
SARS-CoV-2 Evolution among Oncological Population: In-Depth Virological Analysis of a Clinical Cohort

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Setting, Study Population and Design
2.2. Real-Time Reverse-Transcription Polymerase Chain Reaction (RT-PCR) Assay and Unbiased High-Throughput Sequencing (HTS) Analysis
3. Results
3.1. Patients’ Demographics and Clinical Presentation
3.2. RT-PCR Assay Screening
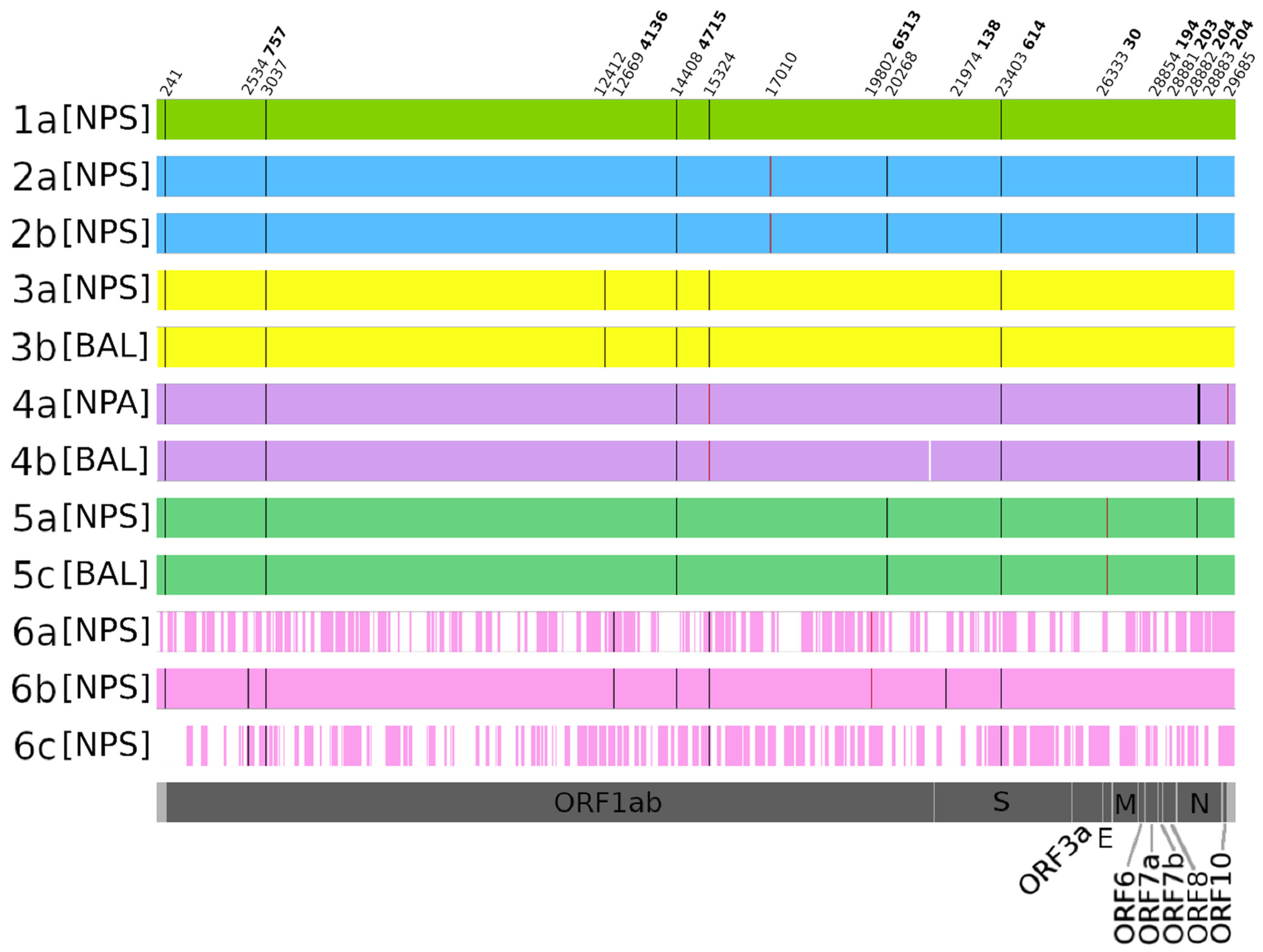
3.3. HTS Investigations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- CDC Guidance for Discontinuation of Transmission-Based Precautions and Disposition of Patients with COVID-19 in Healthcare Settings. Available online: https://www.cdc.gov/coronavirus/2019-ncov/hcp/disposition-hospitalized-patients.html (accessed on 9 August 2021).
