
Cross-Sectional Analysis of the Microbiota of Human Gut and Its Direct Environment in a Household Cohort with High Background of Antibiotic Use

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Community Setting, Study Design, and Sample Collection
2.2. Methods
2.2.1. DNA Extraction and 16S rRNA Amplicon Sequencing
2.2.2. Bioinformatics Analysis
2.2.3. Antimicrobial Resistance Genes
2.2.4. Statistical Tests
3. Results
3.1. Sample Types and Their Microbiome Sequence Information
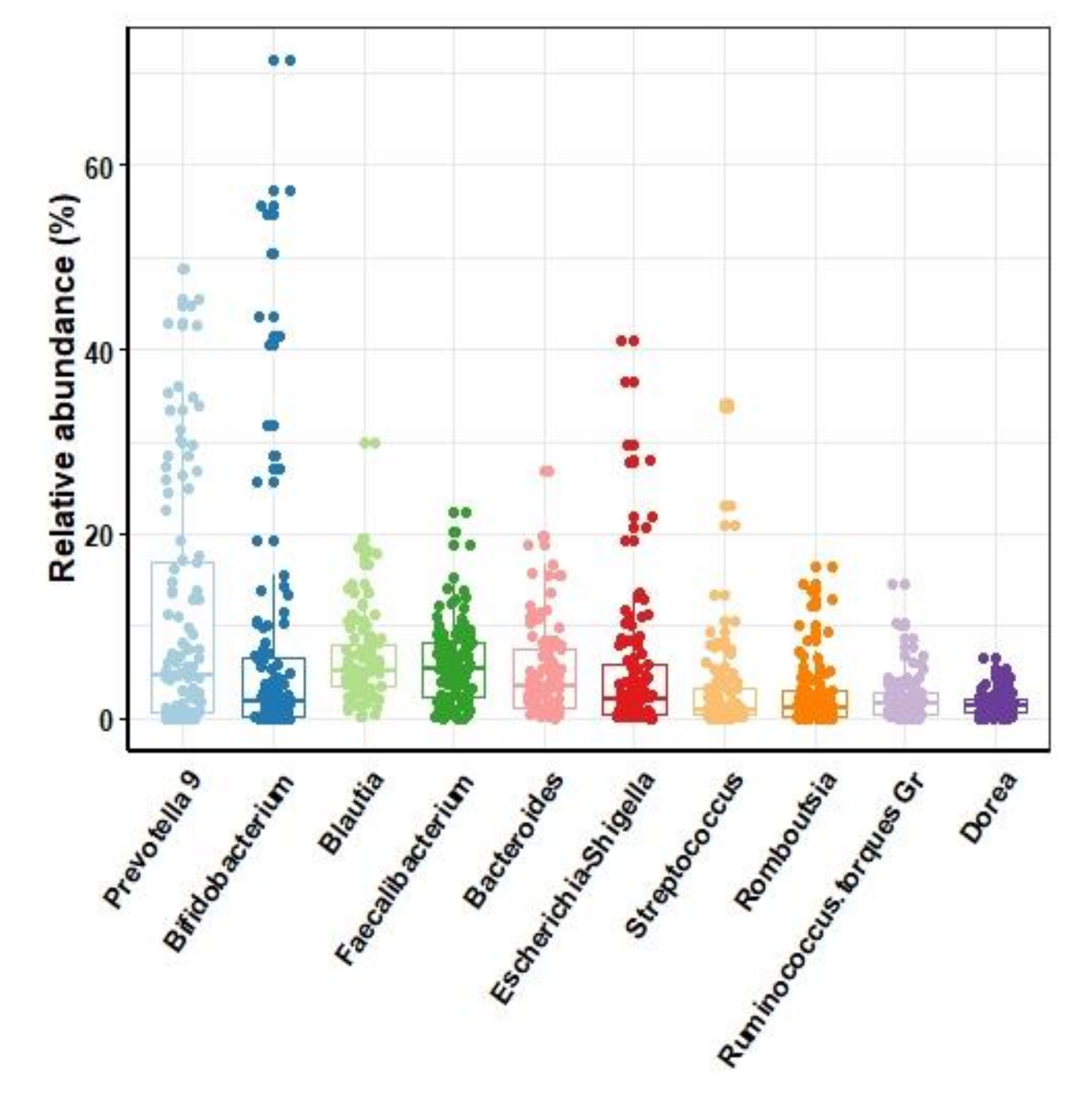
3.2. Microbiota of Human Gut in the Study Cohort
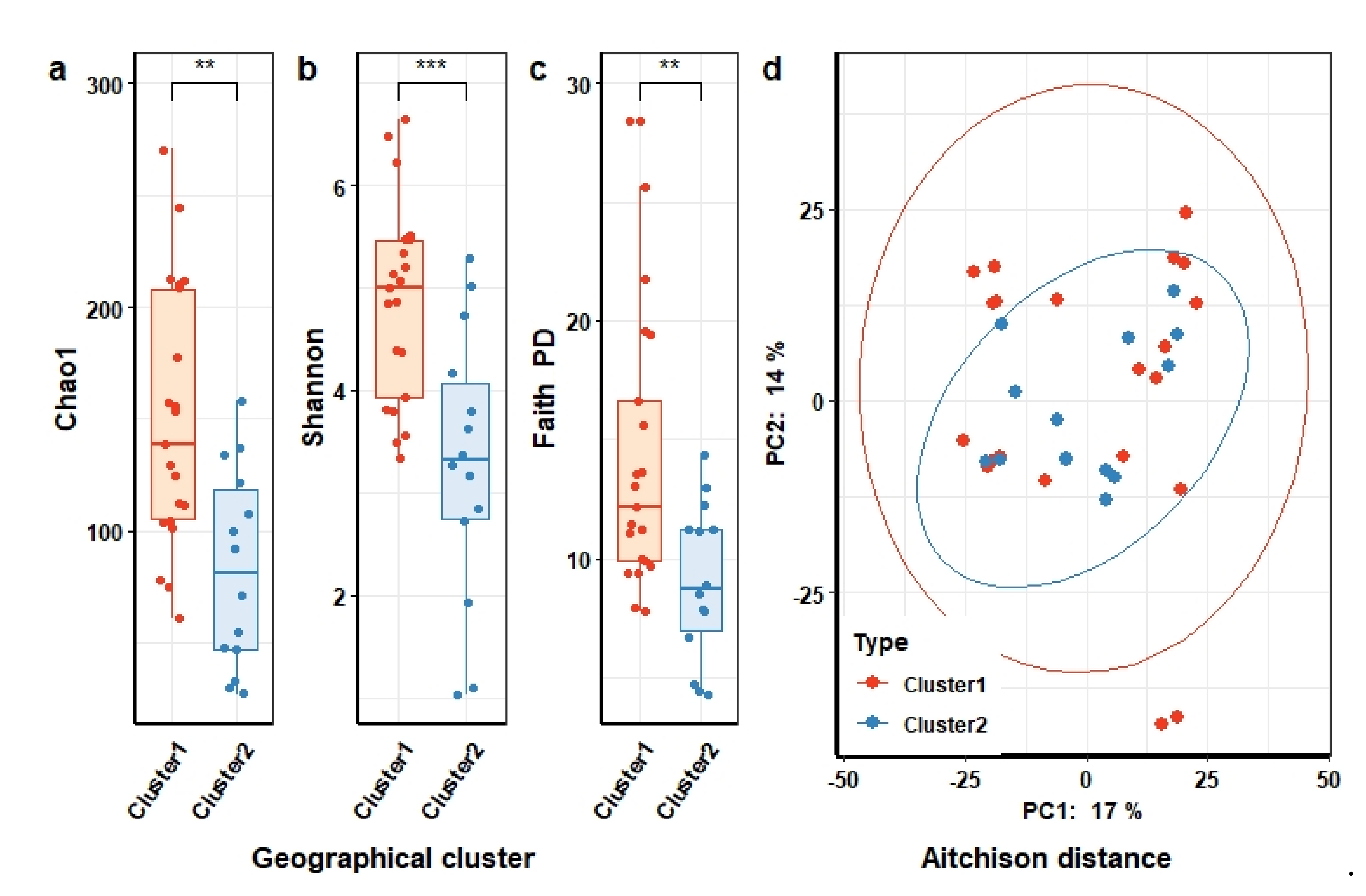
3.2.1. Composition of Gut Microbiota of Individuals in the Context of Demographical Variables of Study Cohort
3.2.2. Composition of Gut Microbiome of Healthy People in the Context of Their Antibiotic Use History
3.3. Microbiota of Nonhuman Samples
3.3.1. The Microbial Composition of Domestic Animals
3.3.2. The Microbial Composition of Water
3.3.3. The Microbial Composition of Processed Food
3.4. Human versus Nonhuman Microbiota
3.4.1. Overall Composition and Diversity of the Human Gut Microbiota in Relation to Microbial Community of Domestic Animal Gut, Water, and Processed Food
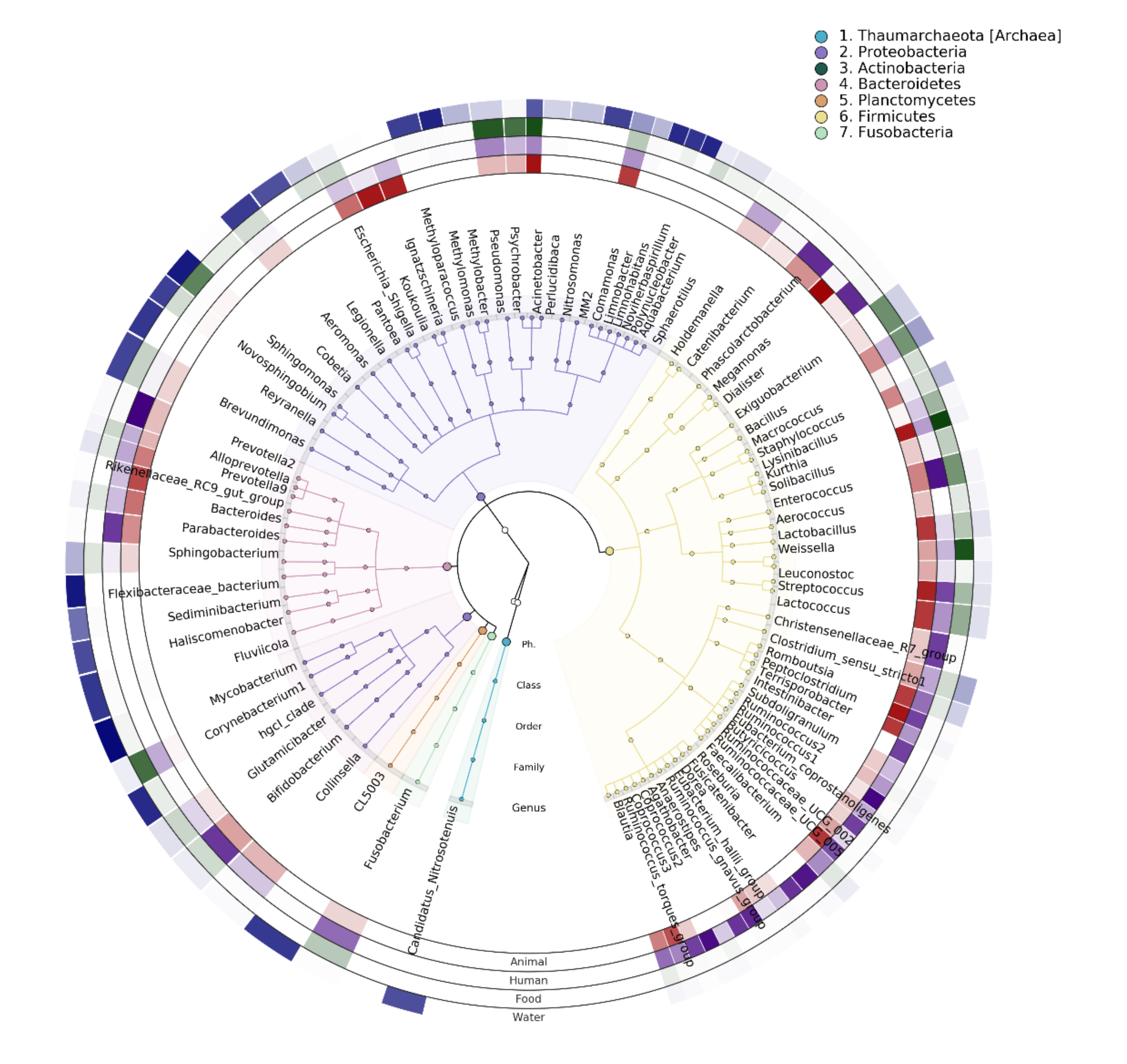
3.4.2. Potential Dispersal of Microbial Taxa between the Microbiota of Humans and Nonhumans
