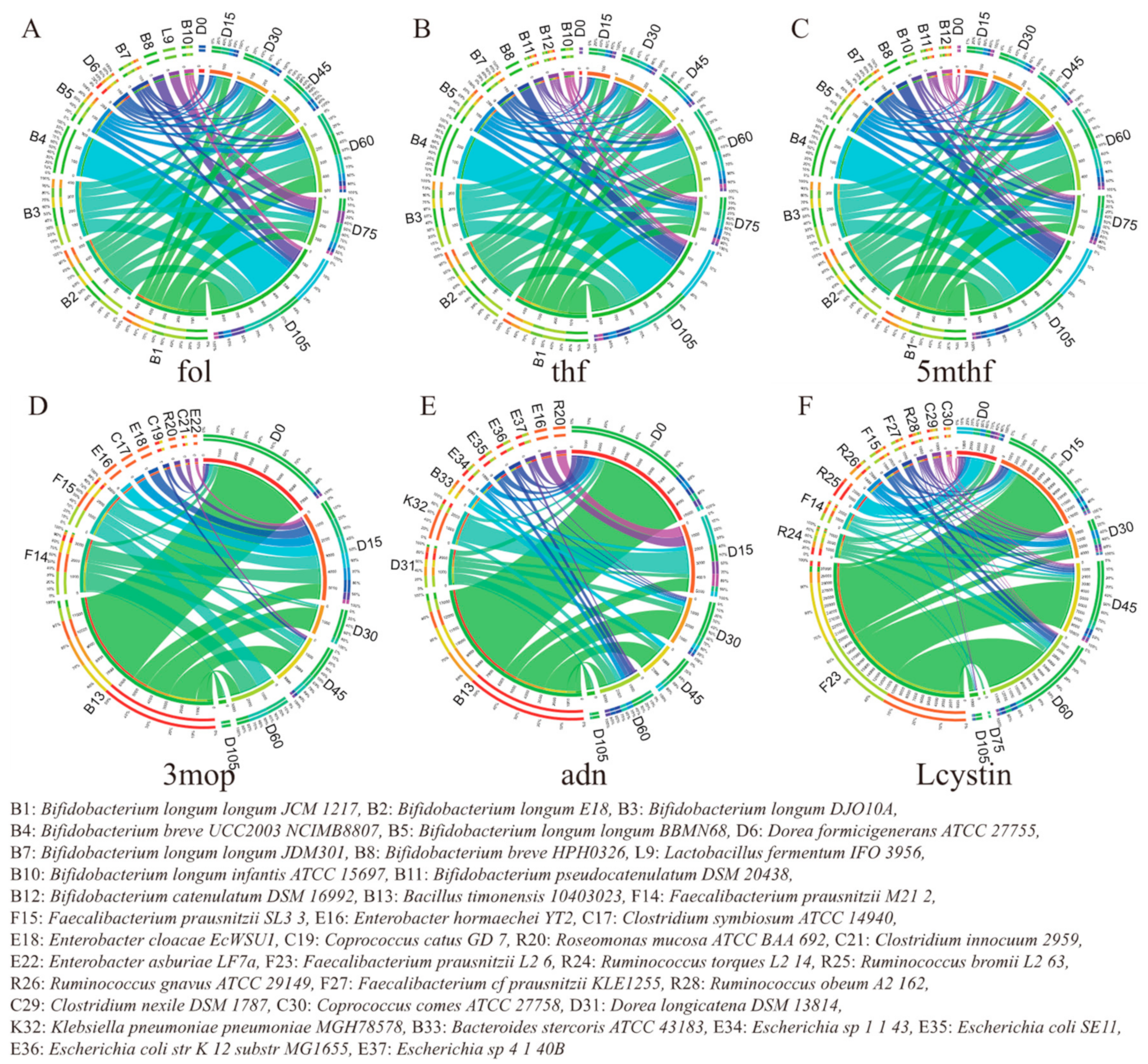
3.3. Key Metabolites Associated with Obesity-Relevant Clinical Parameters
3.4. Microbes Contributing to Key Obesity-Related Metabolites
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blüher, M. Obesity: Global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 2019, 15, 288–298. [Google Scholar] [CrossRef] [PubMed]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Zhang, F.; Ding, X.; Wu, G.; Lam, Y.Y.; Wang, X.; Fu, H.; Xue, X.; Lu, C.; Ma, J.; et al. Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science 2018, 359, 1151–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurung, M.; Li, Z.; You, H.; Rodrigues, R.; Jump, D.B.; Morgun, A.; Shulzhenko, N. Role of gut microbiota in type 2 diabetes pathophysiology. EBioMedicine 2020, 51, 102590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helmink, B.A.; Khan, M.A.W.; Hermann, A.; Gopalakrishnan, V.; Wargo, J.A. The microbiome, cancer, and cancer therapy. Nat. Med. 2019, 25, 377–388. [Google Scholar] [CrossRef]
- Fan, Y.; Pedersen, O. Gut microbiota in human metabolic health and disease. Nat. Rev. Microbiol. 2020, 19, 55–71. [Google Scholar] [CrossRef]
- Sung, J.; Kim, S.; Cabatbat, J.J.T.; Jang, S.; Jin, Y.-S.; Jung, G.Y.; Chia, N.; Kim, P.-J. Global metabolic interaction network of the human gut microbiota for context-specific community-scale analysis. Nat. Commun. 2017, 8, 15393. [Google Scholar] [CrossRef] [Green Version]
- Jansma, J.; El Aidy, S. Understanding the host-microbe interactions using metabolic modeling. Microbiome 2021, 9, 16. [Google Scholar] [CrossRef]
- Diener, C.; Gibbons, S.M.; Resendis-Antonio, O. MICOM: Metagenome-Scale Modeling To Infer Metabolic Interactions in the Gut Microbiota. mSystems 2020, 5. [Google Scholar] [CrossRef] [Green Version]
- Bauer, E.; Thiele, I. From Network Analysis to Functional Metabolic Modeling of the Human Gut Microbiota. mSystems 2018, 3, e00209-17. [Google Scholar] [CrossRef] [Green Version]
- Lewis, N.; Hixson, K.K.; Conrad, T.M.; Lerman, J.; Charusanti, P.; Polpitiya, A.D.; Adkins, J.; Schramm, G.; Purvine, S.; Lopez-Ferrer, D.; et al. Omic data from evolved E. coli are consistent with computed optimal growth from genome-scale models. Mol. Syst. Biol. 2010, 6, 390. [Google Scholar] [CrossRef]
- Orth, J.D.; Thiele, I.; Palsson, B.O. What is flux balance analysis? Nat. Biotechnol. 2010, 28, 245–248. [Google Scholar] [CrossRef]
- Medlock, G.L.; Carey, M.A.; McDuffie, D.G.; Mundy, M.B.; Giallourou, N.; Swann, J.R.; Kolling, G.L.; Papin, J.A. Inferring Metabolic Mechanisms of Interaction within a Defined Gut Microbiota. Cell Syst. 2018, 7, 245–257.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, O.D.; Rocha, M.; Maia, P. A Review of Dynamic Modeling Approaches and Their Application in Computational Strain Optimization for Metabolic Engineering. Front. Microbiol. 2018, 9, 1690. [Google Scholar] [CrossRef] [PubMed]
- Gottstein, W.; Olivier, B.G.; Bruggeman, F.; Teusink, B. Constraint-based stoichiometric modelling from single organisms to microbial communities. J. R. Soc. Interface 2016, 13, 20160627. [Google Scholar] [CrossRef]