- Choi, B.; Choudhary, M.C.; Regan, J.; Sparks, J.A.; Padera, R.F.; Qiu, X.; Solomon, I.H.; Kuo, H.H.; Boucau, J.; Bowman, K.; et al. Persistence and Evolution of SARS-CoV-2 in an Immunocompromised Host. N. Engl. J. Med. 2020, 383, 2291–2293. [Google Scholar] [CrossRef]
- Kemp, S.A.; Collier, D.A.; Datir, R.P.; Ferreira, I.A.; Gayed, S.; Jahun, A.; Hosmillo, M.; Rees-Spear, C.; Mlcochova, P.; Lumb, I.U.; et al. SARS-CoV-2 evolution during treatment of chronic infection. Nature 2021, 592, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Avanzato, V.A.; Matson, M.J.; Seifert, S.N.; Pryce, R.; Williamson, B.N.; Anzick, S.L.; Barbian, K.; Judson, S.D.; Fischer, E.R.; Martens, C.; et al. Case Study: Prolonged Infectious SARS-CoV-2 Shedding from an Asymptomatic Immunocompromised Individual with Cancer. Cell 2020, 183, 1901–1912. [Google Scholar] [CrossRef] [PubMed]
- Aydillo, T.; Gonzalez-Reiche, A.S.; Aslam, S.; van de Guchte, A.; Khan, Z.; Obla, A.; Dutta, J.; van Bakel, H.; Aberg, J.; García-Sastre, A.; et al. Shedding of Viable SARS-CoV-2 after Immunosuppressive Therapy for Cancer. N. Engl. J. Med. 2020, 383, 2586–2588. [Google Scholar] [CrossRef]
- Caillard, S.; Benotmane, I.; Vargas, G.G.; Perrin, P.; Fafi-Kremer, S. SARS-CoV-2 viral dynamics in immunocompromised patients. Am. J. Transpl. 2021, 21, 1667–1669. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.Y.W.; Cazier, J.-B.; Angelis, V.; Arnold, R.; Bisht, V.; Campton, N.A.; Chackathayil, J.; Cheng, V.W.; Curley, H.M.; Fittall, M.W.T.; et al. COVID-19 mortality in patients with cancer on chemotherapy or other anticancer treatments: A prospective cohort study. Lancet 2020, 395, 1919–1926. [Google Scholar] [CrossRef]
- Addeo, A.; Friedlaender, A. Cancer and COVID-19: Unmasking their ties. Cancer Treat. Rev. 2020, 88, 102041. [Google Scholar] [CrossRef]
- Gosain, R.; Abdou, Y.; Singh, A.; Rana, N.; Puzanov, I.; Ernstoff, M.S. COVID-19 and Cancer: A Comprehensive Review. Curr. Oncol. Rep. 2020, 22, 53. [Google Scholar] [CrossRef]
- Kaiser, L.; Vu, D.L.; Eberhardt, C.S. Paradoxes and uncertainties of immunomodulatory treatments in the fight against COVID-19. Clin. Infect. Dis. 2021, 72, e1144–e1145. [Google Scholar] [CrossRef]
- Zapor, M. Persistent Detection and Infectious Potential of SARS-CoV-2 Virus in Clinical Specimens from COVID-19 Patients. Viruses 2020, 12, 1384. [Google Scholar] [CrossRef]
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.; Bleicker, T.; Brünink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro. Surveill. 2020, 25, 2000045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petty, T.J.; Cordey, S.; Padioleau, I.; Docquier, M.; Turin, L.; Preynat-Seauve, O.; Zdobnov, E.M.; Kaiser, L. Comprehensive Human Virus Screening Using High-Throughput Sequencing with a User-Friendly Representation of Bioinformatics Analysis: A Pilot Study. J. Clin. Microbiol. 2014, 52, 3351–3361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Zheng, X.-S.; Shen, X.-R.; Si, H.-R.; Wang, X.; Wang, Q.; Li, B.; Zhang, W.; Zhu, Y.; Jiang, R.-D.; et al. Prolonged shedding of severe acute respiratory syndrome coronavirus 2 in patients with COVID-19. Emerg. Microbes Infect. 2020, 9, 2571–2577. [Google Scholar] [CrossRef] [PubMed]