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Deng, P.; Swanson, K.S. Gut microbiota of humans, dogs and cats: Current knowledge and future opportunities and challenges. Br. J. Nutr. 2015, 113 (Suppl. 1), S6–S17. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef] [Green Version]
- Xiong, W.; Jousset, A.; Li, R.; Delgado-Baquerizo, M.; Bahram, M.; Logares, R.; Wilden, B.; de Groot, G.A.; Amacker, N.; Kowalchuk, G.A.; et al. A global overview of the trophic structure within microbiomes across ecosystems. Environ. Int. 2021, 151, 106438. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, C.C.; Gascon, M.; Osornio-Vargas, A.R.; Shier, C.; Guttman, D.S.; Becker, A.B.; Azad, M.B.; Sears, M.R.; Lefebvre, D.L.; Moraes, T.J.; et al. Natural environments in the urban context and gut microbiota in infants. Environ. Int. 2020, 142, 105881. [Google Scholar] [CrossRef] [PubMed]
- Steury, R.A.; Currey, M.C.; Cresko, W.A.; Bohannan, B.J.M. Population Genetic Divergence and Environment Influence the Gut Microbiome in Oregon Threespine Stickleback. Genes 2019, 10, 484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobko, T.; Liang, S.; Cheng, W.H.G.; Tun, H.M. Impact of outdoor nature-related activities on gut microbiota, fecal serotonin, and perceived stress in preschool children: The Play&Grow randomized controlled trial. Sci. Rep. 2020, 10, 21993. [Google Scholar]
