
Diversity Indices of Plant Communities and Their Rhizosphere Microbiomes: An Attempt to Find the Connection

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. DNA Extraction and Sequencing
2.3. Bioinformatic Processing
3. Results
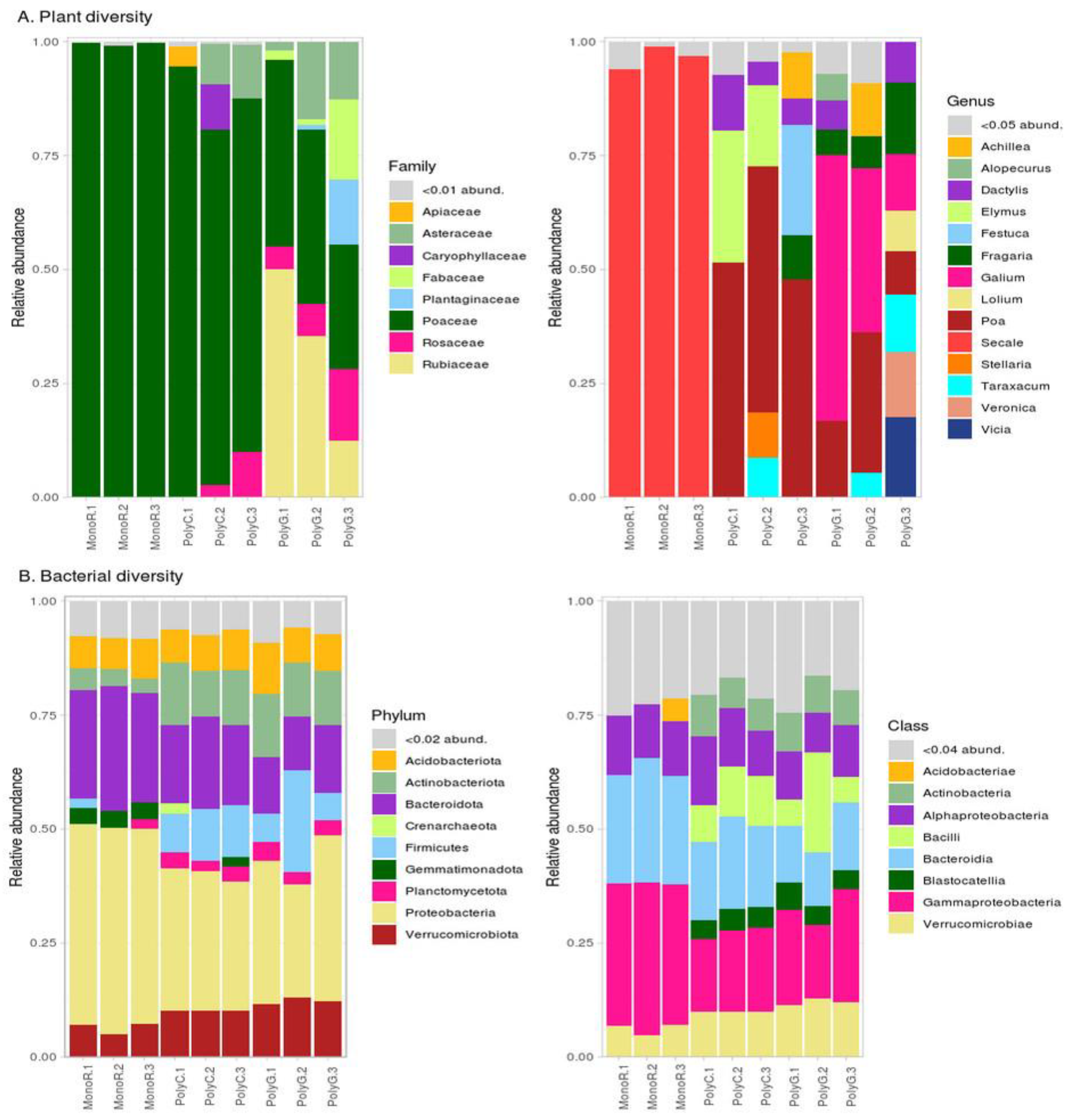
3.1. Taxonomic Composition
3.2. Alpha-Diversity Indices
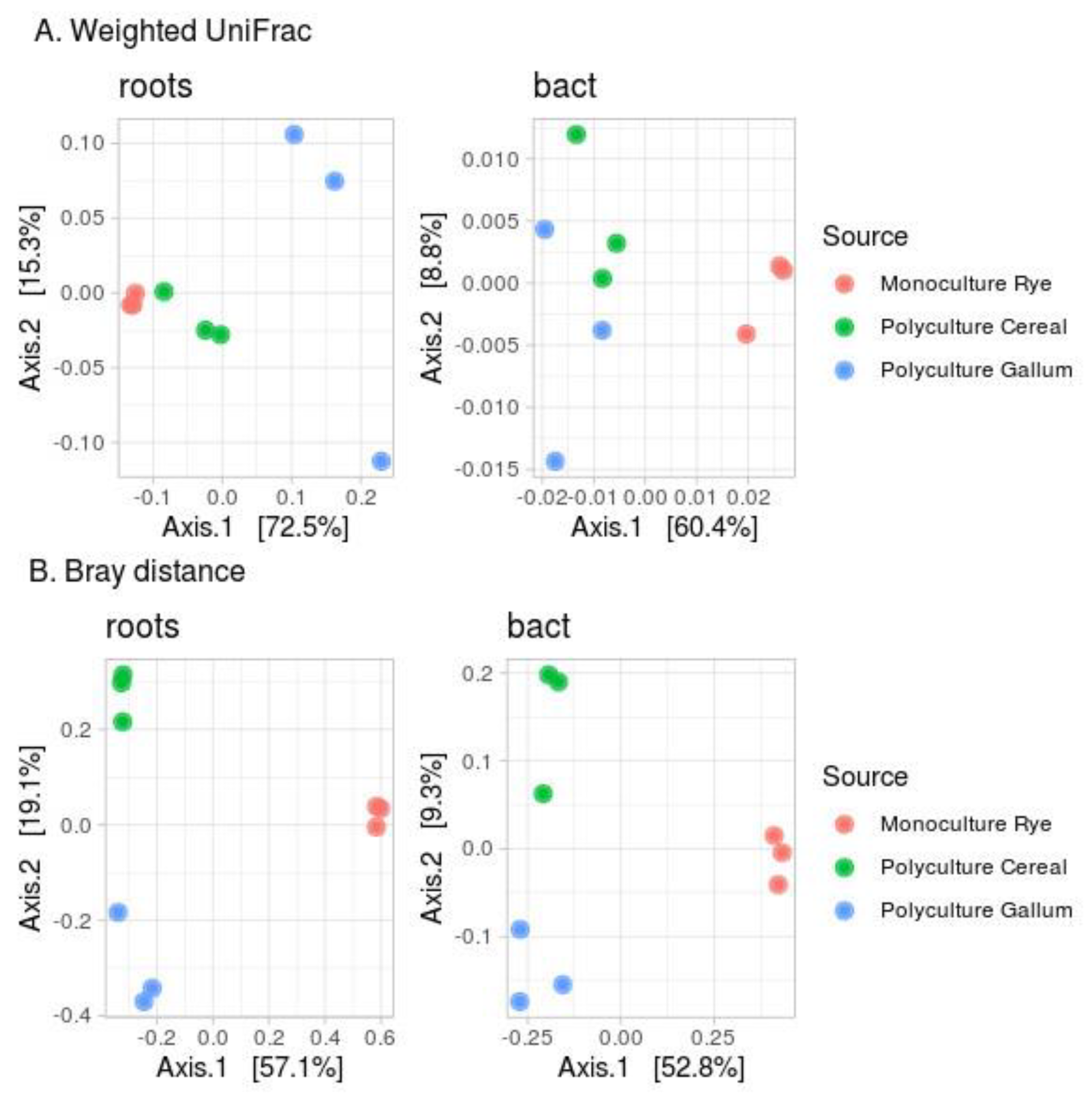
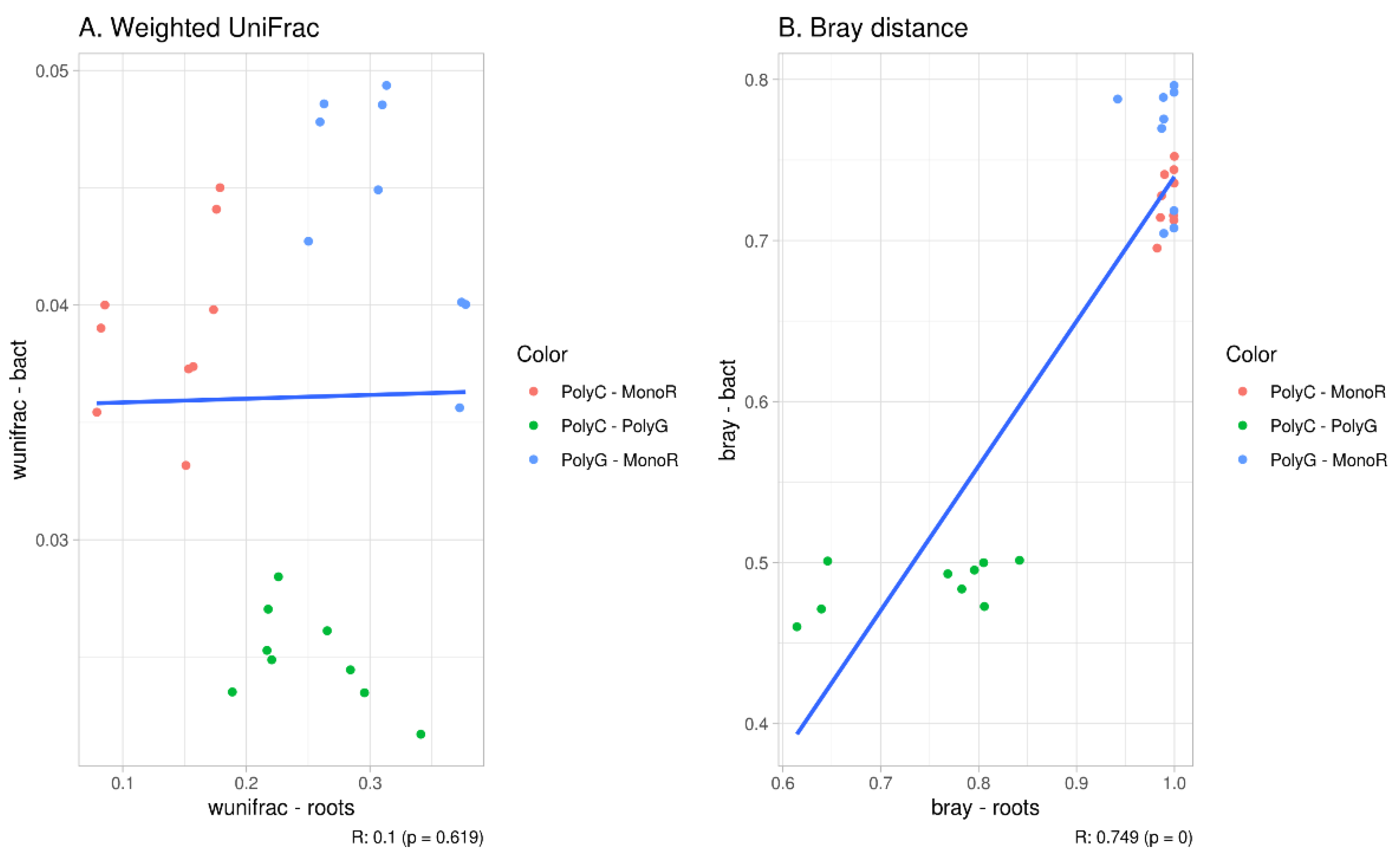
3.3. Beta-Diversity Indices
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gaiero, J.R.; McCall, C.A.; Thompson, K.A.; Day, N.J.; Best, A.S.; Dunfield, K.E. Inside the Root Microbiome: Bacterial Root Endophytes and Plant Growth Promotion. Am. J. Bot. 2013, 100, 1738–1750. [Google Scholar] [CrossRef] [Green Version]
- Philippot, L.; Raaijmakers, J.M.; Lemanceau, P.; Van der Putten, W.H. Going Back to the Roots: The Microbial Ecology of the Rhizosphere. Nat. Rev. Microbiol. 2013, 11, 789–799. [Google Scholar] [CrossRef]
- Kong, H.G.; Song, G.C.; Ryu, C. Inheritance of Seed and Rhizosphere Microbial Communities through Plant–Soil Feedback and Soil Memory. Environ. Microbiol. Rep. 2019, 11, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Bakker, P.A.H.M.; Pieterse, C.M.J.; de Jonge, R.; Berendsen, R.L. The Soil-Borne Legacy. Cell 2018, 172, 1178–1180. [Google Scholar] [CrossRef] [Green Version]
- Berg, G.; Smalla, K. Plant Species and Soil Type Cooperatively Shape the Structure and Function of Microbial Communities in the Rhizosphere: Plant Species, Soil Type and Rhizosphere Communities. FEMS Microbiol. Ecol. 2009, 68, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlaeppi, K.; Bulgarelli, D. The Plant Microbiome at Work. Mol. Plant-Microbe Interact. 