
Whole Genome Sequencing in the Management of Non-Tuberculous Mycobacterial Infections


Abstract
1. Introduction
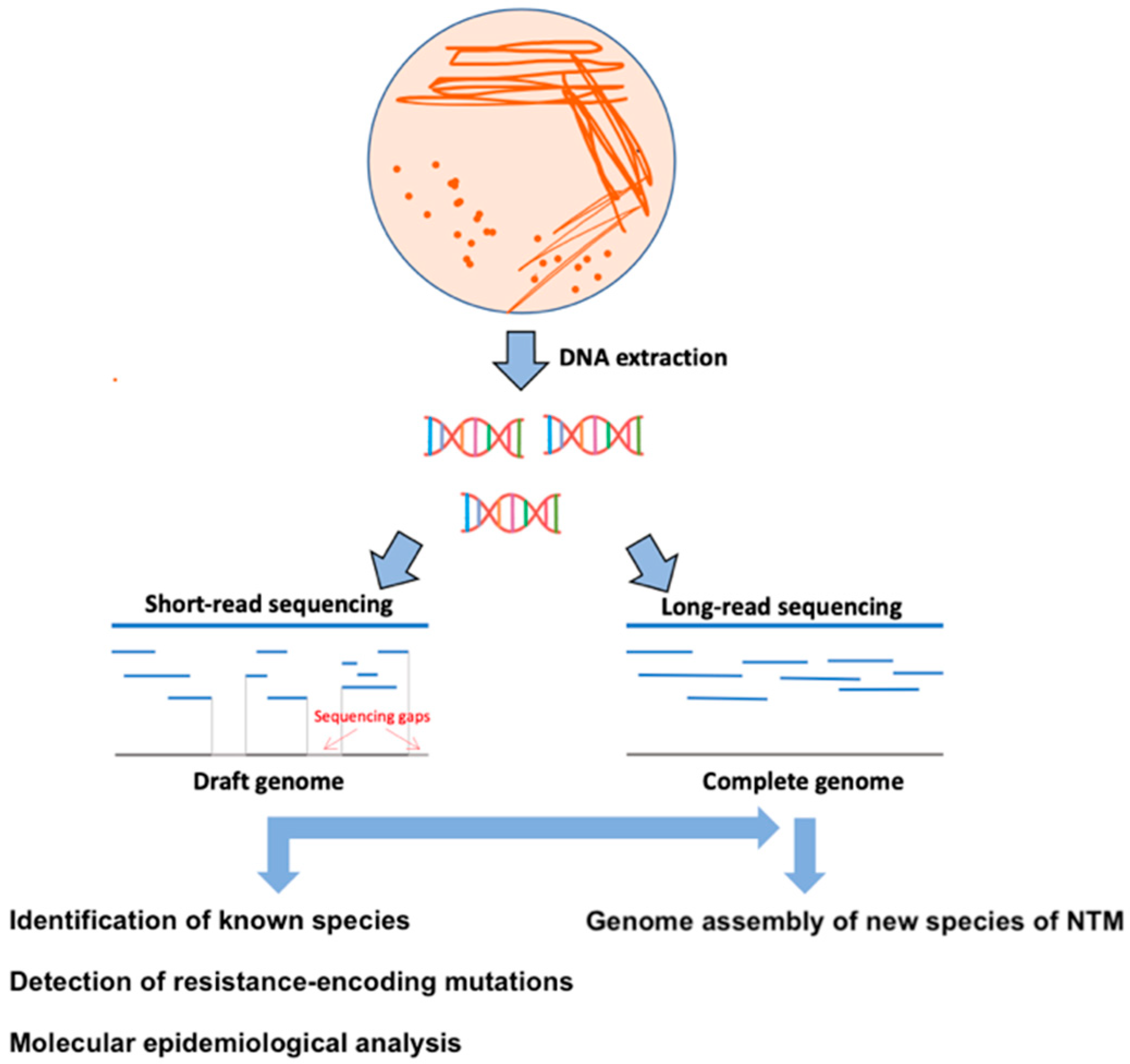
2. WGS Perspectives for Diagnostics and Characterization of Resistance Patterns of NTM
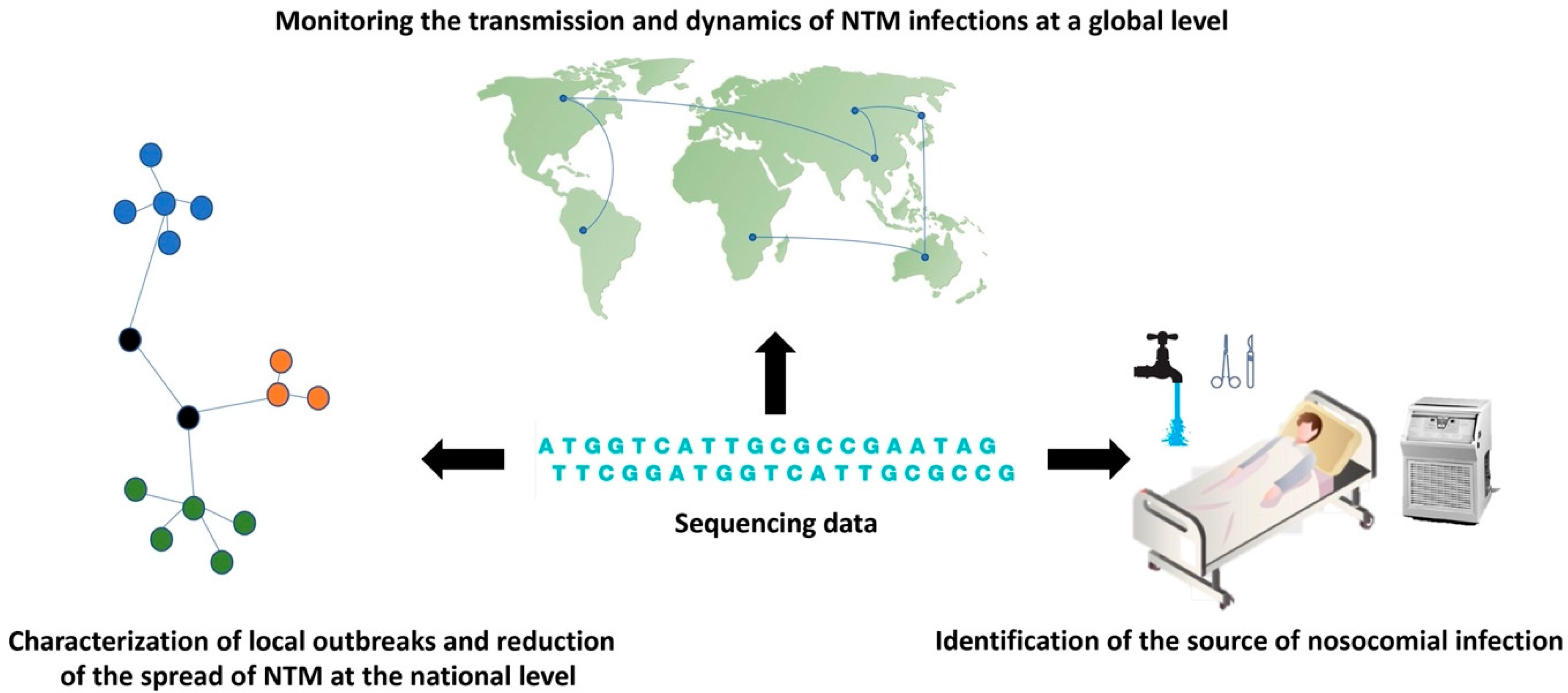
3. WGS Perspectives in Tracing the Transmission of Nosocomial NTM Infection
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Fedrizzi, T.; Meehan, C.; Grottola, A.; Giacobazzi, E.; Serpini, G.F.; Tagliazucchi, S.; Fabio, A.; Bettua, C.; Bertorelli, R.; De Sanctis, V.; et al. Genomic characterization of Nontuberculous Mycobacteria. Sci. Rep. 2017, 7, srep45258. [Google Scholar] [CrossRef]
- Falkinham, I.J. Surrounded by mycobacteria: Nontuberculous mycobacteria in the human environment. J. Appl. Microbiol. 2009, 107, 356–367. [Google Scholar] [CrossRef]
- Falkinham, J.O. Nontuberculous Mycobacteria from Household Plumbing of Patients with Nontuberculous Mycobacteria Disease. Emerg. Infect. Dis. 2011, 17, 419–424. [Google Scholar] [CrossRef]
- Esteban, J.; García-Pedrazuela, M.; Muñoz-Egea, M.C.; Alcaide, F. Current treatment of nontuberculous mycobacteriosis: An update. Expert Opin. Pharm. 2012, 13, 967–986. [Google Scholar] [CrossRef]
- Saxena, S.; Spaink, H.P.; Forn-Cuní, G. Drug Resistance in Nontuberculous Mycobacteria: Mechanisms and Models. Biology 2021, 10, 96. [Google Scholar] [CrossRef]
- Katale, B.Z.; Mbugi, E.V.; Karimuribo, E.D.; Keyyu, J.D.; Kendall, S.; Kibiki, G.S.; Godfrey-Faussett, P.; Michel, A.L.; Kazwala, R.R.; Van Helden, P.; et al. Prevalence and risk factors for infection of bovine tuberculosis in indigenous cattle in the Serengeti ecosystem, Tanzania. BMC Vet. Res. 2013, 9, 1–11. [Google Scholar] [CrossRef]
- Lattos, A.; Giantsis, I.A.; Karagiannis, D.; Theodorou, J.A.; Michaelidis, B. Gut Symbiotic Microbial Communities in the IUCN Critically Endangered Pinna nobilis Suffering from Mass Mortalities, Revealed by 16S rRNA Amplicon NGS. Pathogens 2020, 9, 1002. [Google Scholar] [CrossRef]
- Lattos, A.; Giantsis, I.A.; Karagiannis, D.; Michaelidis, B. First detection of the invasive Haplosporidian and Mycobacteria parasites hosting the endangered bivalve Pinna nobilis in Thermaikos Gulf, North Greece. Mar. Environ. Res. 2020, 155, 104889. [Google Scholar] [CrossRef]
- Prevots, D.R.; Marras, T.K. Epidemiology of Human Pulmonary Infection with Nontuberculous Mycobacteria: A review. Clin. Chest Med. 2015, 36, 13–34. [Google Scholar] [CrossRef]
- Degiacomi, G.; Chiarelli, L.; Recchia, D.; Petricci, E.; Gianibbi, B.; Fiscarelli, E.; Fattorini, L.; Manetti, F.; Pasca, M. The Antimalarial Mefloquine Shows Activity against Mycobacterium abscessus, Inhibiting Mycolic Acid Metabolism. Int. J. Mol. Sci. 2021, 22, 8533. [Google Scholar] [CrossRef]
- Larsson, L.-O.; Polverino, E.; Hoefsloot, W.; Codecasa, L.R.; Diel, R.; Jenkins, S.G.; Loebinger, M.R. Pulmonary disease by non-tuberculous mycobacteria—Clinical management, unmet needs and future perspectives. Expert Rev. Respir. Med. 2017, 11, 977–989. [Google Scholar] [CrossRef] [PubMed]
- Szymanski, E.P.; Leung, J.M.; Fowler, C.J.; Haney, C.; Hsu, A.P.; Chen, F.; Duggal, P.; Oler, A.J.; McCormack, R.; Podack, E.; et al. Pulmonary Nontuberculous Mycobacterial Infection. A Multisystem, Multigenic Disease. Am. J. Respir. Crit. Care Med. 2015, 192, 618–628. [Google Scholar] [CrossRef]
- Mirsaeidi, M.; Farshidpour, M.; Allen, M.B.; Ebrahimi, G.; Falkinham, J.O. Highlight on Advances in Nontuberculous Mycobacterial Disease in North America. BioMed Res. Int. 2014, 2014, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ryu, Y.J.; Koh, W.-J.; Daley, C.L. Diagnosis and Treatment of Nontuberculous Mycobacterial Lung Disease: Clinicians’ Perspectives. Tuberc. Respir. Dis. 2016, 79, 74–84. [Google Scholar] [CrossRef]
- Porvaznik, I.; Solovič, I.; Mokrý, J. Non-Tuberculous Mycobacteria: Classification, Diagnostics, and Therapy. Adv. Exp. Med. Biol. 2017, 944, 19–25. [Google Scholar] [CrossRef]
- Cowman, S.A.; Loebinger, M.R. Diagnosis of Nontuberculous Mycobacteria Lung Disease. Semin. Respir. Crit. Care Med. 2018, 39, 343–350. [Google Scholar] [CrossRef]
- Johnson, M.M.; Odell, J.A. Nontuberculous mycobacterial pulmonary infections. J. Thorac. Dis. 2014, 6, 210–220. [Google Scholar] [CrossRef]
- Chopra, K.; Sidiq, Z.; Hanif, M.; Dwivedi, K.K. Advances in the diagnosis of tuberculosis- Journey from smear microscopy to whole genome sequencing. Indian J. Tuberc. 2020, 67, S61–S68. [Google Scholar] [CrossRef]