- Zomorrodi, A.R.; Maranas, C.D. OptCom: A Multi-Level Optimization Framework for the Metabolic Modeling and Analysis of Microbial Communities. PLoS Comput. Biol. 2012, 8, e1002363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, E.; Zimmermann, J.; Baldini, F.; Thiele, I.; Kaleta, C. BacArena: Individual-based metabolic modeling of heterogeneous microbes in complex communities. PLoS Comput. Biol. 2017, 13, e1005544. [Google Scholar] [CrossRef]
- Gardner, J.J.; Hodge, B.M.S.; Boyle, N.R. Multiscale Multiobjective Systems Analysis (MiMoSA): An advanced metabolic modeling framework for complex systems. Sci. Rep. 2019, 9, 16948. [Google Scholar] [CrossRef] [Green Version]
- García-Jiménez, B.; García, J.L.; Nogales, J. FLYCOP: Metabolic modeling-based analysis and engineering microbial communities. Bioinform. 2018, 34, i954–i963. [Google Scholar] [CrossRef]
- Belcour, A.; Frioux, C.; Aite, M.; Bretaudeau, A.; Hildebrand, F.; Siegel, A. Metage2Metabo, microbiota-scale metabolic complementarity for the identification of key species. eLife 2020, 9, 9. [Google Scholar] [CrossRef]
- Baldini, F.; Heinken, A.; Heirendt, L.; Magnusdottir, S.; Fleming, R.M.T.; Thiele, I. The Microbiome Modeling Toolbox: From microbial interactions to personalized microbial communities. Bioinformatics 2019, 35, 2332–2334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heirendt, L.; Arreckx, S.; Pfau, T.; Mendoza, S.N.; Richelle, A.; Heinken, A.; Haraldsdóttir, H.S.; Wachowiak, J.; Keating, S.M.; Vlasov, V.; et al. Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v.3.0. Nat. Protoc. 2019, 14, 639–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heirendt, L.; Thiele, I.; Fleming, R.M.T. DistributedFBA.jl: High-level, high-performance flux balance analysis in Julia. Bioinformatics 2017, 33, 1421–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendoza, S.N.; Olivier, B.G.; Molenaar, D.; Teusink, B. A systematic assessment of current genome-scale metabolic reconstruction tools. Genome Biol. 2019, 20, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, Z.A.; Lu, J.; Dräger, A.; Miller, P.; Federowicz, S.; Lerman, J.A.; Ebrahim, A.; Palsson, B.O.; Lewis, N.E. BiGG Models: A platform for integrating, standardizing and sharing genome-scale models. Nucleic Acids Res. 2016, 44, D515–D522. [Google Scholar] [CrossRef]
- Arkin, A.P.; Cottingham, R.W.; Henry, C.S.; Harris, N.L.; Stevens, R.L.; Maslov, S.; Dehal, P.; Ware, D.; Perez, F.; Canon, S.; et al. KBase: The United States Department of Energy Systems Biology Knowledgebase. Nat. Biotechnol. 2018, 36, 566–569. [Google Scholar] [CrossRef] [Green Version]
- Machado, D.; Andrejev, S.; Tramontano, M.; Patil, K.R. Fast automated reconstruction of genome-scale metabolic models for microbial species and communities. Nucleic Acids Res. 2018, 46, 7542–7553. [Google Scholar] [CrossRef]
- Magnusdottir, S.; Heinken, A.; Kutt, L.; Ravcheev, D.; Bauer, E.; Noronha, A.; Greenhalgh, K.; Jäger, C.; Baginska, J.; Wilmes, P.; et al. Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat. Biotechnol. 2017, 35, 81–89. [Google Scholar] [CrossRef]