- Song, S.J.; Lauber, C.; Costello, E.K.; Lozupone, C.A.; Humphrey, G.; Berg-Lyons, D.; Caporaso, J.G.; Knights, D.; Clemente, J.C.; Nakielny, S.; et al. Cohabiting family members share microbiota with one another and with their dogs. eLife 2013, 2, e00458. [Google Scholar] [CrossRef] [PubMed]
- Hanski, I.; von Hertzen, L.; Fyhrquist, N.; Koskinen, K.; Torppa, K.; Laatikainen, T.; Karisola, P.; Auvinen, P.; Paulin, L.; Mäkelä, M.J.; et al. Environmental Biodiversity, Human Microbiota, and Allergy Are Interrelated. Proc. Natl. Acad. Sci. USA 2012, 109, 8334–8339. [Google Scholar] [CrossRef] [Green Version]
- van Bruggen, A.H.C.; Goss, E.M.; Havelaar, A.; van Diepeningen, A.D.; Finckh, M.R.; Morris, J.G. One Health-Cycling of diverse microbial communities as a connecting force for soil, plant, animal, human and ecosystem health. Sci. Total Environ. 2019, 664, 927–937. [Google Scholar] [CrossRef]
- Lange, K.; Buerger, M.; Stallmach, A.; Bruns, T. Effects of Antibiotics on Gut Microbiota. Dig. Dis. 2016, 34, 260–268. [Google Scholar] [CrossRef]
- Ng, K.M.; Aranda-Díaz, A.; Tropini, C.; Frankel, M.R.; Van Treuren, W.; O’Loughlin, C.T.; Merrill, B.D.; Yu, F.B.; Pruss, K.M.; Oliveira, R.A.; et al. Recovery of the Gut Microbiota after Antibiotics Depends on Host Diet, Community Context, and Environmental Reservoirs. Cell Host Microbe 2019, 26, 650–665.e4. [Google Scholar]
- Hagan, T.; Cortese, M.; Rouphael, N.; Boudreau, C.; Linde, C.; Maddur, M.S.; Das, J.; Wang, H.; Guthmiller, J.; Zheng, N.Y.; et al. Antibiotics-Driven Gut Microbiome Perturbation Alters Immunity to Vaccines in Humans. Cell 2019, 178, 1313–1328.e13. [Google Scholar] [CrossRef]