2015, 28, 212–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulgarelli, D.; Garrido-Oter, R.; Münch, P.C.; Weiman, A.; Dröge, J.; Pan, Y.; McHardy, A.C.; Schulze-Lefert, P. Structure and Function of the Bacterial Root Microbiota in Wild and Domesticated Barley. Cell Host Microbe 2015, 17, 392–403. [Google Scholar] [CrossRef] [Green Version]
- Edwards, J.; Johnson, C.; Santos-Medellín, C.; Lurie, E.; Podishetty, N.K.; Bhatnagar, S.; Eisen, J.A.; Sundaresan, V. Structure, Variation, and Assembly of the Root-Associated Microbiomes of Rice. Proc. Natl. Acad. Sci. USA 2015, 112, E911–E920. [Google Scholar] [CrossRef] [Green Version]
- Lundberg, D.S.; Lebeis, S.L.; Edgar, R.C.; Eickhorst, T.; Ley, R.E.; Hugenholtz, P.; Tringe, S.G.; Dangl, J.L.; Paredes, S.H.; Yourstone, S.; et al. Defining the Core Arabidopsis Thaliana Root Microbiome. Nature 2012, 488, 86–90. [Google Scholar] [CrossRef] [Green Version]
- Sasse, J.; Martinoia, E.; Northen, T. Feed Your Friends: Do Plant Exudates Shape the Root Microbiome? Trends. Plant. Sci. 2018, 23, 25–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zverev, A.O.; Pershina, E.V.; Shapkin, V.M.; Kichko, A.K.; Mitrofanova, O.P.; Kobylyanskii, V.D.; Yuzikhin, O.S.; Belimov, A.A.; Andronov, E.E. Molecular Analysis of the Rhizosphere Microbial Communities from Gramineous Plants Grown on Contrasting Soils. Microbiology 2020, 89, 231–241. [Google Scholar] [CrossRef]
- Vives-Peris, V.; de Ollas, C.; Gómez-Cadenas, A.; Pérez-Clemente, R.M. Root Exudates: From Plant to Rhizosphere and Beyond. Plant. Cell Rep. 2020, 39, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Goberna, M.; Navarro-Cano, J.A.; Verdú, M. Opposing Phylogenetic Diversity Gradients of Plant and Soil Bacterial Communities. Proc. R. Soc. B Biol. Sci. 2016, 283, 20153003. [Google Scholar] [CrossRef] [Green Version]