- Hertel, J.; Heinken, A.; Martinelli, F.; Thiele, I. Integration of constraint-based modeling with fecal metabolomics reveals large deleterious effects of Fusobacterium spp. on community butyrate production. Gut Microbes 2021, 13, 1915673. [Google Scholar] [CrossRef]
- Henson, M.A. Computational modeling of the gut microbiota reveals putative metabolic mechanisms of recurrent Clostridioides difficile infection. PLoS Comput. Biol. 2021, 17, e1008782. [Google Scholar] [CrossRef]
- Heinken, A.; Hertel, J.; Thiele, I. Metabolic modelling reveals broad changes in gut microbial metabolism in inflammatory bowel disease patients with dysbiosis. npj Syst. Biol. Appl. 2021, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Heinken, A.; Ravcheev, D.A.; Baldini, F.; Heirendt, L.; Fleming, R.M.T.; Thiele, I. Systematic assessment of secondary bile acid metabolism in gut microbes reveals distinct metabolic capabilities in inflammatory bowel disease. Microbiome 2019, 7, 75. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yin, A.; Li, H.; Wang, R.; Wu, G.; Shen, J.; Zhang, M.; Wang, L.; Hou, Y.; Ouyang, H.; et al. Dietary Modulation of Gut Microbiota Contributes to Alleviation of Both Genetic and Simple Obesity in Children. EBioMedicine 2015, 2, 968–984. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhao, L.; Zhang, M. Gut Microbial SNPs Induced by High-Fiber Diet Dominate Nutrition Metabolism and Environmental Adaption of Faecalibacterium prausnitzii in Obese Children. Front. Microbiol. 2021, 12, 683714. [Google Scholar] [CrossRef]
- Li, H.; Wu, G.; Zhao, L.; Zhang, M. Suppressed inflammation in obese children induced by a high-fiber diet is associated with the attenuation of gut microbial virulence factor genes. Virulence 2021, 12, 1754–1770. [Google Scholar] [CrossRef]
- Wu, G.; Zhang, C.; Wu, H.; Wang, R.; Shen, J.; Wang, L.; Zhao, Y.; Pang, X.; Zhang, X.; Zhao, L.; et al. Genomic Microdiversity of Bifidobacterium pseudocatenulatum Underlying Differential Strain-Level Responses to Dietary Carbohydrate Intervention. mBio 2017, 8, e02348-16. [Google Scholar] [CrossRef] [Green Version]
- Zweigner, J.; Schumann, R.R.; Weber, J.R. The role of lipopolysaccharide-binding protein in modulating the innate immune response. Microbes Infect. 2006, 8, 946–952. [Google Scholar] [CrossRef]
- Xiao, S.; Fei, N.; Pang, X.; Shen, J.; Wang, L.; Zhang, B.; Zhang, M.; Zhang, X.; Zhang, C.; Li, M.; et al. A gut microbiota-targeted dietary intervention for amelioration of chronic inflammation underlying metabolic syndrome. FEMS Microbiol. Ecol. 2014, 87, 357–367. [Google Scholar] [CrossRef]
- Fei, N.; Zhao, L. An opportunistic pathogen isolated from the gut of an obese human causes obesity in germfree mice. ISME J. 2012, 7, 880–884. [Google Scholar] [CrossRef]
- Li, H.; Xiao, Z. Study on the Status of Nutrition and Active Ingredients of Coix. Farm Products Process. 2018, 20, 54–56. [Google Scholar] [CrossRef]
- Liu, X. Rapid Determination Main Nutritional Components of Coix Seeds Using near Infrared Spectroscopy Technique and Study on Their Physicochemical Properties. Ph.D. Thesis, Shanghai Jiao Tong University, Shanghai, China, 2016. [Google Scholar]