- Palleja, A.; Mikkelsen, K.H.; Forslund, S.K.; Kashani, A.; Allin, K.H.; Nielsen, T.; Hansen, T.H.; Liang, S.; Feng, Q.; Zhang, C.; et al. Recovery of gut microbiota of healthy adults following antibiotic exposure. Nat. Microbiol. 2018, 3, 1255–1265. [Google Scholar] [CrossRef]
- Dethlefsen, L.; Relman, D.A. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4554–4561. [Google Scholar] [CrossRef] [Green Version]
- Konstantinidis, T.; Tsigalou, C.; Karvelas, A.; Stavropoulou, E.; Voidarou, C.; Bezirtzoglou, E. Effects of Antibiotics upon the Gut Microbiome: A Review of the Literature. Biomedicines 2020, 8, 502. [Google Scholar] [CrossRef]
- Martin-Laurent, F.; Topp, E.; Billet, L.; Batisson, I.; Malandain, C.; Besse-Hoggan, P.; Morin, S.; Artigas, J.; Bonnineau, C.; Kergoat, L.; et al. Environmental risk assessment of antibiotics in agroecosystems: Ecotoxicological effects on aquatic microbial communities and dissemination of antimicrobial resistances and antibiotic biodegradation potential along the soil-water continuum. Environ. Sci. Pollut Res. Int. 2019, 26, 18930–18937. [Google Scholar] [CrossRef]
- Gilbert, J.A.; Blaser, M.J.; Caporaso, J.G.; Jansson, J.K.; Lynch, S.V.; Knight, R. Current understanding of the human microbiome. Nat. Med. 2018, 24, 392–400. [Google Scholar] [CrossRef]