- Somoskovi, A.; Salfinger, M. Nontuberculous Mycobacteria in Respiratory Infections: Advances in diagnosis and identification. Clin. Lab. Med. 2014, 34, 271–295. [Google Scholar] [CrossRef]
- Dohál, M.; Porvazník, I.; Pršo, K.; Rasmussen, E.M.; Solovič, I.; Mokrý, J. Whole-genome sequencing and Mycobacterium tuberculosis: Challenges in sample preparation and sequencing data analysis. Tuberculosis 2020, 123, 101946. [Google Scholar] [CrossRef]
- Yoon, J.-K.; Kim, T.S.; Kim, J.-I.; Yim, J.-J. Whole genome sequencing of Nontuberculous Mycobacterium (NTM) isolates from sputum specimens of co-habiting patients with NTM pulmonary disease and NTM isolates from their environment. BMC Genom. 2020, 21, 1–7. [Google Scholar] [CrossRef]
- Hirabayashi, R.; Nakagawa, A.; Takegawa, H.; Tomii, K. A case of pleural effusion caused by Mycobacterium fortuitum and Mycobacterium mageritense coinfection. BMC Infect. Dis. 2019, 19, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Walker, T.M.; Merker, M.; Knoblauch, A.; Helbling, P.; Schoch, O.D.; van der Werf, M.J.; Kranzer, K.; Fiebig, L.; Kröger, S.; Haas, W.; et al. A cluster of multidrug-resistant Mycobacterium tuberculosis among patients arriving in Europe from the Horn of Africa: A molecular epidemiological study. Lancet Infect. Dis. 2018, 18, 431–440. [Google Scholar] [CrossRef]
- Genestet, C.; Hodille, E.; Berland, J.-L.; Ginevra, C.; Bryant, J.E.; Ader, F.; Lina, G.; Dumitrescu, O. Whole-genome sequencing in drug susceptibility testing of Mycobacterium tuberculosis in routine practice in Lyon, France. Int. J. Antimicrob. Agents 2020, 55, 105912. [Google Scholar] [CrossRef]
- Mehta, M.; Marras, T.K. Impaired health-related quality of life in pulmonary nontuberculous mycobacterial disease. Respir. Med. 2011, 105, 1718–1725. [Google Scholar] [CrossRef]
- Nishiuchi, Y.; Iwamoto, T.; Maruyama, F. Infection Sources of a Common Non-tuberculous Mycobacterial Pathogen, Mycobacterium avium Complex. Front. Med. 2017, 4, 27. [Google Scholar] [CrossRef]
- Advani, J.; Verma, R.; Chatterjee, O.; Pachouri, P.K.; Upadhyay, P.; Singh, R.; Yadav, J.; Naaz, F.; Ravikumar, R.; Buggi, S.; et al. Whole Genome Sequencing of Mycobacterium tuberculosis Clinical Isolates from India Reveals Genetic Heterogeneity and Region-Specific Variations That Might Affect Drug Susceptibility. Front. Microbiol. 2019, 10, 309. [Google Scholar] [CrossRef]
- Tortoli, E. Microbiological Features and Clinical Relevance of New Species of the Genus Mycobacterium. Clin. Microbiol. Rev. 2014, 27, 727–752. [Google Scholar] [CrossRef]
- Quan, T.P.; Bawa, Z.; Foster, D.; Walker, T.; Elías, C.D.O.; Rathod, P.; Iqbal, Z.; Bradley, P.; Mowbray, J.; Walker, A.S.; et al. Evaluation of Whole-Genome Sequencing for Mycobacterial Species Identification and Drug Susceptibility Testing in a Clinical Setting: A Large-Scale Prospective Assessment of Performance against Line Probe Assays and Phenotyping. J. Clin. Microbiol. 2018, 56, e01480-17. [Google Scholar] [CrossRef]
- Hoshino, Y.; Suzuki, K. Differential diagnostic assays for discriminating mycobacteria, especially for nontuberculous mycobacteria: What does the future hold? Future Microbiol. 2015, 10, 205–216. [Google Scholar] [CrossRef]
- Morimoto, K.; Aono, A.; Murase, Y.; Sekizuka, T.; Kurashima, A.; Takaki, A.; Sasaki, Y.; Igarashi, Y.; Chikamatsu, K.; Goto, H.; et al. Prevention of aerosol isolation of nontuberculous Mycobacterium from the patient’s bathroom. ERJ Open Res. 2018, 4, 00150–02017. [Google Scholar] [CrossRef] [PubMed]
- Bryant, J.; Grogono, D.; Greaves, D.; Foweraker, J.; Roddick, I.; Inns, T.; Reacher, M.; Haworth, C.S.; Curran, M.D.; Harris, S.; et al. Whole-genome sequencing to identify transmission of Mycobacterium abscessus between patients with cystic fibrosis: A retrospective cohort study. Lancet 2013, 381, 1551–1560. [Google Scholar] [CrossRef]
- Fukushima, K.; Kitada, S.; Matsumoto, Y.; Komukai, S.; Kuge, T.; Kawasaki, T.; Matsuki, T.; Motooka, D.; Tsujino, K.; Miki, M.; et al. Serum GPL core antibody levels are associated with disease activity and treatment outcomes in Mycobacterium avium complex lung disease following first line antibiotic treatment. Respir. Med. 2021, 187, 106585. [Google Scholar] [CrossRef] [PubMed]
- Flohr, S.; Ramette, A.; Agyeman, P.K.A.; Duppenthaler, A.; Scherer, C.; Keller, P.M.; Aebi, C. Recurrent Mycobacterium chelonae Skin Infection Unmasked as Factitious Disorder Using Bacterial Whole Genome Sequence Analysis. Open Forum Infect. Dis. 2020, 7, ofaa506. [Google Scholar] [CrossRef]
