
Phylosymbiosis in the Rhizosphere Microbiome Extends to Nitrogen Cycle Functional Potential

 , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
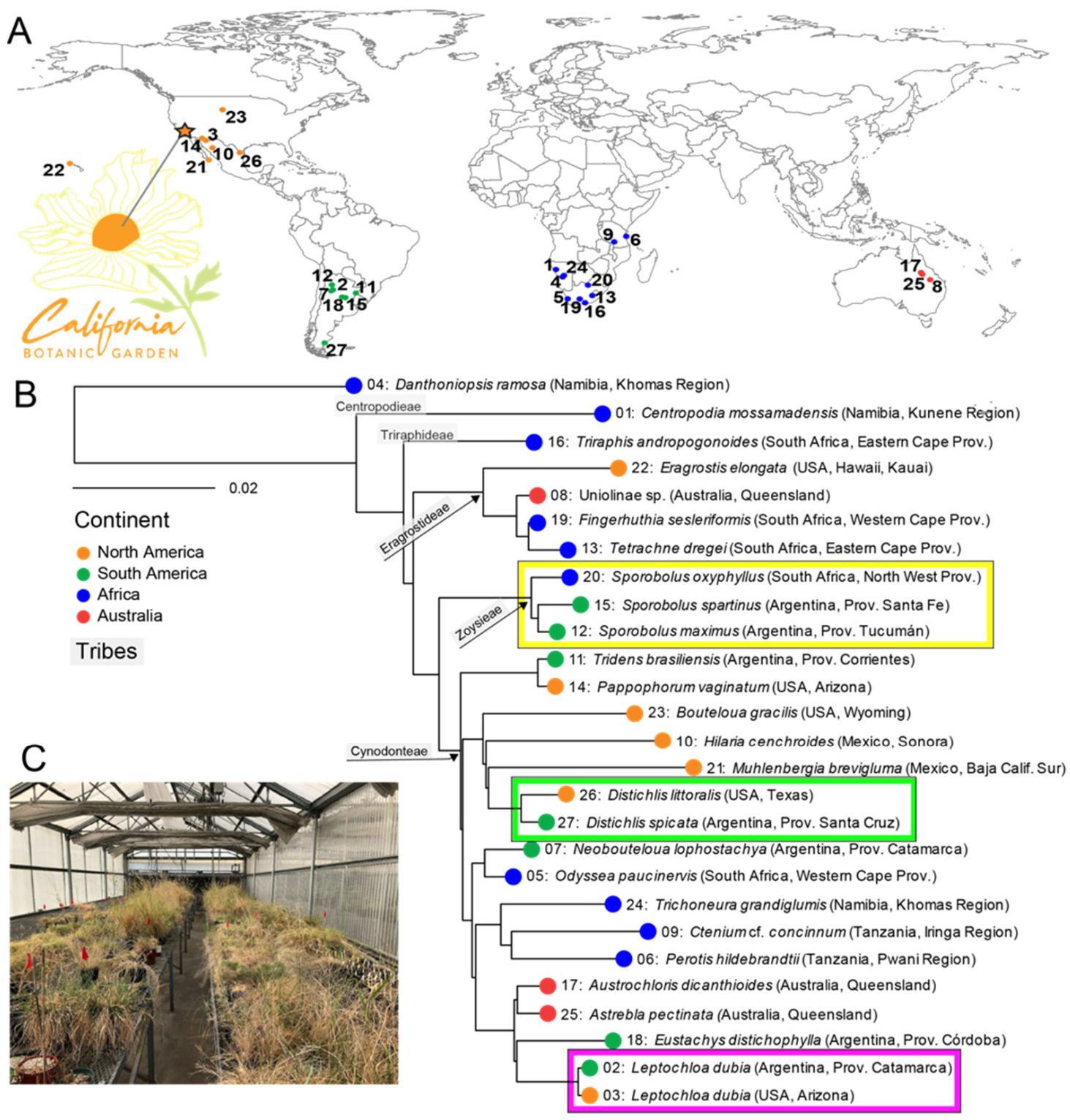
2.1. Plant Collection and Phylogeny
2.2. Rhizosphere Metagenome Sequencing
2.3. Nitrogen Cycle Protein Database
2.4. Metagenome Annotation
2.5. Data Analysis and Statistics
3. Results
3.1. Plant Phylogeny
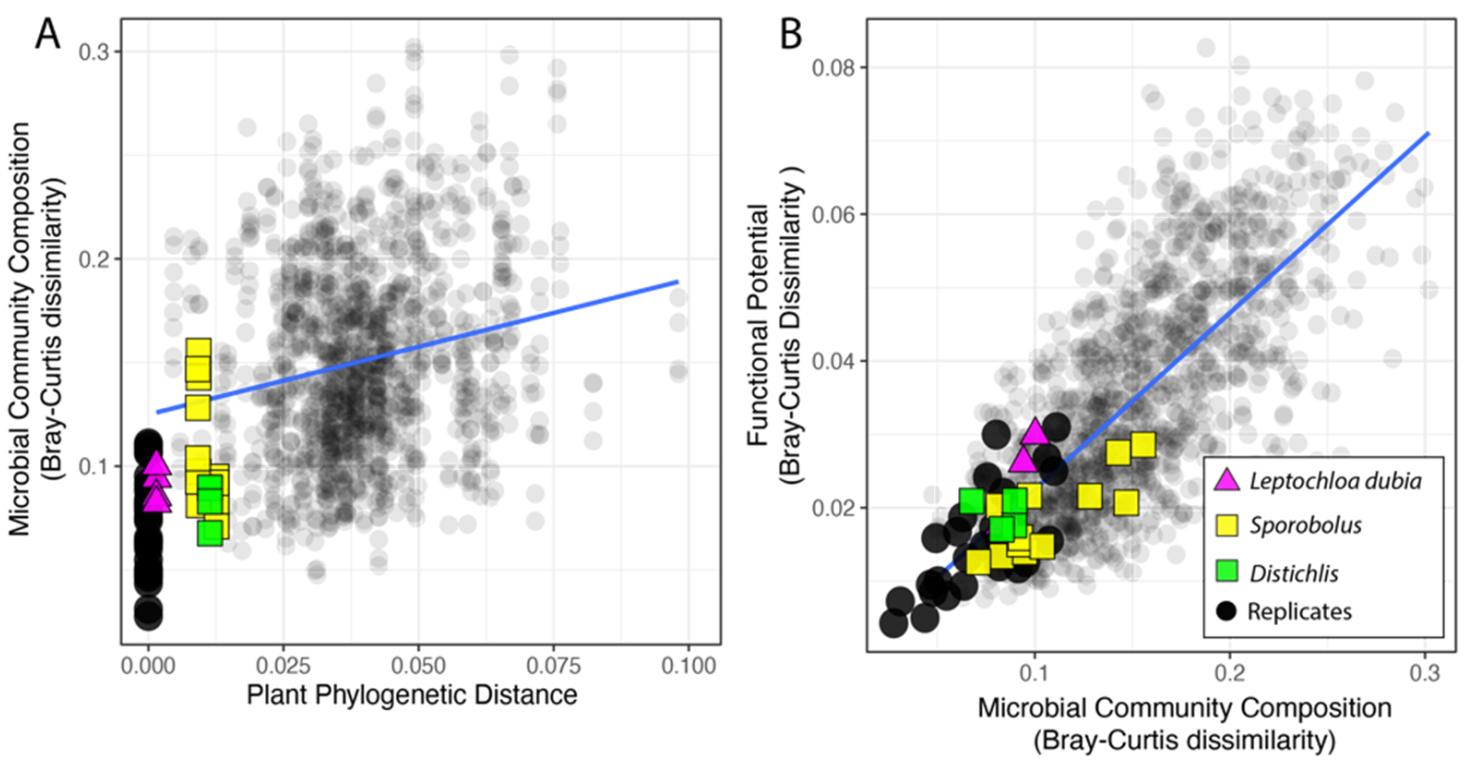
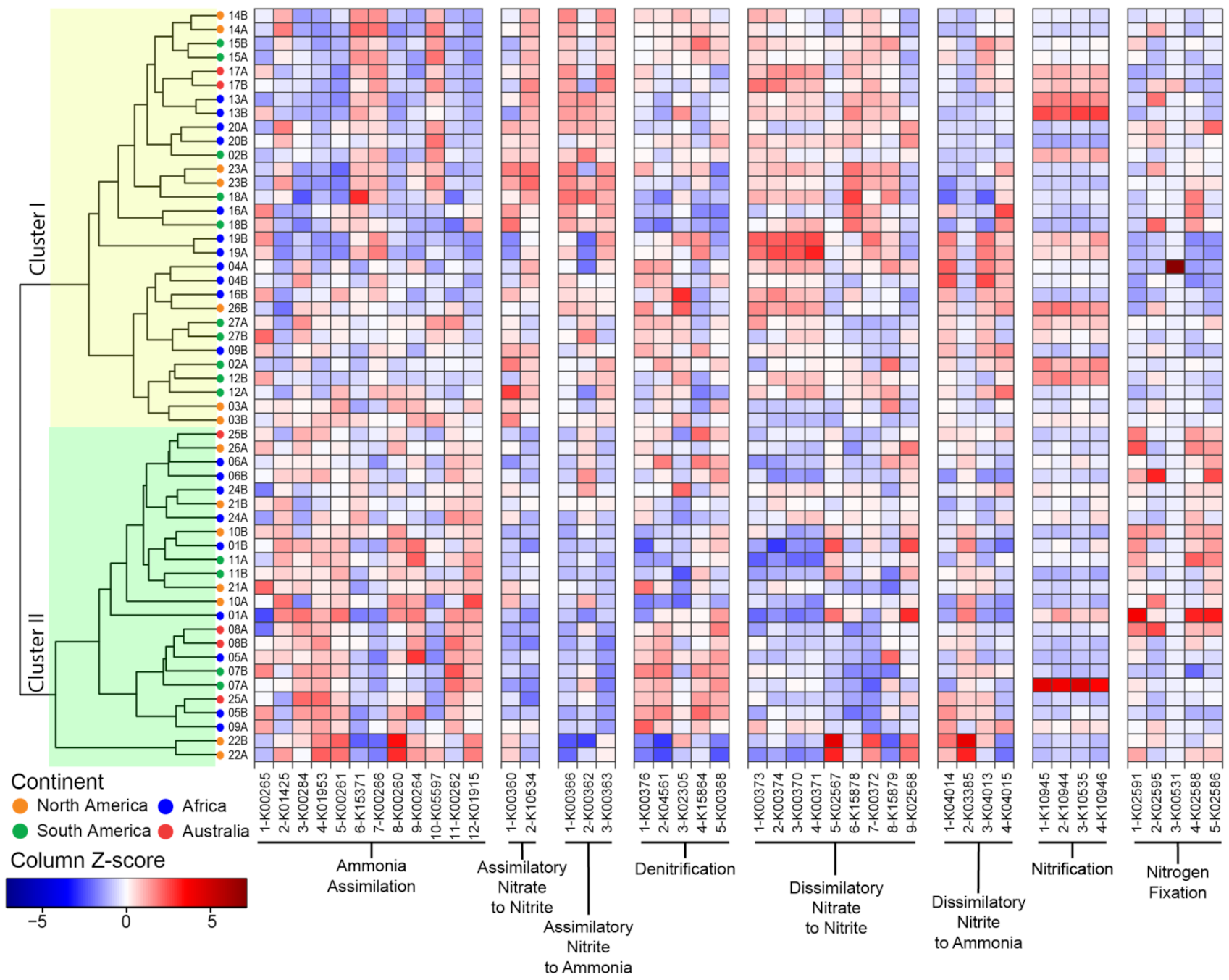
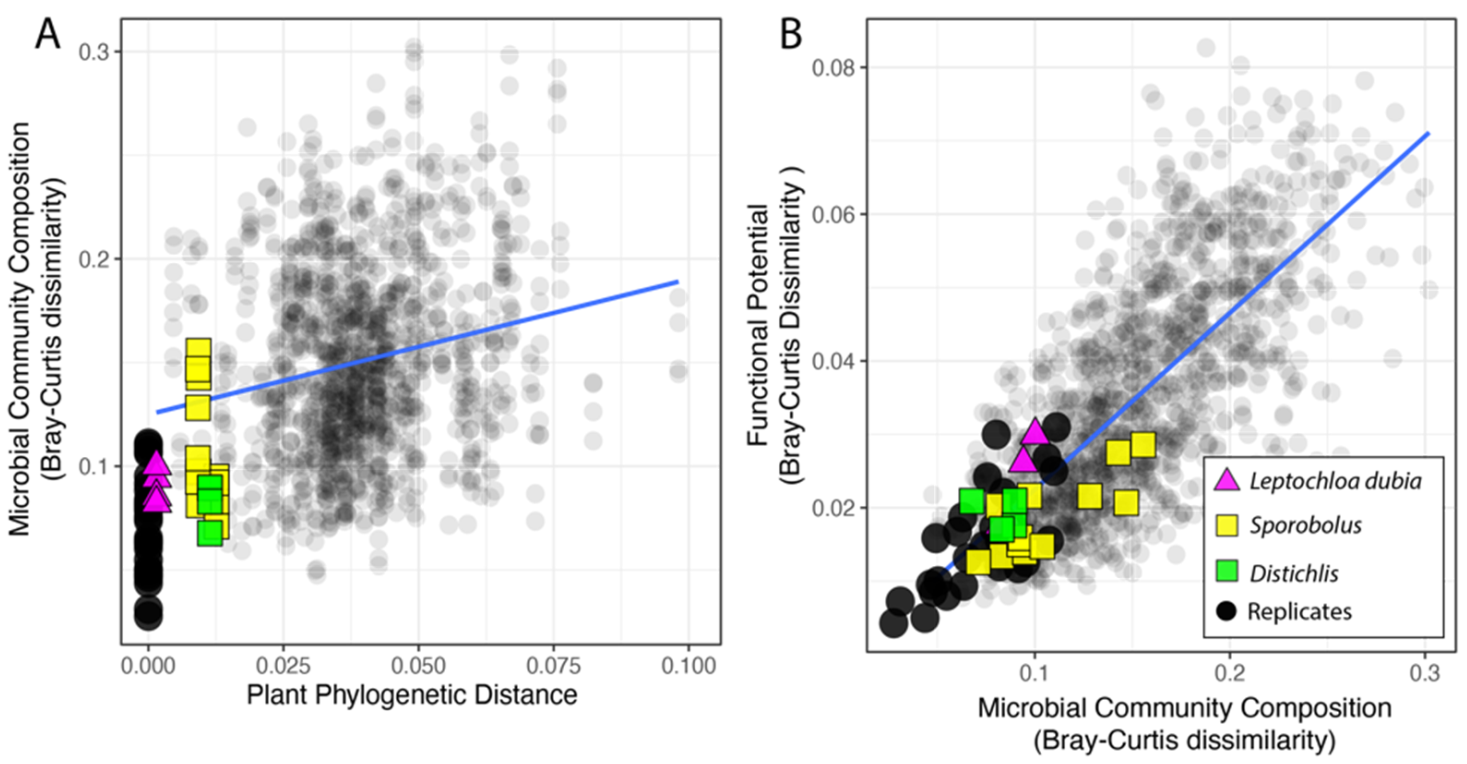
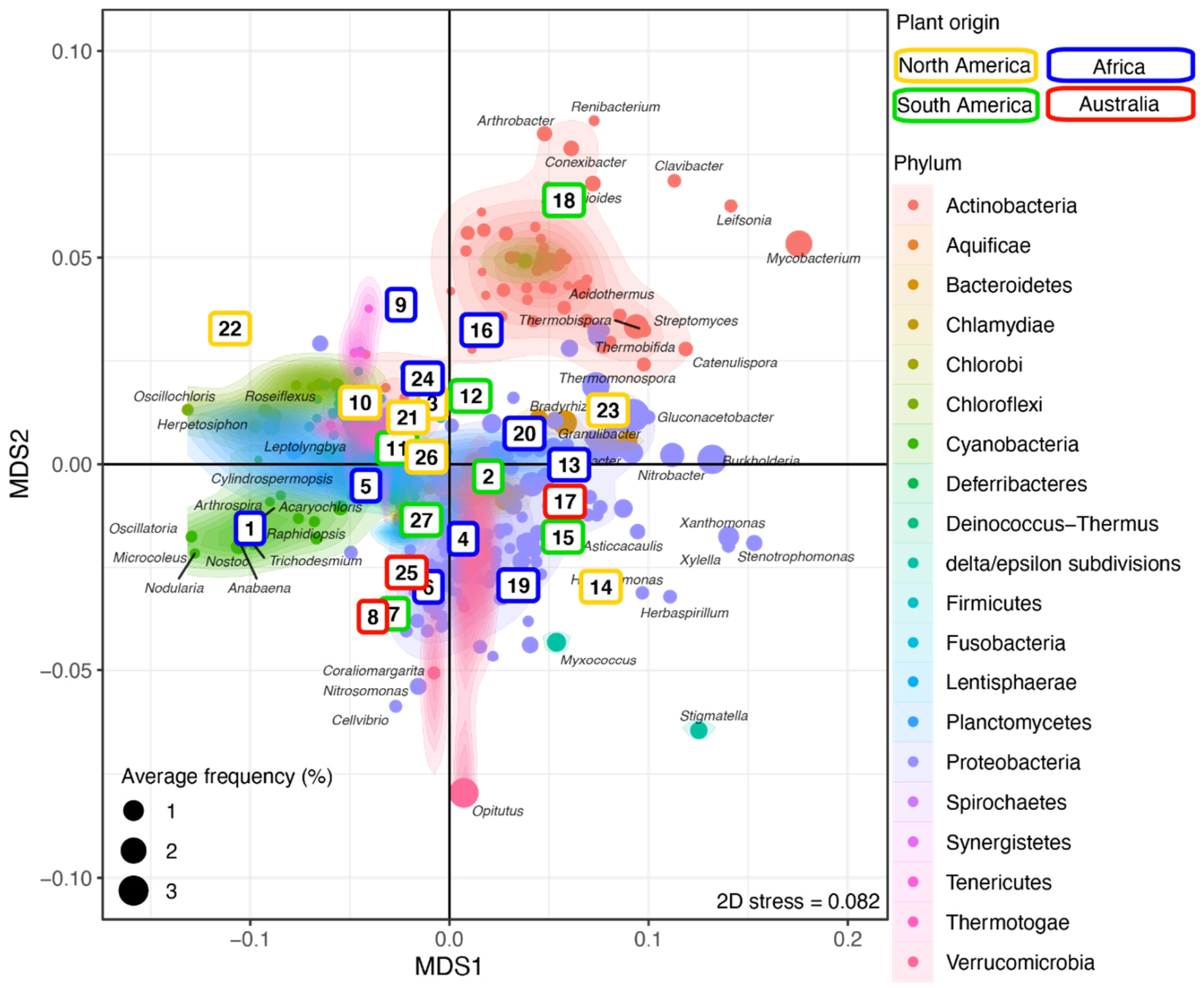
3.2. Microbiome Composition
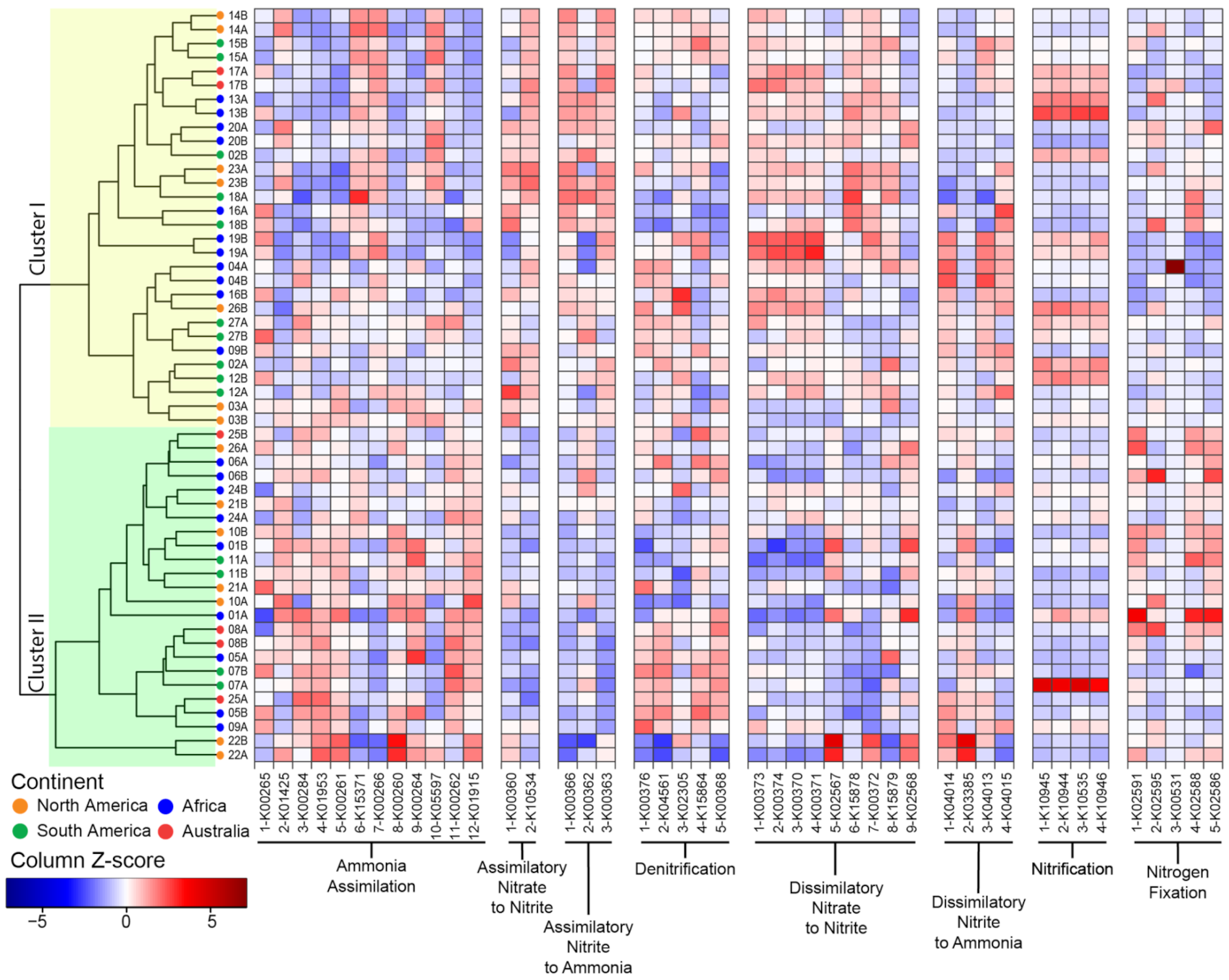
3.3. Functional Potential for Nitrogen Cycling
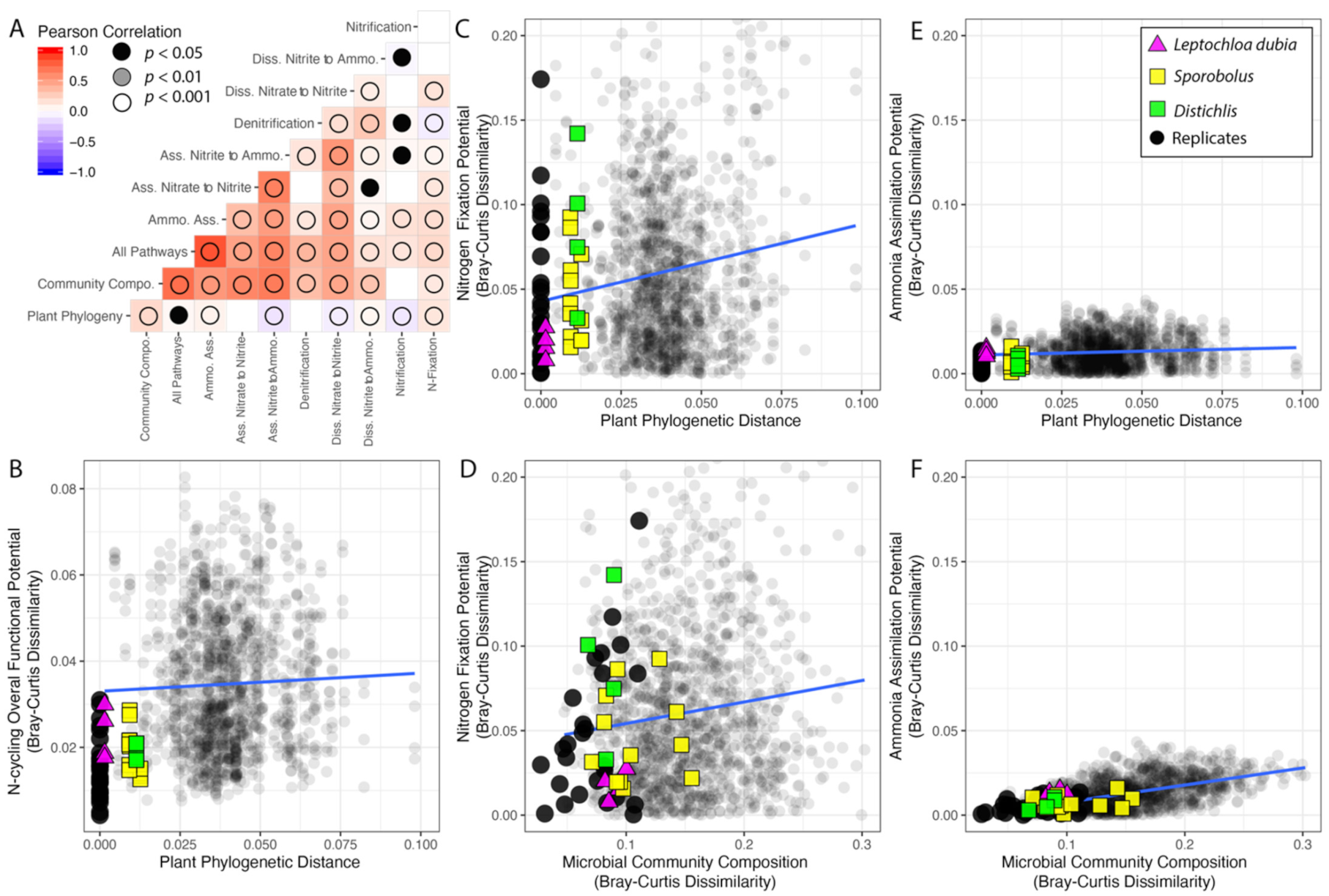
3.4. Phylosymbiosis and N Cycling Traits
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cleland, E.E.; Harpole, W.S. Nitrogen Enrichment and Plant Communities. Ann. N. Y. Acad. Sci. 2010, 1195, 46–61. [Google Scholar] [CrossRef] [PubMed]
- Fowler, D.; Coyle, M.; Skiba, U.; Sutton, M.A.; Cape, J.N.; Reis, S.; Sheppard, L.J.; Jenkins, A.; Grizzetti, B.; Galloway, J.N.; et al. The Global Nitrogen Cycle in the Twentyfirst Century. Philos. Trans. R. Soc. B Biol. Sci. 2013. [Google Scholar] [CrossRef]
- Fitzpatrick, C.R.; Copeland, J.; Wang, P.W.; Guttman, D.S.; Kotanen, P.M.; Johnson, M.T.J. Assembly and Ecological Function of the Root Microbiome across Angiosperm Plant Species. Proc. Natl. Acad. Sci. USA 2018, 115, E1157–E1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, M.B.; Berlemont, R.; Martiny, A.C.; Martiny, J.B. Nitrogen Cycling Potential of a Grassland Litter Microbial Community. Appl. Environ. Microbiol. 2015, 81, 7012–7022. [Google Scholar] [CrossRef] [Green Version]
- Philippot, L.; Raaijmakers, J.M.; Lemanceau, P.; van der Putten, W.H. Going Back to the Roots: The Microbial Ecology of the Rhizosphere. Nat. Rev. Microbiol. 2013, 11, 789–799. [Google Scholar] [CrossRef]