- Dassen, S.; Cortois, R.; Martens, H.; De Hollander, M.; Kowalchuk, G.A.; van der Putten, W.H.; De Deyn, G.B. Differential Responses of Soil Bacteria, Fungi, Archaea and Protists to Plant Species Richness and Plant Functional Group Identity. Mol. Ecol. 2017, 26, 4085–4098. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Wang, X.; Liang, C.; Hao, Z.; Zhou, L.; Ma, S.; Li, X.; Yang, S.; Yao, F.; Jiang, Y. Aboveground-Belowground Biodiversity Linkages Differ in Early and Late Successional Temperate Forests. Sci. Rep. 2015, 5, 12234. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.-Q.; Wang, J.; Shen, C.; He, J.-Z.; Ge, Y. Plant Evenness Modulates the Effect of Plant Richness on Soil Bacterial Diversity. Sci. Total Environ. 2019, 662, 8–14. [Google Scholar] [CrossRef]
- Prober, S.M.; Leff, J.W.; Bates, S.T.; Borer, E.T.; Firn, J.; Harpole, W.S.; Lind, E.M.; Seabloom, E.W.; Adler, P.B.; Bakker, J.D.; et al. Plant Diversity Predicts Beta but Not Alpha Diversity of Soil Microbes across Grasslands Worldwide. Ecol. Lett. 2015, 18, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Sharma, T.R.; Devanna, B.N.; Kiran, K.; Singh, P.K.; Arora, K.; Jain, P.; Tiwari, I.M.; Dubey, H.; Saklani, B.; Kumari, M.; et al. Status and Prospects of Next Generation Sequencing Technologies in Crop Plants. Curr. Issues Mol. Biol. 2018, 27, 1–36. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.; Helmstetter, A.J.; Devey, D.; Wilkinson, T.; Papadopulos, A.S.T. Field-Based Species Identification of Closely-Related Plants Using Real-Time Nanopore Sequencing. Sci. Rep. 2017, 7, 8345–8348. [Google Scholar] [CrossRef] [PubMed]