- Lee, B.Y.; Kim, S.; Hong, Y.; Lee, S.-D.; Kim, W.S.; Kim, D.S.; Shim, T.S.; Jo, K.-W. Risk Factors for Recurrence after Successful Treatment of Mycobacterium avium Complex Lung Disease. Antimicrob. Agents Chemother. 2015, 59, 2972–2977. [Google Scholar] [CrossRef] [PubMed]
- Boyle, D.P.; Zembower, T.R.; Qi, C. Relapse versus Reinfection of Mycobacterium avium Complex Pulmonary Disease. Patient Characteristics and Macrolide Susceptibility. Ann. Am. Thorac. Soc. 2016, 13, 1956–1961. [Google Scholar] [CrossRef] [PubMed]
- Floto, R.A.; Olivier, K.N.; Saiman, L.; Daley, C.L.; Herrmann, J.-L.; Nick, J.A.; Noone, P.G.; Bilton, D.; Corris, P.; Gibson, R.L.; et al. US Cystic Fibrosis Foundation and European Cystic Fibrosis Society consensus recommendations for the management of non-tuberculous mycobacteria in individuals with cystic fibrosis. Thorax 2015, 71, i1–i22. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.D.; Greenberg, D.E.; Ehrmantraut, M.E.; Guide, S.V.; Ding, L.; Shea, Y.; Brown, M.R.; Chernick, M.; Steagall, W.K.; Glasgow, C.G.; et al. Pulmonary Nontuberculous Mycobacterial Disease: Prospective study of a distinct preexisting syndrome. Am. J. Respir. Crit. Care Med. 2008, 178, 1066–1074. [Google Scholar] [CrossRef]
- Lake, M.A.; Ambrose, L.R.; Lipman, M.C.I.; Lowe, D.M. “Why me, why now?” Using clinical immunology and epidemiology to explain who gets nontuberculous mycobacterial infection. BMC Med. 2016, 14, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Aiello, T.B.; Levy, C.; Zaccariotto, T.R.; Paschoal, I.A.; Pereira, M.C.; Da Silva, M.T.N.; Ribeiro, J.D.; Ribeiro, A.F.; Toro, A.D.C.; Mauch, R.M. Prevalence and clinical outcomes of nontuberculous mycobacteria in a Brazilian cystic fibrosis reference center. Pathog. Dis. 2018, 76. [Google Scholar] [CrossRef]
- Neulat-Ripoll, F.; Pasek, S.; Schenowitz, C.; Dossat, C.; Barbe, V.; Rottman, M.; Macheras, E.; Heym, B.; Herrmann, J.-L.; Daffé, M.; et al. Non Mycobacterial Virulence Genes in the Genome of the Emerging Pathogen Mycobacterium abscessus. PLoS ONE 2009, 4, e5660. [Google Scholar] [CrossRef]
- Tortoli, E.; Trovato, A.; Baldan, R.; Costa, D.; Simonetti, T.M.; Cirillo, D.M. Molecular typing of Mycobacterium abscessus isolated from cystic fibrosis patients. Int. J. Mycobacteriol. 2017, 6, 138–141. [Google Scholar] [CrossRef] [PubMed]
- Redondo, N.; Mok, S.; Montgomery, L.; Flanagan, P.; McNamara, E.; Smyth, E.G.; O’Sullivan, N.; Schaffer, K.; Rogers, T.R.; Fitzgibbon, M.M. Genomic Analysis of Mycobacterium abscessus Complex Isolates Collected in Ireland between 2006 and 2017. J. Clin. Microbiol. 2020, 58, e00295-20. [Google Scholar] [CrossRef] [PubMed]
- Gilljam, M.; Scherstén, H.; Silverborn, M.; Jönsson, B.; Hollsing, A.E. Lung transplantation in patients with cystic fibrosis and Mycobacterium abscessus infection. J. Cyst. Fibros. 2010, 9, 272–276. [Google Scholar] [CrossRef]
- Lobo, L.J.; Chang, L.C.; Esther, C.R.; Gilligan, P.H.; Tulu, Z.; Noone, P.G. Lung transplant outcomes in cystic fibrosis patients with pre-operative Mycobacterium abscessus respiratory infections. Clin. Transplant. 2013, 27, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Qvist, T.; Pressler, T.; Thomsen, V.; Skov, M.; Iversen, M.; Katzenstein, T. Nontuberculous Mycobacterial Disease Is Not a Contraindication to Lung Transplantation in Patients with Cystic Fibrosis: A Retrospective Analysis in a Danish Patient Population. Transplant. Proc. 2013, 45, 342–345. [Google Scholar] [CrossRef]
- Kavaliunaite, E.; Harris, K.A.; Aurora, P.; Dixon, G.; Shingadia, D.; Muthialu, N.; Spencer, H. Outcome according to subspecies following lung transplantation in cystic fibrosis pediatric patients infected with Mycobacterium abscessus. Transpl. Infect. Dis. 2020, 22, e13274. [Google Scholar] [CrossRef] [PubMed]
- Keating, M.R.; Daly, J.S. Nontuberculous mycobacterial infections in solid organ transplantation. Am. J. Transplant. 2013, 13, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Friedman, D.Z.P.; Cervera, C.; Halloran, K.; Tyrrell, G.; Doucette, K. Non-tuberculous mycobacteria in lung transplant recipients: Prevalence, risk factors, and impact on survival and chronic lung allograft dysfunction. Transpl. Infect. Dis. 2020, 22, e13229. [Google Scholar] [CrossRef]