- Philippot, L.; Spor, A.; Hénault, C.; Bru, D.; Bizouard, F.; Jones, C.M.; Sarr, A.; Maron, P.-A.A.; He, C. Loss in Microbial Diversity Affects Nitrogen Cycling in Soil. ISME J. 2013, 7, 1609–1619. [Google Scholar] [CrossRef]
- Steenhoudt, O.; Vanderleyden, J. Azospirillum, a Free-Living Nitrogen-Fixing Bacterium Closely Associated with Grasses: Genetic, Biochemical and Ecological Aspects. FEMS Microbiol. Rev. 2000, 24, 487–506. [Google Scholar] [CrossRef]
- Masson-Boivin, C.; Sachs, J.L. Symbiotic Nitrogen Fixation by Rhizobia—The Roots of a Success Story. Curr. Opin. Plant Biol. 2018, 44, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Sellstedt, A.; Richau, K.H. Aspects of Nitrogen-Fixing Actinobacteria, in Particular Free-Living and Symbiotic Frankia. FEMS Microbiol. Lett. 2013, 342, 179–186. [Google Scholar] [CrossRef] [Green Version]
- Paerl, H. The Cyanobacterial Nitrogen Fixation Paradox in Natural Waters. F1000Research 2017, 6, 244. [Google Scholar] [CrossRef] [Green Version]
- Santi, C.; Bogusz, D.; Franche, C. Biological Nitrogen Fixation in Non-Legume Plants. Ann. Bot. 2013, 111, 743–767. [Google Scholar] [CrossRef] [Green Version]
- Gaimster, H.; Alston, M.; Richardson, D.J.; Gates, A.J.; Rowley, G. Transcriptional and Environmental Control of Bacterial Denitrification and N2O Emissions. FEMS Microbiol. Lett. 2018, 365, fnx277. [Google Scholar] [CrossRef]
- Stein, L.Y.; Klotz, M.G. The Nitrogen Cycle. Curr. Biol. 2016, 26, R94–R98. [Google Scholar] [CrossRef] [Green Version]
- Louca, S.; Polz, M.F.; Mazel, F.; Albright, M.B.N.; Huber, J.A.; O’Connor, M.I.; Ackermann, M.; Hahn, A.S.; Srivastava, D.S.; Crowe, S.A.; et al. Function and Functional Redundancy in Microbial Systems. Nat. Ecol. Evol. 2018, 2, 936–943. [Google Scholar] [CrossRef]
- Martiny, J.B.H.; Jones, S.E.; Lennon, J.T.; Martiny, A.C. Microbiomes in Light of Traits: A Phylogenetic Perspective. Science 2015, 350, aac9323. [Google Scholar] [CrossRef] [Green Version]
- Zimmerman, A.E.; Martiny, A.C.; Allison, S.D. Microdiversity of Extracellular Enzyme Genes among Sequenced Prokaryotic Genomes. ISME J. 2013, 7, 1187–1199. [Google Scholar] [CrossRef] [Green Version]