- Tepasse, P.R.; Hafezi, W.; Lutz, M.; Kühn, J.; Wilms, C.; Wiewrodt, R.; Sackarnd, J.; Keller, M.; Schmidt, H.H.; Vollenberg, R. Persisting SARS-CoV-2 viraemia after rituximab therapy: Two cases with fatal outcome and a review of the literature. Br. J. Haematol. 2020, 190, 185–188. [Google Scholar] [CrossRef]
- Sepulcri, C.; Dentone, C.; Mikulska, M.; Bruzzone, B.; Lai, A.; Fenoglio, D.; Bozzano, F.; Bergna, A.; Parodi, A.; Altosole, T.; et al. The longest persistence of viable SARS-CoV-2 with recurrence of viremia and relapsing symptomatic COVID-19 in an immunocompromised patient—A case study. Open Forum Infect. Dis. 2021, ofab217. [Google Scholar] [CrossRef]
- Benotmane, I.; Gautier-Vargas, G.; Wendling, M.; Perrin, P.; Velay, A.; Bassand, X.; Bedo, D.; Baldacini, C.; Sagnard, M.; Bozman, D.; et al. In-depth virological assessment of kidney transplant recipients with COVID-19. Arab. Archaeol. Epigr. 2020, 20, 3162–3172. [Google Scholar] [CrossRef]
- Siqueira, J.D.; Goes, L.R.; Alves, B.M.; Carvalho, P.S.D.; Cicala, C.; Arthos, J.; Viola, J.P.; de Melo, A.C.; Soares, M.A. SARS-CoV-2 genomic analyses in cancer patients reveal elevated intrahost genetic diversity. Virus Evol. 2021, 7, veab013. [Google Scholar] [CrossRef]
- Rueca, M.; Bartolini, B.; Gruber, C.; Piralla, A.; Baldanti, F.; Giombini, E.; Messina, F.; Marchioni, L.; Ippolito, G.; Di Caro, A.; et al. Compartmentalized Replication of SARS-CoV-2 in Upper vs. Lower Respiratory Tract Assessed by Whole Genome Quasispecies Analysis. Microorganisms 2020, 8, 1302. [Google Scholar] [CrossRef]
- Moustafa, A.M.; Planet, P.J. Jumping a Moving Train: SARS-CoV-2 Evolution in Real Time. J. Pediatric Infect. Dis. Soc. 2021. [Google Scholar] [CrossRef]
- Sun, F.; Wang, X.; Tan, S.; Dan, Y.; Lu, Y.; Zhang, J.; Xu, J.; Tan, Z.; Xiang, X.; Zhou, Y.; et al. SARS-CoV-2 Quasispecies Provides an Advantage Mutation Pool for the Epidemic Variants. Microbiol. Spectr. 2021, 9, e0026121. [Google Scholar] [CrossRef]
- Pérez-Lago, L.; Aldámiz-Echevarría, T.; García-Martínez, R.; Pérez-Latorre, L.; Herranz, M.; Sola-Campoy, P.J.; Suárez-González, J.; Martínez-Laperche, C.; Comas, I.; González-Candelas, F.; et al. Different Within-Host Viral Evolution Dynamics in Severely Immunosuppressed Cases with Persistent SARS-CoV-2. Biomedicines 2021, 9, 808. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Nb of patients with ≥1 NPS | 16 |
| Median nb of NPS (range) | 3 (2–9) |
| Nb of ≥1 positive NPS (%) | 5 (31.2) |