- Huang, H.C.; Weigt, S.S.; Derhovanessian, A.; Palchevskiy, V.; Ardehali, A.; Saggar, R.; Saggar, R.; Kubak, B.; Gregson, A.; Ross, D.J.; et al. Non-tuberculous Mycobacterium infection after lung transplantation is associated with increased mortality. J. Heart Lung Transplant. 2011, 30, 790–798. [Google Scholar] [CrossRef] [PubMed]
- Daniel-Wayman, S.; Abate, G.; Barber, D.L.; Bermudez, L.E.; Coler, R.N.; Cynamon, M.H.; Daley, C.L.; Davidson, R.M.; Dick, T.; Floto, R.A.; et al. Advancing Translational Science for Pulmonary Nontuberculous Mycobacterial Infections. A Road Map for Research. Am. J. Respir. Crit. Care Med. 2019, 199, 947–951. [Google Scholar] [CrossRef]
- Shaw, L.P.; Doyle, R.M.; Kavaliunaite, E.; Spencer, H.; Balloux, F.; Dixon, G.; Harris, K. Children with Cystic Fibrosis Are Infected with Multiple Subpopulations of Mycobacterium abscessus With Different Antimicrobial Resistance Profiles. Clin. Infect. Dis. 2019, 69, 1678–1686. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, W.; Braun, J.D.; Burchell, J.; Witte, C.L.; Rideout, B.A. Whole-genome analysis of mycobacteria from birds at the San Diego Zoo. PLoS ONE 2017, 12, e0173464. [Google Scholar] [CrossRef]
- Khieu, V.; Ananta, P.; Kaewprasert, O.; Laohaviroj, M.; Namwat, W.; Faksri, K. Whole-Genome Sequencing Analysis to Identify Infection with Multiple Species of Nontuberculous Mycobacteria. Pathogens 2021, 10, 879. [Google Scholar] [CrossRef] [PubMed]
- Operario, D.J.; Pholwat, S.; Koeppel, A.F.; Prorock, A.; Bao, Y.; Sol-Church, K.; Scheurenbrand, M.; Poulter, M.; Turner, S.; Parikh, H.I.; et al. Mycobacterium avium Complex Diversity within Lung Disease, as Revealed by Whole-Genome Sequencing. Am. J. Respir. Crit. Care Med. 2019, 200, 393–396. [Google Scholar] [CrossRef]
- Greninger, A.; Langelier, C.; Cunningham, G.; Keh, C.; Melgar, M.; Chiu, C.; Miller, S. Two Rapidly Growing Mycobacterial Species Isolated from a Brain Abscess: First Whole-Genome Sequences of Mycobacterium immunogenum and Mycobacterium llatzerense. J. Clin. Microbiol. 2015, 53, 2374–2377. [Google Scholar] [CrossRef]
- Tortoli, E.; Kohl, T.A.; Brown-Elliott, B.A.; Trovato, A.; Leao, S.; Garcia, M.J.; Vasireddy, S.; Turenne, C.Y.; Griffith, D.E.; Philley, J.V.; et al. Emended description of Mycobacterium abscessus, Mycobacterium abscessus subsp. abscessus and Mycobacterium abscessus subsp. bolletii and designation of Mycobacterium abscessus subsp. massiliense comb. nov. Int. J. Syst. Evol. Microbiol. 2016, 66, 4471–4479. [Google Scholar] [CrossRef]
- Kuge, T.; Fukushima, K.; Matsumoto, Y.; Abe, Y.; Akiba, E.; Haduki, K.; Saito, H.; Nitta, T.; Kawano, A.; Kawasaki, T.; et al. Pulmonary disease caused by a newly identified mycobacterium: Mycolicibacterium toneyamachuris: A case report. BMC Infect. Dis. 2020, 20, 1–5. [Google Scholar] [CrossRef]
- Bouso, J.M.; Planet, P.J. Complete nontuberculous mycobacteria whole genomes using an optimized DNA extraction protocol for long-read sequencing. BMC Genom. 2019, 20, 793. [Google Scholar] [CrossRef]
- Bainomugisa, A.; Duarte, T.; Lavu, E.; Pandey, S.; Coulter, C.; Marais, B.J.; Coin, L. A complete high-quality MinION nanopore assembly of an extensively drug-resistant Mycobacterium tuberculosis Beijing lineage strain identifies novel variation in repetitive PE/PPE gene regions. Microb. Genom. 2018, 4, e000188. [Google Scholar] [CrossRef]
- Tyson, J.R.; O’Neil, N.; Jain, M.; Olsen, H.E.; Hieter, P.; Snutch, T.P. MinION-based long-read sequencing and assembly extends the Caenorhabditis elegans reference genome. Genome Res. 2018, 28, 266–274. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Kinjo, T.; Motooka, D.; Nabeya, D.; Jung, N.; Uechi, K.; Horii, T.; Iida, T.; Fujita, J.; Nakamura, S. Comprehensive subspecies identification of 175 nontuberculous mycobacteria species based on 7547 genomic profiles. Emerg. Microbes Infect. 2019, 8, 1043–1053. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, J.; Epperson, L.E.; Durbin, D.; Honda, J.R.; Strong, M. Intraspecies plasmid and genomic variation of Mycobacterium kubicae revealed by the complete genome sequences of two clinical isolates. Microb. Genom. 2021, 7, 000497. [Google Scholar] [CrossRef]
- Votintseva, A.A.; Bradley, P.; Pankhurst, L.; Elías, C.D.O.; Loose, M.; Nilgiriwala, K.; Chatterjee, A.; Smith, E.G.; Sanderson, N.; Walker, T.M.; et al. Same-Day Diagnostic and Surveillance Data for Tuberculosis via Whole-Genome Sequencing of Direct Respiratory Samples. J. Clin. Microbiol. 2017, 55, 1285–1298. [Google Scholar] [CrossRef] [PubMed]