- Berlemont, R.; Allison, S.D.; Weihe, C.; Lu, Y.; Brodie, E.L.; Martiny, J.B.H.; Martiny, A.C. Cellulolytic Potential under Environmental Changes in Microbial Communities from Grassland Litter. Front. Microbiol. 2014, 5, 639. [Google Scholar] [CrossRef]
- Treseder, K.K.; Berlemont, R.; Allison, S.D.; Martiny, A.C. Nitrogen Enrichment Shifts Functional Genes Related to Nitrogen and Carbon Acquisition in the Fungal Community. Soil Biol. Biochem. 2018, 123, 87–96. [Google Scholar] [CrossRef] [Green Version]
- Amend, A.S.; Martiny, A.C.; Allison, S.D.; Berlemont, R.; Goulden, M.L.; Lu, Y.; Treseder, K.K.; Weihe, C.; Martiny, J.B.H. Microbial Response to Simulated Global Change Is Phylogenetically Conserved and Linked with Functional Potential. ISME J. 2015, 10, 109–118. [Google Scholar] [CrossRef] [Green Version]
- Berlemont, R.; Martiny, A.C. Glycoside Hydrolases across Environmental Microbial Communities. PLoS Comput. Biol. 2016, 12, e1005300. [Google Scholar] [CrossRef] [Green Version]
- Emmett, B.D.; Youngblut, N.D.; Buckley, D.H.; Drinkwater, L.E. Plant Phylogeny and Life History Shape Rhizosphere Bacterial Microbiome of Summer Annuals in an Agricultural Field. Front. Microbiol. 2017, 8, 2414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzpatrick, C.R.; Salas-González, I.; Conway, J.M.; Finkel, O.M.; Gilbert, S.; Russ, D.; Teixeira, P.J.P.L.; Dangl, J.L. The Plant Microbiome: From Ecology to Reductionism and Beyond. Annu. Rev. Microbiol. 2020, 74, 81–100. [Google Scholar] [CrossRef] [PubMed]
- Kohl, K.D.; Dearing, M.D.; Bordenstein, S.R. Microbial Communities Exhibit Host Species Distinguishability and Phylosymbiosis along the Length of the Gastrointestinal Tract. Mol. Ecol. 2018, 27, 1874–1883. [Google Scholar] [CrossRef] [PubMed]
- Durán, P.; Thiergart, T.; Garrido-Oter, R.; Agler, M.; Kemen, E.; Schulze-Lefert, P.; Hacquard, S. Microbial Interkingdom Interactions in Roots Promote Arabidopsis Survival. Cell 2018, 175, 973–983.e14. [Google Scholar] [CrossRef]