- Skuodienė, R.; Tomchuk, D. Root Mass and Root to Shoot Ratio of Different Perennial Forage Plants under Western Lithuania Climatic Conditions. Rom. Agric. Res. 2015, 32, 1–11. [Google Scholar]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global Patterns of 16S RRNA Diversity at a Depth of Millions of Sequences per Sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef] [Green Version]
- Doyle, J.J.; Doyle, J.L. Isolation of Plant DNA from Fresh Tissue. Focus 1990, 12, 13–15. [Google Scholar]
- Cheng, T.; Xu, C.; Lei, L.; Li, C.; Zhang, Y.; Zhou, S. Barcoding the Kingdom Plantae: New PCR Primers for ITS Regions of Plants with Improved Universality and Specificity. Mol. Ecol. Resour. 2016, 16, 138–149. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. Embnet. J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Banchi, E.; Ametrano, C.G.; Greco, S.; Stanković, D.; Muggia, L.; Pallavicini, A. PLANiTS: A Curated Sequence Reference Dataset for Plant ITS DNA Metabarcoding. Database J. Biol. Databases Curation 2020, 2020, baz155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kembel, S.W.; Cowan, P.D.; Helmus, M.R.; Cornwell, W.K.; Morlon, H.; Ackerly, D.D.; Blomberg, S.P.; Webb, C.O. Picante: R Tools for Integrating Phylogenies and Ecology. Bioinformatics 2010, 26, 1463–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genomebiol. Com. 2014, 15. [Google Scholar] [CrossRef] [Green Version]