- Johansen, M.D.; Herrmann, J.-L.; Kremer, L. Non-tuberculous mycobacteria and the rise of Mycobacterium abscessus. Nat. Rev. Microbiol. 2020, 18, 392–407. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, I.; Tiberi, S.; Farooqi, J.; Jabeen, K.; Yeboah-Manu, D.; Migliori, G.B.; Hasan, R. Non-tuberculous mycobacterial infections—A neglected and emerging problem. Int. J. Infect. Dis. 2020, 92, S46–S50. [Google Scholar] [CrossRef]
- Munita, J.M.; Arias, C.A. Mechanisms of Antibiotic Resistance. Microbiol. Spectrum 2021, 4. [Google Scholar] [CrossRef]
- Jarlier, V.; Nikaido, H. Mycobacterial cell wall: Structure and role in natural resistance to antibiotics. FEMS Microbiol. Lett. 1994, 123, 11–18. [Google Scholar] [CrossRef]
- Nessar, R.; Cambau, E.; Reyrat, J.M.; Murray, A.; Gicquel, B. Mycobacterium abscessus: A new antibiotic nightmare. J. Antimicrob. Chemother. 2012, 67, 810–818. [Google Scholar] [CrossRef]
- Richard, M.; Gutiérrez, A.V.; Viljoen, A.; Rodriguez-Rincon, D.; Roquet-Baneres, F.; Blaise, M.; Everall, I.; Parkhill, J.; Floto, R.A.; Kremer, L. Mutations in the MAB_2299c TetR Regulator Confer Cross-Resistance to Clofazimine and Bedaquiline in Mycobacterium abscessus. Antimicrob. Agents Chemother. 2019, 63, e01316-18. [Google Scholar] [CrossRef]
- Rodrigues, L.; Sampaio, D.; Couto, I.; Machado, D.; Kern, W.V.; Amaral, L.; Viveiros, M. The role of efflux pumps in macrolide resistance in Mycobacterium avium complex. Int. J. Antimicrob. Agents 2009, 34, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.C.; Vasireddy, R.; Vasireddy, S.; Philley, J.V.; Brown-Elliott, B.A.; Perry, B.J.; Griffith, D.E.; Benwill, J.L.; Cameron, A.; Wallace, R.J. Emergence of mmpT5 Variants during Bedaquiline Treatment of Mycobacterium intracellulare Lung Disease. J. Clin. Microbiol. 2017, 55, 574–584. [Google Scholar] [CrossRef]
- Nasiri, M.J.; Haeili, M.; Ghazi, M.; Goudarzi, H.; Pormohammad, A.; Fooladi, A.A.I.; Feizabadi, M.M. New Insights in to the Intrinsic and Acquired Drug Resistance Mechanisms in Mycobacteria. Front. Microbiol. 2017, 8, 681. [Google Scholar] [CrossRef]
- Dymova, M.; Alkhovik, O.; Evdokimova, L.; Cherednichenko, A.; Petrenko, T.; Batyrshina, Y. Whole genome-sequencing of non-tuberculous mycobacteria. Eur. Respir. J. 2016, 48, PA891. [Google Scholar] [CrossRef]
- Wetzstein, N.; Kohl, T.A.; Andres, S.; Schultze, T.G.; Geil, A.; Kim, E.; Biciusca, T.; Hügel, C.; Hogardt, M.; Lehn, A.; et al. Comparative analysis of phenotypic and genotypic antibiotic susceptibility patterns in Mycobacterium avium complex. Int. J. Infect. Dis. 2020, 93, 320–328. [Google Scholar] [CrossRef]
- Lipworth, S.; Hough, N.; Leach, L.; Morgan, M.; Jeffery, K.; Andersson, M.; Robinson, E.; Smith, E.G.; Crook, D.; Peto, T.; et al. Whole-Genome Sequencing for Predicting Clarithromycin Resistance in Mycobacterium abscessus. Antimicrob. Agents Chemother. 2019, 63, e01204-18. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, J.; Zhang, S.; Shi, W.; Zhang, W.; Zhu, M.; Zhang, Y. Novel Mutations Associated with Clofazimine Resistance in Mycobacterium abscessus. Antimicrob. Agents Chemother. 2018, 62, e00544-18. [Google Scholar] [CrossRef]
- Bronson, R.A.; Gupta, C.; Manson, A.L.; Nguyen, J.A.; Bahadirli-Talbott, A.; Parrish, N.M.; Earl, A.M.; Cohen, K.A. Global phylogenomic analyses of Mycobacterium abscessus provide context for non cystic fibrosis infections and the evolution of antibiotic resistance. Nat. Commun. 2021, 12, 1–10. [Google Scholar] [CrossRef]
- Yoshida, M.; Sano, S.; Chien, J.-Y.; Fukano, H.; Suzuki, M.; Asakura, T.; Morimoto, K.; Murase, Y.; Miyamoto, S.; Kurashima, A.; et al. A novel DNA chromatography method to discriminate Mycobacterium abscessus subspecies and macrolide susceptibility. EBioMedicine 2021, 64, 103187. [Google Scholar] [CrossRef]
- Realegeno, S.; Mirasol, R.; Garner, O.B.; Yang, S. Clinical Whole Genome Sequencing for Clarithromycin and Amikacin Resistance Prediction and Subspecies Identification of Mycobacterium abscessus. J. Mol. Diagn. 2021. [Google Scholar] [CrossRef]
- Dedrick, R.M.; Smith, B.E.; Garlena, R.A.; Russell, D.A.; Aull, H.G.; Mahalingam, V.; Divens, A.M.; Guerrero-Bustamante, C.A.; Zack, K.M.; Abad, L.; et al. Mycobacterium abscessus Strain Morphotype Determines Phage Susceptibility, the Repertoire of Therapeutically Useful Phages, and Phage Resistance. mBio 2021, 12, e03431-20. [Google Scholar] [CrossRef]