- Huang, A.C.; Jiang, T.; Liu, Y.X.; Bai, Y.C.; Reed, J.; Qu, B.; Goossens, A.; Nützmann, H.W.; Bai, Y.; Osbourn, A. A Specialized Metabolic Network Selectively Modulates Arabidopsis Root Microbiota. Science 2019, 364, eaau6389. [Google Scholar] [CrossRef]
- Lundberg, D.S.; Lebeis, S.L.; Paredes, S.H.; Yourstone, S.; Gehring, J.; Malfatti, S.; Tremblay, J.; Engelbrektson, A.; Kunin, V.; del Rio, T.G.; et al. Defining the Core Arabidopsis Thaliana Root Microbiome. Nature 2013, 488, 86–90. [Google Scholar] [CrossRef] [Green Version]
- Brooks, A.W.; Kohl, K.D.; Brucker, R.M.; van Opstal, E.J.; Bordenstein, S.R. Phylosymbiosis: Relationships and Functional Effects of Microbial Communities across Host Evolutionary History. PLoS Biol. 2016, 14, e2000225. [Google Scholar] [CrossRef]
- Kim, H.; Lee, K.K.; Jeon, J.; Harris, W.A.; Lee, Y.H. Domestication of Oryza Species Eco-Evolutionarily Shapes Bacterial and Fungal Communities in Rice Seed. Microbiome 2020, 8, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Kohl, K.D. Ecological and Evolutionary Mechanisms Underlying Patterns of Phylosymbiosis in Host-Associated Microbial Communities. Philos. Trans. R. Soc. B Biol. Sci. 2020, 375, 20190251. [Google Scholar] [CrossRef] [Green Version]
- Mazel, F.; Davis, K.M.; Loudon, A.; Kwong, W.K.; Groussin, M.; Parfrey, L.W. Is Host Filtering the Main Driver of Phylosymbiosis across the Tree of Life? mSystems 2018, 3, e00097-18. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.J.; Bordenstein, S.R. An Introduction to Phylosymbiosis. Proc. R. Soc. B Biol. Sci. 2020, 287, 20192900. [Google Scholar] [CrossRef] [Green Version]
- Haycock, N.E.; Pinay, G. Groundwater Nitrate Dynamics in Grass and Poplar Vegetated Riparian Buffer Strips during the Winter. J. Environ. Qual. 1993, 22, 273–278. [Google Scholar] [CrossRef]
- DeAngelis, K.M.; Silver, W.L.; Thompson, A.W.; Firestone, M.K. Microbial Communities Acclimate to Recurring Changes in Soil Redox Potential Status. Environ. Microbiol. 2010, 12, 3137–3149. [Google Scholar] [CrossRef]