- Flandroy, L.; Poutahidis, T.; Berg, G.; Clarke, G.; Dao, M.-C.; Decaestecker, E.; Furman, E.; Haahtela, T.; Massart, S.; Plovier, H.; et al. The impact of human activities and lifestyles on the interlinked microbiota and health of humans and of ecosystems. Sci. Total Environ. 2018, 627, 1018–1038. [Google Scholar] [CrossRef]
- Wang, C.; Li, P.; Yan, Q.; Chen, L.; Li, T.; Zhang, W.; Li, H.; Chen, C.; Han, X.; Zhang, S.; et al. Characterization of the Pig Gut Microbiome and Antibiotic Resistome in Industrialized Feedlots in China. mSystems 2019, 4, e00206-19. [Google Scholar] [CrossRef] [Green Version]
- Binh, V.N.; Dang, N.; Anh, N.; Ky, L.X.; Thai, P.K. Antibiotics in the aquatic environment of Vietnam: Sources, concentrations, risk and control strategy. Chemosphere 2018, 197, 438–450. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.P.; Saegerman, C.; Douny, C.; Dinh, T.V.; Xuan, B.H.; Vu, B.D.; Hong, N.P.; Scippo, M.L. First Survey on the Use of Antibiotics in Pig and Poultry Production in the Red River Delta Region. of Vietnam. Food Public Health 2013, 3, 247–256. [Google Scholar]
- Kinh, N.V.; Khue, L.N.; Cuong, T.Q.; Kinh, L.N.; Ha, N.T.B.; Mai, H.T.; Ha, N.H.; Trung, N.V.; Ca, P.V.; Chuc, N.T.K.; et al. Situation Analysis: Antibiotic Use and Resistance in Vietnam; CDDEP: Washington, DC, USA; New Delphi, India, 2010; Available online: https://cddep.org/wp-content/uploads/2017/06/vn_report_web_1_8.pdf (accessed on 25 July 2021).