- Phillips, M.; Von Reyn, C.F. Nosocomial Infections Due to Nontuberculous Mycobacteria. Clin. Infect. Dis. 2001, 33, 1363–1374. [Google Scholar] [CrossRef]
- Wallace, R.J.; Brown, B.A.; Griffith, D.E. Nosocomial outbreaks/pseudo outbreaks caused by nontuberculous mycobacteria. Annu. Rev. Microbiol. 1998, 52, 453–490. [Google Scholar] [CrossRef]
- Shakoor, S.; Owais, M.; Hasan, R.; Irfan, S. Nosocomial and Healthcare-Associated NTM Infections and Their Control. In Nontuberculous Mycobacteria (NTM): Microbiological, Clinical and Geographical Distribution; Elsevier: Amsterdam, The Netherlands, 2019; pp. 177–190. ISBN 9780128146934. [Google Scholar]
- Jamieson, F.B.; Teatero, S.; Guthrie, J.L.; Neemuchwala, A.; Fittipaldi, N.; Mehaffy, C. Whole-Genome Sequencing of the Mycobacterium tuberculosis Manila Sublineage Results in Less Clustering and Better Resolution than Mycobacterial Interspersed Repetitive-Unit-Variable-Number Tandem-Repeat (MIRU-VNTR) Typing and Spoligotyping. J. Clin. Microbiol. 2014, 52, 3795–3798. [Google Scholar] [CrossRef]
- Trewby, H.; Wright, D.; Breadon, E.L.; Lycett, S.J.; Mallon, T.R.; McCormick, C.; Johnson, P.; Orton, R.J.; Allen, A.R.; Galbraith, J.; et al. Use of bacterial whole-genome sequencing to investigate local persistence and spread in bovine tuberculosis. Epidemics 2016, 14, 26–35. [Google Scholar] [CrossRef]
- Price-Carter, M.; Brauning, R.; De Lisle, G.W.; Livingstone, P.; Neill, M.; Sinclair, J.; Paterson, B.; Atkinson, G.; Knowles, G.; Crews, K.; et al. Whole Genome Sequencing for Determining the Source of Mycobacterium bovis Infections in Livestock Herds and Wildlife in New Zealand. Front. Vet. Sci. 2018, 5, 272. [Google Scholar] [CrossRef] [PubMed]
- Black, A.T.; Hamblion, E.L.; Buttivant, H.; Anderson, S.R.; Stone, M.; Casali, N.; Drobniewski, F.; Nwoguh, F.; Marshall, B.G.; Booth, L. Tracking and responding to an outbreak of tuberculosis using MIRU-VNTR genotyping and whole genome sequencing as epidemiological tools. J. Public Health 2018, 40, e66–e73. [Google Scholar] [CrossRef]
- Ahlstrom, C.; Barkema, H.W.; Stevenson, K.; Zadoks, R.N.; Biek, R.; Kao, R.; Trewby, H.; Haupstein, D.; Kelton, D.F.; Fecteau, G.; et al. Limitations of variable number of tandem repeat typing identified through whole genome sequencing of Mycobacterium avium subsp. paratuberculosis on a national and herd level. BMC Genom. 2015, 16, 161. [Google Scholar] [CrossRef]
- Harris, K.A.; Underwood, A.; Kenna, D.T.D.; Brooks, A.; Kavaliunaite, E.; Kapatai, G.; Tewolde, R.; Aurora, P.; Dixon, G. Whole-genome sequencing and epidemiological analysis do not provide evidence for cross-transmission of Mycobacterium abscessus in a cohort of pediatric cystic fibrosis patients. Clin. Infect. Dis. 2015, 60, 1007–1016. [Google Scholar] [CrossRef]
- Ratnatunga, C.; Lutzky, V.P.; Kupz, A.; Doolan, D.L.; Reid, D.W.; Field, M.; Bell, S.; Thomson, R.M.; Miles, J.J. The Rise of Non-Tuberculosis Mycobacterial Lung Disease. Front. Immunol. 2020, 11, 303. [Google Scholar] [CrossRef]
- Aitken, M.L.; Limaye, A.; Pottinger, P.; Whimbey, E.; Goss, C.; Tonelli, M.R.; Cangelosi, G.A.; Dirac, M.A.; Olivier, K.N.; Brown-Elliott, B.A.; et al. Respiratory Outbreak of Mycobacterium abscessus Subspecies massiliensein a Lung Transplant and Cystic Fibrosis Center. Am. J. Respir. Crit. Care Med. 2012, 185, 231–232. [Google Scholar] [CrossRef]
- Tortoli, E.; Kohl, T.A.; Trovato, A.; Baldan, R.; Campana, S.; Cariani, L.; Colombo, C.; Costa, D.; Cristadoro, S.; Di Serio, M.C.; et al. Mycobacterium abscessus in patients with cystic fibrosis: Low impact of inter-human transmission in Italy. Eur. Respir. J. 2017, 50, 1602525. [Google Scholar] [CrossRef] [PubMed]
- Hasan, N.A.; Davidson, R.M.; Epperson, L.E.; Kammlade, S.M.; Rodger, R.R.; Levin, A.R.; Sherwood, A.; Sagel, S.D.; Martiniano, S.L.; Daley, C.L.; et al. Population genomics of nontuberculous mycobacteria recovered from United States cystic fibrosis patients. bioRxiv 2019, 663559. [Google Scholar] [CrossRef]
- Yan, J.; Kevat, A.; Martinez, E.; Teese, N.; Johnson, K.; Ranganathan, S.; Harrison, J.; Massie, J.; Daley, A. Investigating transmission of Mycobacterium abscessus amongst children in an Australian cystic fibrosis centre. J. Cyst. Fibros. 2020, 19, 219–224. [Google Scholar] [CrossRef]