- Walters, W.A.; Jin, Z.; Youngblut, N.; Wallace, J.G.; Sutter, J.; Zhang, W.; González-Peña, A.; Peiffer, J.; Koren, O.; Shi, Q.; et al. Large-Scale Replicated Field Study of Maize Rhizosphere Identifies Heritable Microbes. Proc. Natl. Acad. Sci. USA 2018, 14, 609–622. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Tavaré, S. Some Probabilistic and Statistical Problems in the Analysis of DNA Sequences. Am. Math. Soc. Lect. Math. Life Sci. 1986, 17, 57–86. [Google Scholar]
- Keegan, K.P.; Glass, E.M.; Meyer, F. MG-RAST, a Metagenomics Service for Analysis of Microbial Community Structure and Function. In Microbial Environmental Genomics (MEG); Humana Press: New York, NY, USA, 2016; pp. 207–233. [Google Scholar]
- Kanehisa, M.; Sato, Y.; Furumichi, M.; Morishima, K.; Tanabe, M. New Approach for Understanding Genome Variations in KEGG. Nucleic Acids Res. 2019, 47, D590–D595. [Google Scholar] [CrossRef] [Green Version]
- The UniProt Consortium UniProt: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [CrossRef]
- Kanehisa, M. Subramaniam The KEGG Database. Proc. Novartis Found. Symp. 2002, 247, 91–103. [Google Scholar]
- Berlemont, R.; Winans, N.; Talamantes, D.; Dang, H.; Tsai, H.W. MetaGeneHunt for protein domain annotation in short-read metagenomes. Sci. Rep. 2020. [Google Scholar] [CrossRef]
- Wilke, A.; Harrison, T.; Wilkening, J.; Field, D.; Glass, E.M.; Kyrpides, N.; Mavrommatis, K.; Meyer, F. The M5nr: A novel non-redundant database containing protein sequences and annotations from multiple sources and associated tools. BMC Bioinform. 2012, 13, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilke, A.; Bischof, J.; Harrison, T.; Brettin, T.; D’Souza, M.; Gerlach, W.; Matthews, H.; Paczian, T.; Wilkening, J.; Glass, E.M.; et al. A RESTful API for Accessing Microbial Community Data for MG-RAST. PLoS Comput. Biol. 2015, 11, e1004008. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.; Bisseling, T. The Evolutionary Aspects of Legume Nitrogen–Fixing Nodule Symbiosis. Results Probl. Cell Differ. 2020, 69, 387–408. [Google Scholar] [CrossRef] [PubMed]