| Median positivity duration, d (range) | 28 (15–114) |
| Nb of patients with BAL | 4 |
| Median delay from NPS, d (range) | 13 (7–22) |
| Nb positive (%) | 4 (100) |
| Median CT value (range) | 24 (19.8–25) |
| Nb of patients with viremia performed | 21 |
| Median delay from NPS, d (range) | 9 (0–51) |
| Nb positive (%) | 8 (38.1) |
| Median delay from NPS of positive samples, d (range) | 8 (0–25) |
| Median CT value (range) | 35.2 (32.4–39.3) |
| ID Patients | Sample | Sample Date | CT Value | Delay from First Sample (d) | Type of Investigation |
|---|---|---|---|---|---|
| 1a | NPS | 29.03.2020 | 22.2 | intra-host temporal variability | |
| 1b | NPA | 19.04.2020 | 26.22 | 21 | |
| 2a | NPS | 27.03.2020 | 17 | intra-host temporal variability | |
| 2b | NPS | 15.04.2020 | 17.7 | 19 | |
| 2c | NPS | 13.05.2020 | 31.7 | 47 | |
| 3a | NPS | 13.03.2020 | 12.6 | intra-host compartmental variability | |
| 3b | BAL | 26.03.2020 | 19.8 | 13 | |
| 4a | NPA | 01.04.2020 | 16.9 | intra-host compartmental variability | |
| 4b | BAL | 08.04.2020 | 23 | 7 | |
| 5a | NPS | 31.03.2020 | 17 | intra-host compartmental variability | |
| 5b | plasma | 19.04.2020 | 30.6 | 19 | |
| 5c | BAL | 22.04.2020 | 25 | 22 | |
| 5d | plasma | 27.04.2020 | 30.5 | 27 | |
| 5e | BAL | 03.05.2020 | 31 | 33 | |
| 6a | NPS | 02.04.2020 | 26.3 | intra-host temporal variability | |
| 6b | NPS | 12.04.2020 | 20.7 | 10 | |
| 6c | NPS | 24.04.2020 | 27 | 22 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laubscher, F.; Cordey, S.; Friedlaender, A.; Schweblin, C.; Noetzlin, S.; Simand, P.-F.; Bordry, N.; De Sousa, F.; Pigny, F.; Baggio, S.; et al. SARS-CoV-2 Evolution among Oncological Population: In-Depth Virological Analysis of a Clinical Cohort. Microorganisms 2021, 9, 2145. https://doi.org/10.3390/microorganisms9102145
Laubscher F, Cordey S, Friedlaender A, Schweblin C, Noetzlin S, Simand P-F, Bordry N, De Sousa F, Pigny F, Baggio S, et al. SARS-CoV-2 Evolution among Oncological Population: In-Depth Virological Analysis of a Clinical Cohort. Microorganisms. 2021; 9(10):2145. https://doi.org/10.3390/microorganisms9102145
Chicago/Turabian StyleLaubscher, Florian, Samuel Cordey, Alex Friedlaender, Cecilia Schweblin, Sarah Noetzlin, Pierre-François Simand, Natacha Bordry, Filipe De Sousa, Fiona Pigny, Stephanie Baggio, and et al. 2021. "SARS-CoV-2 Evolution among Oncological Population: In-Depth Virological Analysis of a Clinical Cohort" Microorganisms 9, no. 10: 2145. https://doi.org/10.3390/microorganisms9102145
APA StyleLaubscher, F., Cordey, S., Friedlaender, A., Schweblin, C., Noetzlin, S., Simand, P.-F., Bordry, N., De Sousa, F., Pigny, F., Baggio, S., Getaz, L., Dietrich, P.-Y., Kaiser, L., & Vu, D.-L. (2021). SARS-CoV-2 Evolution among Oncological Population: In-Depth Virological Analysis of a Clinical Cohort. Microorganisms, 9(10), 2145. https://doi.org/10.3390/microorganisms9102145