- Davidson, R.M.; Benoit, J.B.; Kammlade, S.M.; Hasan, N.A.; Epperson, L.E.; Smith, T.; Vasireddy, S.; Brown-Elliott, B.A.; Nick, J.A.; Olivier, K.N.; et al. Genomic characterization of sporadic isolates of the dominant clone of Mycobacterium abscessus subspecies massiliense. Sci. Rep. 2021, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bryant, J.M.; Grogono, D.M.; Rodriguez-Rincon, D.; Everall, I.; Brown, K.P.; Moreno, P.; Verma, D.; Hill, E.; Drijkoningen, J.; Gilligan, P.; et al. Emergence and spread of a human-transmissible multidrug-resistant nontuberculous mycobacterium. Science 2016, 354, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Ravnholt, C.; Kolpen, M.; Skov, M.; Moser, C.; Katzenstein, T.L.; Pressler, T.; Høiby, N.; Qvist, T. The importance of early diagnosis of Mycobacterium abscessus complex in patients with cystic fibrosis. APMIS 2018, 126, 885–891. [Google Scholar] [CrossRef] [PubMed]
- Davidson, R.M. A Closer Look at the Genomic Variation of Geographically Diverse Mycobacterium abscessus Clones That Cause Human Infection and Disease. Front. Microbiol. 2018, 9, 2988. [Google Scholar] [CrossRef]
- Lipworth, S.; Hough, N.; Weston, N.; Muller-Pebody, B.; Phin, N.; Myers, R.; Chapman, S.; Flight, W.; Alexander, E.; Smith, E.G.; et al. Epidemiology of Mycobacterium abscessus in England: An observational study. Lancet Microbe 2021, 2, e498–e507. [Google Scholar] [CrossRef]
- Saiman, L.; Siegel, J.D.; Lipuma, J.J.; Brown, R.; Bryson, E.A.; Chambers, M.J.; Downer, V.S.; Fliege, J.; Hazle, L.A.; Jain, M.; et al. Infection Prevention and Control Guideline for Cystic Fibrosis: 2013 Update. Infect. Control Hosp. Epidemiol. 2014, 35, s1–s67. [Google Scholar] [CrossRef]
- Haller, S.; Höller, C.; Jacobshagen, A.; Hamouda, O.; Abu Sin, M.; Monnet, D.L.; Plachouras, D.; Eckmanns, T. Contamination during production of heater-cooler units by Mycobacterium chimaera potential cause for invasive cardiovascular infections: Results of an outbreak investigation in Germany, April 2015 to February 2016. Eurosurveillance 2016, 21, 1–6. [Google Scholar] [CrossRef]
- Schreiber, P.W.; Kohl, T.A.; Kuster, S.P.; Niemann, S.; Sax, H. The global outbreak of Mycobacterium chimaera infections in cardiac surgery—A systematic review of whole-genome sequencing studies and joint analysis. Clin. Microbiol. Infect. 2021. [Google Scholar] [CrossRef] [PubMed]
- van Ingen, J.; Kohl, T.A.; Kranzer, K.; Hasse, B.; Keller, P.M.; Szafrańska, A.K.; Hillemann, D.; Chand, M.; Schreiber, P.W.; Sommerstein, R.; et al. Global outbreak of severe Mycobacterium chimaera disease after cardiac surgery: A molecular epidemiological study. Lancet Infect. Dis. 2017, 17, 1033–1041. [Google Scholar] [CrossRef]
- Svensson, E.; Jensen, E.T.; Rasmussen, E.M.; Folkvardsen, D.B.; Norman, A.; Lillebaek, T. Mycobacterium chimaera in Heater–Cooler Units in Denmark Related to Isolates from the United States and United Kingdom. Emerg. Infect. Dis. 2017, 23, 507–509. [Google Scholar] [CrossRef]
- Diekema, D.J. Mycobacterium chimaera infections after cardiovascular surgery: Lessons from a global outbreak. Trans. Am. Clin. Climatol. Assoc. 2019, 130, 136–144. [Google Scholar]
- Ghodousi, A.; Borroni, E.; Peracchi, M.; Palù, G.; Fallico, L.; Rassu, M.; Manfrin, V.; Mantegani, P.; Monzillo, V.; Manganelli, R.; et al. Genomic analysis of cardiac surgery-associated Mycobacterium chimaera infections in Italy. PLoS ONE 2020, 15, e0239273. [Google Scholar] [CrossRef]
- Lecorche, E.; Daniau, C.; La, K.; Mougari, F.; Benmansour, H.; Kumanski, S.; Robert, J.; Fournier, S.; Lebreton, G.; Carbonne, A.; et al. Mycobacterium chimaera Genomics with Regard to Epidemiological and Clinical Investigations Conducted for an Open Chest Postsurgical Mycobacterium chimaera Infection Outbreak. Open Forum Infect. Dis. 2021, 8, ofab192. [Google Scholar] [CrossRef]
- Götting, T.; Klassen, S.; Jonas, D.; Benk, C.; Serr, A.; Wagner, D.; Ebner, W. Heater–cooler units: Contamination of crucial devices in cardiothoracic surgery. J. Hosp. Infect. 2016, 93, 223–228. [Google Scholar] [CrossRef]
- Kohler, P.; Kuster, S.; Bloemberg, G.; Schulthess, B.; Frank, M.; Tanner, F.C.; Rössle, M.; Böni, C.; Falk, V.; Wilhelm, M.J.; et al. Healthcare-associated prosthetic heart valve, aortic vascular graft, and disseminated Mycobacterium chimaera infections subsequent to open heart surgery. Eur. Heart J. 2015, 36, 2745–2753. [Google Scholar] [CrossRef] [PubMed]
- Health Departments | NTM | HAI | CDC. Available online: https://www.cdc.gov/hai/organisms/ntm/health-departments.html (accessed on 6 September 2021).