- Lie, T.A.; Akkermans, A.D.L.; van Egeraat, A.W.S.M. Natural Variation in Symbiotic Nitrogen-Fixing Rhizobium and Frankia Spp. Antonie Van Leeuwenhoek 1984, 50, 489–503. [Google Scholar] [CrossRef]
- Peterson, P.; Columbus, T.; Pennington, S. Classification and Biogeography of New World Grasses: Chloridoideae. Aliso 2007, 23, 580–594. [Google Scholar] [CrossRef] [Green Version]
- Duvall, M.R.; Fisher, A.E.; Columbus, J.T.; Ingram, A.L.; Wysocki, W.P.; Burke, S.V.; Clark, L.G.; Kelchner, S.A. Phylogenomics and Plastome Evolution of the Chloridoid Grasses (Chloridoideae: Poaceae). Int. J. Plant Sci. 2016, 177, 235–246. [Google Scholar] [CrossRef]
- Fisher, A.E.; Hasenstab, K.M.; Bell, H.L.; Blaine, E.; Ingram, A.L.; Columbus, J.T. Evolutionary History of Chloridoid Grasses Estimated from 122 Nuclear Loci. Mol. Phylogenetics Evol. 2016, 105, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Dos Santos, P.C.; Fang, Z.; Mason, S.W.; Setubal, J.C.; Dixon, R. Distribution of Nitrogen Fixation and Nitrogenase-like Sequences amongst Microbial Genomes. BMC Genom. 2012, 13, 162. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Quensen, J.F.; Sun, Y.; Wang, Q.; Brown, C.T.; Cole, J.R.; Tiedje, J.M. Review, Evaluation, and Directions for Gene-Targeted Assembly for Ecological Analyses of Metagenomes. Front. Genet. 2019, 10, 957. [Google Scholar] [CrossRef] [Green Version]
- Martiny, A.C.; Treseder, K.; Pusch, G. Phylogenetic Conservatism of Functional Traits in Microorganisms. ISME J. 2013, 7, 830–838. [Google Scholar] [CrossRef]
- Houlton, B.Z.; Wang, Y.P.; Vitousek, P.M.; Field, C.B. A Unifying Framework for Dinitrogen Fixation in the Terrestrial Biosphere. Nature 2008, 454, 327–330. [Google Scholar] [CrossRef]