- Nga, D.T.T.; Chuc, N.T.K.; Hoa, N.P.; Hoa, N.Q.; Nguyen, N.T.T.; Loan, H.T.; Toan, T.K.; Phuc, H.D.; Horby, P.; Van, Y.; et al. Antibiotic sales in rural and urban pharmacies in northern Vietnam: An observational study. BMC Pharmacol. Toxicol. 2014, 15, 24555709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, D.K.; Chu, J.; Do, N.T.; Brose, F.; Degand, G.; Delahaut, P.; De Pauw, E.; Douny, C.; Van, N.K.; Vu, T.D.; et al. Monitoring Antibiotic Use and Residue in Freshwater Aquaculture for Domestic Use in Vietnam. Ecohealth 2015, 12, 480–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raph, L.; Hamers, H.R.v.D. Antibiotic consumption in low-income and middle-income countries. Lancet Glob. Health 2018, 6, e732. [Google Scholar] [CrossRef] [Green Version]
- Bich, V.T.N.; Thanh, L.V.; Thai, P.D.; Van Phuong, T.T.; Oomen, M.; Driessen, C.; Beuken, E.; Hoang, T.H.; van Doorn, H.R.; Penders, J.; et al. exploration of the gut and environmental resistome in a community in northern Vietnam in relation to antibiotic use. Antimicrob. Resist. Infect. Control 2019, 8, 194. [Google Scholar] [CrossRef]
- Galazzo, G.; van Best, N.; Benedikter, B.J.; Janssen, K.; Bervoets, L.; Driessen, C.; Oomen, M.; Lucchesi, M.; van Eijck, P.H.; Becker, H.E.F.; et al. How to Count Our Microbes? The Effect of Different Quantitative Microbiome Profiling Approaches. Front. Cell. Infect. Microbiol. 2020, 10, 403. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High.-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Holmes, S.P. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 2017, 11, 2639–2643. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Chao, A. Nonparametric Estimation of the Number of Classes in a Population. Scand. J. Stat. 1984, 11, 265–270. [Google Scholar]
- Faith, D.P. Conservation evaluation and phylogenetic diversity. Biol. Conserv. 1992, 61, 1–10. [Google Scholar] [CrossRef]
- Martino, C.; Morton, J.T.; Marotz, C.A.; Thompson, L.R.; Tripathi, A.; Knight, R.; Zengler, K. A Novel Sparse Compositional Technique Reveals Microbial Perturbations. mSystems 2019, 4, e00016-19. [Google Scholar] [CrossRef] [Green Version]
- Aitchison, J.; Shen, S.M. Logistic-Normal Distributions: Some Properties and Uses. Biometrika 1980, 67, 261–272. [Google Scholar] [CrossRef]
- Lozupone, C.; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001, 26, 32–46. [Google Scholar]
- Mandal, S.; Van Treuren, W.; White, R.A.; Eggesbø, M.; Knight, R.; Peddada, S.D. Analysis of composition of microbiomes: A novel method for studying microbial composition. Microb. Ecol. Health Dis. 2015, 26, 27663. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Duan, Y.; Xu, Z.; Lyu, N.; Liu, F.; Liang, S.; Zhu, B. An examination of data from the American Gut Project reveals that the dominance of the genus Bifidobacterium is associated with the diversity and robustness of the gut microbiota. MicrobiologyOpen 2019, 8, e939. [Google Scholar] [CrossRef]