- Hartman, K.; van der Heijden, M.G.A.; Roussely-Provent, V.; Walser, J.-C.; Schlaeppi, K. Deciphering Composition and Function of the Root Microbiome of a Legume Plant. Microbiome 2017, 5, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarria-Guzmán, Y.; Chávez-Romero, Y.; Gómez-Acata, S.; Montes-Molina, J.A.; Morales-Salazar, E.; Dendooven, L.; Navarro-Noya, Y.E. Bacterial Communities Associated with Different Anthurium Andraeanum L. Plant Tissues. Microbes Environ. 2016, 31, 321–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascual, J.; Wüst, P.K.; Geppert, A.; Foesel, B.U.; Huber, K.J.; Overmann, J. Novel Isolates Double the Number of Chemotrophic Species and Allow the First Description of Higher Taxa in Acidobacteria Subdivision 4. Syst. Appl. Microbiol. 2015, 38, 534–544. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zverev, A.O.; Kichko, A.A.; Pinaev, A.G.; Provorov, N.A.; Andronov, E.E. Diversity Indices of Plant Communities and Their Rhizosphere Microbiomes: An Attempt to Find the Connection. Microorganisms 2021, 9, 2339. https://doi.org/10.3390/microorganisms9112339
Zverev AO, Kichko AA, Pinaev AG, Provorov NA, Andronov EE. Diversity Indices of Plant Communities and Their Rhizosphere Microbiomes: An Attempt to Find the Connection. Microorganisms. 2021; 9(11):2339. https://doi.org/10.3390/microorganisms9112339
Chicago/Turabian StyleZverev, Aleksei O., Arina A. Kichko, Aleksandr G. Pinaev, Nikolay A. Provorov, and Evgeny E. Andronov. 2021. "Diversity Indices of Plant Communities and Their Rhizosphere Microbiomes: An Attempt to Find the Connection" Microorganisms 9, no. 11: 2339. https://doi.org/10.3390/microorganisms9112339
APA StyleZverev, A. O., Kichko, A. A., Pinaev, A. G., Provorov, N. A., & Andronov, E. E. (2021). Diversity Indices of Plant Communities and Their Rhizosphere Microbiomes: An Attempt to Find the Connection. Microorganisms, 9(11), 2339. https://doi.org/10.3390/microorganisms9112339