- Menge, D.N.L.; Levin, S.A.; Hedin, L.O. Evolutionary Tradeoffs Can Select against Nitrogen Fixation and Thereby Maintain Nitrogen Limitation. Proc. Natl. Acad. Sci. USA 2008, 105, 1573–1578. [Google Scholar] [CrossRef] [Green Version]
- Elhady, A.; Hallmann, J.; Heuer, H. Symbiosis of Soybean with Nitrogen Fixing Bacteria Affected by Root Lesion Nematodes in a Density-Dependent Manner. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- van Schreven, D.A. The Effect of Some Actinomycetes on the Growth and Nitrogen-Fixing Power of Azotobacter Chroococcum. Plant Soil 1963, 19, 1–18. [Google Scholar] [CrossRef]
- Patel, J.J. Antagonism of Actinomycetes against Rhizobia. Plant Soil 1974, 41, 395–402. [Google Scholar] [CrossRef]
- Nelson, M.B.; Martiny, A.C.; Martiny, J.B.H. Global Biogeography of Microbial Nitrogen-Cycling Traits in Soil. Proc. Natl. Acad. Sci. USA 2016, 113, 8033–8040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bothe, H.; Schmitz, O.; Yates, M.G.; Newton, W.E. Nitrogen Fixation and Hydrogen Metabolism in Cyanobacteria. Microbiol. Mol. Biol. Rev. 2010, 74, 529–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mus, F.; Alleman, A.B.; Pence, N.; Seefeldt, L.C.; Peters, J.W. Exploring the Alternatives of Biological Nitrogen Fixation. Metallomics 2018, 10, 523–538. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, T.L.; Ludwig, M.; Dixon, R.; Boyd, E.S.; Dos Santos, P.C.; Setubal, J.C.; Bryant, D.A.; Dean, D.R.; Peters, J.W. Transcriptional Profiling of Nitrogen Fixation in Azotobacter Vinelandii. J. Bacteriol. 2011, 193, 4477–4486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaby, J.C.; Buckley, D.H. A Comprehensive Aligned NifH Gene Database: A Multipurpose Tool for Studies of Nitrogen-Fixing Bacteria. Database 2014, 2014, bau001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choo, Q.C.; Samian, M.R.; Najimudin, N. Phylogeny and Characterization of Three NifH-Homologous Genes from Paenibacillus Azotofixans. Appl. Environ. Microbiol. 2003, 69, 3658–3662. [Google Scholar] [CrossRef] [Green Version]