- Mueller, S.; Saunier, K.; Hanisch, C.; Norin, E.; Alm, L.; Midtvedt, T.; Cresci, A.; Silvi, S.; Orpianesi, C.; Verdenelli, M.C.; et al. Differences in fecal microbiota in different European study populations in relation to age, gender, and country: A cross-sectional study. Appl. Environ. Microbiol. 2006, 72, 1027–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escobar, J.S.; Klotz, B.; Valdes, B.E.; Agudelo, G.M. The gut microbiota of Colombians differs from that of Americans, Europeans and Asians. BMC Microbiol. 2014, 14, 311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagar, S.; Vos, A.P.; Morgan, M.E.; Garssen, J.; Georgiou, N.A.; Boon, L.; Kraneveld, A.D.; Folkerts, G. The combination of Bifidobacterium breve with non-digestible oligosaccharides suppresses airway inflammation in a murine model for chronic asthma. Biochim. Biophys. Acta 2014, 1842, 573–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangin, I.; Lévêque, C.; Magne, F.; Suau, A.; Pochart, P. Long-Term Changes in Human Colonic Bifidobacterium Populations Induced by a 5-Day Oral Amoxicillin-Clavulanic Acid Treatment. PLoS ONE 2012, 7, e50257. [Google Scholar] [CrossRef] [Green Version]
- Khan, T.J.; Hasan, M.N.; Azhar, E.I.; Yasir, M. Association of gut dysbiosis with intestinal metabolites in response to antibiotic treatment. Hum. Microbiome J. 2019, 11, 100054. [Google Scholar] [CrossRef]
- Smits, S.A.; Leach, J.; Sonnenburg, E.D.; Gonzalez, C.G.; Lichtman, J.S.; Reid, G.; Knight, R.; Manjurano, A.; Changalucha, J.; Elias, J.E.; et al. Seasonal cycling in the gut microbiome of the Hadza hunter-gatherers of Tanzania. Science 2017, 357, 802–806. [Google Scholar] [CrossRef] [Green Version]
- Clemente, J.C.; Pehrsson, E.C.; Blaser, M.J.; Sandhu, K.; Gao, Z.; Wang, B.; Magris, M.; Hidalgo, G.; Contreras, M.; Noya-Alarcón, Ó.; et al. The microbiome of uncontacted Amerindians. Sci. Adv. 2015, 1, e1500183. [Google Scholar] [CrossRef] [Green Version]
- Gibson, M.K.; Wang, B.; Ahmadi, S.; Burnham, C.A.; Tarr, P.I.; Warner, B.B.; Dantas, G. Developmental dynamics of the preterm infant gut microbiota and antibiotic resistome. Nat. Microbiol. 2016, 1, 16024. [Google Scholar] [CrossRef]
- Odamaki, T.; Kato, K.; Sugahara, H.; Hashikura, N.; Takahashi, S.; Xiao, J.Z.; Abe, F.; Osawa, R. Age-related changes in gut microbiota composition from newborn to centenarian: A cross-sectional study. BMC Microbiol. 2016, 16, 90. [Google Scholar] [CrossRef] [Green Version]
- O’Callaghan, A.; van Sinderen, D. Bifidobacteria and Their Role as Members of the Human Gut Microbiota. Front. Microbiol. 2016, 7, 925. [Google Scholar] [CrossRef] [Green Version]
- Seo, B.; Yoo, J.E.; Lee, Y.M.; Ko, G. Sellimonas intestinalis gen. nov., sp. nov., isolated from human faeces. Int. J. Syst. Evol. Microbiol. 2016, 66, 951–956. [Google Scholar] [CrossRef] [Green Version]
- Loughman, A.; Ponsonby, A.L.; O’Hely, M.; Symeonides, C.; Collier, F.; Tang, M.; Carlin, J.; Ranganathan, S.; Allen, K.; Pezic, A.; et al. Gut microbiota composition during infancy and subsequent behavioural outcomes. EBioMedicine 2020, 52, 102640. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Li, D.; Ke, W.; Liang, D.; Hu, X.; Chen, F. Resveratrol-induced gut microbiota reduces obesity in high-fat diet-fed mice. Int. J. Obes. 2020, 44, 213–225. [Google Scholar] [CrossRef]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef] [Green Version]
- Raymond, F.; Déraspe, M.; Boissinot, M.; Bergeron, M.G.; Corbeil, J. Partial recovery of microbiomes after antibiotic treatment. Gut Microbes 2016, 7, 428–434. [Google Scholar] [CrossRef] [Green Version]
- Jakobsson, H.E.; Jernberg, C.; Andersson, A.F.; Sjölund-Karlsson, M.; Jansson, J.K.; Engstrand, L. Short-Term Antibiotic Treatment Has Differing Long-Term Impacts on the Human Throat and Gut Microbiome. PLoS ONE 2010, 5, e9836. [Google Scholar] [CrossRef] [Green Version]