- Chand, M.; Lamagni, T.; Kranzer, K.; Hedge, J.; Moore, G.; Parks, S.; Collins, S.; Elías, C.D.O.; Ahmed, N.; Brown, T.; et al. Insidious Risk of Severe Mycobacterium chimaera Infection in Cardiac Surgery Patients. Clin. Infect. Dis. 2017, 64, 335–342. [Google Scholar] [CrossRef]
- Sommerstein, R.; Schreiber, P.W.; Diekema, D.; Edmond, M.; Hasse, B.; Marschall, J.; Sax, H. Mycobacterium chimaera Outbreak Associated with Heater-Cooler Devices: Piecing the Puzzle Together. Infect. Control. Hosp. Epidemiol. 2017, 38, 103–108. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Decalonne, M.; Lecorche, E.; Hau, E.; Petiteau, A.; Moreau, C.; Milan, O.; Lanotte, P.; Mereghetti, L.; Cambau, E.; Van Der Mee-Marquet, N. Cause Analysis of an Infection in Facelift Surgery Due to Mycobacterium chelonae. Front. Med. 2019, 6, 243. [Google Scholar] [CrossRef]
- Hammond, S.E.; Al-Bayati, A.; Joumblat, N.; Salgado, C.J. Mycobacterium chelonae Infection of the Buttocks Secondary to Lipofilling: A Case Report and Review of the Literature. Aesthetic Plast. Surg. 2017, 41, 1150–1154. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, H.; Viana-Niero, C.; Nogueira, C.L.; Bispo, P.; Pinto, F.; Uzam, C.D.P.P.; Matsumoto, C.K.; Machado, A.M.O.; Leao, S.; Hofling-Lima, A.L.; et al. Identification of the Infection Source of an Outbreak of Mycobacterium chelonae Keratitis After Laser in Situ Keratomileusis. Cornea 2018, 37, 116–122. [Google Scholar] [CrossRef]
- Labuda, S.M.; Garner, K.; Cima, M.; Moulton-Meissner, H.; Halpin, A.L.; Charles-Toney, N.; Yu, P.; Bolton, E.; Pierce, R.; Crist, M.B.; et al. Bloodstream Infections with a Novel Nontuberculous Mycobacterium Involving 52 Outpatient Oncology Clinic Patients―Arkansas, 2018. Clin. Infect. Dis. 2020, 71, e178–e185. [Google Scholar] [CrossRef]
- Philley, J.V.; Kannan, A.; Griffith, D.E.; Devine, M.S.; Benwill, J.L.; WallaceJr, R.J.; Brown-Elliott, B.A.; Thakkar, F.; Taskar, V.; Fox, J.G.; et al. Exosome secretome and mediated signaling in breast cancer patients with nontuberculous mycobacterial disease. Oncotarget 2017, 8, 18070–18081. [Google Scholar] [CrossRef]
- Inkster, T.; Peters, C.; Seagar, A.; Holden, M.; Laurenson, I. Investigation of two cases of Mycobacterium chelonae infection in haemato-oncology patients using whole-genome sequencing and a potential link to the hospital water supply. J. Hosp. Infect. 2021, 114, 111–116. [Google Scholar] [CrossRef]
- Yoshida, S.; Iwamoto, T.; Kobayashi, T.; Nomoto, R.; Inoue, Y.; Tsuyuguchi, K.; Suzuki, K. Two New Cases of Pulmonary Infection by Mycobacterium shigaense, Japan. Emerg. Infect. Dis. 2020, 26, 2728–2732. [Google Scholar] [CrossRef]
- Brown-Elliott, B.A.; Woods, G.L. Antimycobacterial Susceptibility Testing of Nontuberculous Mycobacteria. J. Clin. Microbiol. 2019, 57, 834–853. [Google Scholar] [CrossRef]
- de Moura, V.C.N.; Verma, D.; Everall, I.; Brown, K.P.; Belardinelli, J.M.; Shanley, C.; Stapleton, M.; Parkhill, J.; Floto, R.A.; Ordway, D.J.; et al. Increased Virulence of Outer Membrane Porin Mutants of Mycobacterium abscessus. Front. Microbiol. 2021, 12, 1976. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Sequencing Platform | Read-Length (bp) | Output | Accuracy | Advantage |
|---|---|---|---|---|
| Illumina MiSeq | 25–300 | >50 Gb | >99% | Higher read quality A lower amount of input DNA Lower cost per sample |
| Ion PGMTM | <400 | 30–2 Gb | <98% | Higher read quality Cost-effective |
| MinION | short to ultra-long (>4 Mb) reads | 300–15 Gb | >95% | Resolving repetitive regions, G + C rich regions, and indels Portable |
| PacBio RS II (Single molecule real-time) | >20,000 | 1–10 Gb | >99.999% | Higher read quality Resolving repetitive regions, G + C rich regions, and indels |
| Accurate diagnostics of NTM infection within a clinically relevant time frame |
| Unrestricted classification of NTM subspecies compared to other genotyping methods |
| Characterization of the resistance profile and identification of novel gene mutation encoding resistance |
| Setting the appropriate combination of antibiotics and increasing the effectiveness of the treatment regimen |
| Monitoring of transmission dynamics and clustering of NTM in hospital settings to prevent nosocomial infections |
| Description of new genes affecting the pathogenesis of NTM infections |
| Development of new drugs |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dohál, M.; Porvazník, I.; Solovič, I.; Mokrý, J. Whole Genome Sequencing in the Management of Non-Tuberculous Mycobacterial Infections. Microorganisms 2021, 9, 2237. https://doi.org/10.3390/microorganisms9112237
Dohál M, Porvazník I, Solovič I, Mokrý J. Whole Genome Sequencing in the Management of Non-Tuberculous Mycobacterial Infections. Microorganisms. 2021; 9(11):2237. https://doi.org/10.3390/microorganisms9112237
Chicago/Turabian StyleDohál, Matúš, Igor Porvazník, Ivan Solovič, and Juraj Mokrý. 2021. "Whole Genome Sequencing in the Management of Non-Tuberculous Mycobacterial Infections" Microorganisms 9, no. 11: 2237. https://doi.org/10.3390/microorganisms9112237
APA StyleDohál, M., Porvazník, I., Solovič, I., & Mokrý, J. (2021). Whole Genome Sequencing in the Management of Non-Tuberculous Mycobacterial Infections. Microorganisms, 9(11), 2237. https://doi.org/10.3390/microorganisms9112237