- Oda, Y.; Samanta, S.K.; Rey, F.E.; Wu, L.; Liu, X.; Yan, T.; Zhou, J.; Harwood, C.S. Functional Genomic Analysis of Three Nitrogenase Isozymes in the Photosynthetic Bacterium Rhodopseudomonas Palustris. J. Bacteriol. 2005, 187, 7784–7794. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # of Traits | Plant Phylogeny Slope (Std. Error) | Community Composition Slope (Std. Error) | |
|---|---|---|---|
| Model | lm(Pathway~PlantPhylo) | lm(Pathway~Com.Comp.) | |
| All pathways | 44 | 0.042 * (0.021) | 0.241 *** (0.004) |
| Ammo. Ass. | 12 | 0.044 *** (0.012) | 0.101 *** (0.003) |
| Ass. Nitrate to Nitrite | 2 | NS | NS |
| Ass. Nitrite to Ammonia | 3 | −0.197 *** (0.033) | 0.335 *** (0.007) |
| Denitrification | 5 | NS | 0.236 *** (0.013) |
| Diss. Nitrate to Nitrite | 9 | −0.126 *** (0.031) | 0.219 *** (0.008) |
| Diss. Nitrite to Ammo. | 4 | 0.141 ** (0.052) | 0.249 *** (0.015) |
| Nitrification | 4 | −1.470 (0.299) | NS |
| N-Fixation | 5 | 0.456 *** (0.065) | 0.127 *** (0.020) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Bel, M.; Fisher, A.E.; Ball, L.; Columbus, J.T.; Berlemont, R. Phylosymbiosis in the Rhizosphere Microbiome Extends to Nitrogen Cycle Functional Potential. Microorganisms 2021, 9, 2476. https://doi.org/10.3390/microorganisms9122476
Van Bel M, Fisher AE, Ball L, Columbus JT, Berlemont R. Phylosymbiosis in the Rhizosphere Microbiome Extends to Nitrogen Cycle Functional Potential. Microorganisms. 2021; 9(12):2476. https://doi.org/10.3390/microorganisms9122476
Chicago/Turabian StyleVan Bel, Mikayla, Amanda E. Fisher, Laymon Ball, J. Travis Columbus, and Renaud Berlemont. 2021. "Phylosymbiosis in the Rhizosphere Microbiome Extends to Nitrogen Cycle Functional Potential" Microorganisms 9, no. 12: 2476. https://doi.org/10.3390/microorganisms9122476
APA StyleVan Bel, M., Fisher, A. E., Ball, L., Columbus, J. T., & Berlemont, R. (2021). Phylosymbiosis in the Rhizosphere Microbiome Extends to Nitrogen Cycle Functional Potential. Microorganisms, 9(12), 2476. https://doi.org/10.3390/microorganisms9122476