- Shaw, L.P.; Bassam, H.; Barnes, C.P.; Walker, A.S.; Klein, N.; Balloux, F. Modelling microbiome recovery after antibiotics using a stability landscape framework. ISME J. 2019, 13, 1845–1856. [Google Scholar] [CrossRef] [Green Version]
- Raymond, F.; Ouameur, A.A.; Déraspe, M.; Iqbal, N.; Gingras, H.; Dridi, B.; Leprohon, P.; Plante, P.L.; Giroux, R.; Bérubé, È.; et al. The initial state of the human gut microbiome determines its reshaping by antibiotics. ISME J. 2016, 10, 707–720. [Google Scholar] [CrossRef]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. What is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet., and Diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef] [Green Version]
- Hendriksen, R.S.; Munk, P.; Njage, P.; van Bunnik, B.; McNally, L.; Lukjancenko, O.; Röder, T.; Nieuwenhuijse, D.; Pedersen, S.K.; Kjeldgaard, J.; et al. Global monitoring of antimicrobial resistance based on metagenomics analyses of urban sewage. Nat. Commun. 2019, 10, 1124. [Google Scholar] [CrossRef]
- Cao, J.; Hu, Y.; Liu, F.; Wang, Y.; Bi, Y.; Lv, N.; Li, J.; Zhu, B.; Gao, G.F. Metagenomic analysis reveals the microbiome and resistome in migratory birds. Microbiome 2020, 8, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cinquepalmi, V.; Monno, R.; Fumarola, L.; Ventrella, G.; Calia, C.; Greco, M.F.; Vito, D.d.; Soleo, L. Environmental contamination by dog’s faeces: A public health problem? Int. J. Environ. Res. Public Health 2012, 10, 72–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, P.; Prasoodanan, P.k.V.; Dhakan, D.B.; Kumar, S.; Sharma, V.K. Metagenome of a polluted river reveals a reservoir of metabolic and antibiotic resistance genes. Environ. Microbiome 2019, 14, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vu Thi Ngoc, B.; Ho Bich, H.; Galazzo, G.; Vu Tien Viet, D.; Oomen, M.; Nghiem Nguyen Minh, T.; Tran Huy, H.; van Doorn, H.R.; Wertheim, H.F.L.; Penders, J. Cross-Sectional Analysis of the Microbiota of Human Gut and Its Direct Environment in a Household Cohort with High Background of Antibiotic Use. Microorganisms 2021, 9, 2115. https://doi.org/10.3390/microorganisms9102115
Vu Thi Ngoc B, Ho Bich H, Galazzo G, Vu Tien Viet D, Oomen M, Nghiem Nguyen Minh T, Tran Huy H, van Doorn HR, Wertheim HFL, Penders J. Cross-Sectional Analysis of the Microbiota of Human Gut and Its Direct Environment in a Household Cohort with High Background of Antibiotic Use. Microorganisms. 2021; 9(10):2115. https://doi.org/10.3390/microorganisms9102115
Chicago/Turabian StyleVu Thi Ngoc, Bich, Hai Ho Bich, Gianluca Galazzo, Dung Vu Tien Viet, Melissa Oomen, Trang Nghiem Nguyen Minh, Hoang Tran Huy, Hindrik Rogier van Doorn, Heiman F. L. Wertheim, and John Penders. 2021. "Cross-Sectional Analysis of the Microbiota of Human Gut and Its Direct Environment in a Household Cohort with High Background of Antibiotic Use" Microorganisms 9, no. 10: 2115. https://doi.org/10.3390/microorganisms9102115
APA StyleVu Thi Ngoc, B., Ho Bich, H., Galazzo, G., Vu Tien Viet, D., Oomen, M., Nghiem Nguyen Minh, T., Tran Huy, H., van Doorn, H. R., Wertheim, H. F. L., & Penders, J. (2021). Cross-Sectional Analysis of the Microbiota of Human Gut and Its Direct Environment in a Household Cohort with High Background of Antibiotic Use. Microorganisms, 9(10), 2115. https://doi.org/10.3390/microorganisms9102115