- Virtual Metabolic Human Nutriton Design. Available online: https://www.vmh.life/#nutrition/dietdesigner (accessed on 15 October 2021).
- Read Coverage Calculator for Metagenomics (CoverM). Available online: https://github.com/wwood/CoverM (accessed on 15 October 2021).
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Interactive Online Version of Circos. Available online: http://mkweb.bcgsc.ca/tableviewer/ (accessed on 15 October 2021).
- Labruna, G.; Pasanisi, F.; Nardelli, C.; Caso, R.; Vitale, D.F.; Contaldo, F.; Sacchetti, L. High Leptin/Adiponectin Ratio and Serum Triglycerides Are Associated With an “At-Risk” Phenotype in Young Severely Obese Patients. Obesity 2011, 19, 1492–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, K.A.; Brown, S.A. Searching for Evidence of an Anti-Inflammatory Diet in Children: A Systematic Review of Randomized Controlled Trials for Pediatric Obesity Interventions with a Focus on Leptin, Ghrelin, and Adiponectin. Biol. Res. Nurs. 2017, 19, 511–530. [Google Scholar] [CrossRef]
- Bird, J.; Ronnenberg, A.G.; Choi, S.-W.; Du, F.; Mason, J.B.; Liu, Z. Obesity Is Associated with Increased Red Blood Cell Folate Despite Lower Dietary Intakes and Serum Concentrations. J. Nutr. 2014, 145, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Mlodzik-Czyzewska, M.A.; Malinowska, A.M.; Chmurzynska, A. Low folate intake and serum levels are associated with higher body mass index and abdominal fat accumulation: A case control study. Nutr. J. 2020, 19, 53. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.J.; Chen, Y.; Lu, M.; Liu, L.; Shi, L.X. Association between serum Fe levels and obesity: A meta-analysis. Nutr. Hosp. 2015, 31, 2451–2454. [Google Scholar] [CrossRef]
- Huang, J.; Qi, S.-J. Childhood obesity and food intake. World J. Pediatr. 2015, 11, 101–107. [Google Scholar] [CrossRef]
- Fukunaka, A.; Fujitani, Y. Role of Zinc Homeostasis in the Pathogenesis of Diabetes and Obesity. Int. J. Mol. Sci. 2018, 19, 476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noronha, A.; Modamio, J.; Jarosz, Y.; Guerard, E.; Sompairac, N.; Preciat, G.; Daníelsdóttir, A.D.; Krecke, M.; Merten, D.; Haraldsdóttir, H.S.; et al. The Virtual Metabolic Human database: Integrating human and gut microbiome metabolism with nutrition and disease. Nucleic Acids Res. 2019, 47, D614–D624. [Google Scholar] [CrossRef]
- Go, Y.-M.; Jones, D.P. Cysteine/cystine redox signaling in cardiovascular disease. Free. Radic. Biol. Med. 2011, 50, 495–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardo, F.; Villalobos-Labra, R.; Chiarello, D.I.; Salsoso, R.; Toledo, F.; Gutierrez, J.; Leiva, A.; Sobrevia, L. Molecular implications of adenosine in obesity. Mol. Asp. Med. 2017, 55, 90–101. [Google Scholar] [CrossRef] [Green Version]
- Meriño, M.; Briones, L.; Palma, V.; Herlitz, K.; Escudero, C. Role of adenosine receptors in the adipocyte–macrophage interaction during obesity. Endocrinología, Diabetes y Nutrición 2017, 64, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Pompei, A.; Cordisco, L.; Amaretti, A.; Zanoni, S.; Matteuzzi, D.; Rossi, M. Folate Production by Bifidobacteria as a Potential Probiotic Property. Appl. Environ. Microbiol. 2007, 73, 179–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugahara, H.; Odamaki, T.; Hashikura, N.; Abe, F.; Xiao, J.-Z. Differences in folate production by bifidobacteria of different origins. Biosci. Microbiota Food Health 2015, 34, 87–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shulpekova, Y.; Nechaev, V.; Kardasheva, S.; Sedova, A.; Kurbatova, A.; Bueverova, E.; Kopylov, A.; Malsagova, K.; Dlamini, J.; Ivashkin, V. The Concept of Folic Acid in Health and Disease. Molecules 2021, 26, 3731. [Google Scholar] [CrossRef]
- Thiele, I.; Sahoo, S.; Heinken, A.; Hertel, J.; Heirendt, L.; Aurich, M.K.; Fleming, R.M. Personalized whole-body models integrate metabolism, physiology, and the gut microbiome. Mol. Syst. Biol. 2020, 16, e8982. [Google Scholar] [CrossRef]
- Geng, J.; Ji, B.; Li, G.; López-Isunza, F.; Nielsen, J. CODY enables quantitatively spatiotemporal predictions on in vivo gut microbial variability induced by diet intervention. Proc. Natl. Acad. Sci. USA 2021, 118, e2019336118. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Food Name | Day 0, 15, 30 | Day 45, 60 | Day 75, 105 |
|---|---|---|---|
| Formula 1 | |||
| Adlay | 222 | 128 | 123 |
| Hyacinth beans | 111 | 64.3 | 61.8 |
| Buckwheat | 111 | 64.3 | 61.8 |
| Oats | 166 | 96.5 | 92.7 |
| Yam | 111 | 64.3 | 61.8 |
| Soybean | 55.6 | 32.2 | 30.9 |
| Red bean | 55.6 | 32.2 | 30.9 |
| Peanut | 55.6 | 32.2 | 30.9 |
| Goji berries | 55.6 | 32.2 | 30.9 |
| Yellow corn | 55.6 | 32.2 | 30.9 |
| Lotus seed | 55.6 | 32.2 | 30.9 |
| Big jujube | 55.6 | 32.2 | 30.9 |
| Olive oil | 16.2 | 14.8 | 14.6 |
| Formula 2 | |||
| Bitter gourd | 36.6 | 43.1 | 43.1 |
| Fibersol-2 | 2.44 | 2.87 | 2.87 |
| Oligosaccharides | 0.61 | 0.72 | 0.72 |
| Isomaltose | 1.02 | 1.20 | 1.20 |
| Formula 3 | |||
| Fibersol-2 | 16.1 | 16.1 | 53.6 |
| Oligosaccharides | 4.02 | 4.02 | 13.4 |
| Isomaltose | 6.70 | 6.70 | 22.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiang, B.; Zhao, L.; Zhang, M. Metagenome-Scale Metabolic Network Suggests Folate Produced by Bifidobacterium longum Might Contribute to High-Fiber-Diet-Induced Weight Loss in a Prader–Willi Syndrome Child. Microorganisms 2021, 9, 2493. https://doi.org/10.3390/microorganisms9122493
Xiang B, Zhao L, Zhang M. Metagenome-Scale Metabolic Network Suggests Folate Produced by Bifidobacterium longum Might Contribute to High-Fiber-Diet-Induced Weight Loss in a Prader–Willi Syndrome Child. Microorganisms. 2021; 9(12):2493. https://doi.org/10.3390/microorganisms9122493
Chicago/Turabian StyleXiang, Baoyu, Liping Zhao, and Menghui Zhang. 2021. "Metagenome-Scale Metabolic Network Suggests Folate Produced by Bifidobacterium longum Might Contribute to High-Fiber-Diet-Induced Weight Loss in a Prader–Willi Syndrome Child" Microorganisms 9, no. 12: 2493. https://doi.org/10.3390/microorganisms9122493
APA StyleXiang, B., Zhao, L., & Zhang, M. (2021). Metagenome-Scale Metabolic Network Suggests Folate Produced by Bifidobacterium longum Might Contribute to High-Fiber-Diet-Induced Weight Loss in a Prader–Willi Syndrome Child. Microorganisms, 9(12), 2493. https://doi.org/10.3390/microorganisms9122493