Abstract
Vibrio mimicus is an emerging pathogen, mainly associated with contaminated seafood consumption. However, little is known about its evolution, biodiversity, and pathogenic potential. This study analyzes the pan-, core, and accessory genomes of nine V. mimicus strains. The core genome yielded 2424 genes in chromosome I (ChI) and 822 genes in chromosome II (ChII), with an accessory genome comprising an average of 10.9% of the whole genome for ChI and 29% for ChII. Core genome phylogenetic trees were obtained, and V. mimicus ATCC-33654 strain was the closest to the outgroup in both chromosomes. Additionally, a phylogenetic study of eight conserved genes (ftsZ, gapA, gyrB, topA, rpoA, recA, mreB, and pyrH), including Vibrio cholerae, Vibrio parilis, Vibrio metoecus, and Vibrio caribbenthicus, clearly showed clade differentiation. The main virulence genes found in ChI corresponded with type I secretion proteins, extracellular components, flagellar proteins, and potential regulators, while, in ChII, the main categories were type-I secretion proteins, chemotaxis proteins, and antibiotic resistance proteins. The accessory genome was characterized by the presence of mobile elements and toxin encoding genes in both chromosomes. Based on the genome atlas, it was possible to characterize differential regions between strains. The pan-genome of V. mimicus encompassed 3539 genes for ChI and 2355 genes for ChII. These results give us an insight into the virulence and gene content of V. mimicus, as well as constitute the first approach to its diversity.
1. Introduction
The genus Vibrio contains more than 100 species typically isolated from aquatic environments, and several of them may cause infections in humans and animals [1,2]. Vibrio mimicus has been recognized as an emergent pathogen in human diseases, and it has been isolated from cases of gastroenteritis, ear infections, and severe cholera-like diarrhoea, as well as from several marine products, aquatic plants, sediments, and water (marine, brackish, and freshwater) [3,4,5]. V. mimicus strains encode a wide variety of virulence factors, such as he, hemagglutinins, pili, metalloproteases, enterotoxins, and siderophores, which are mediated by several different mechanisms [5,6,7,8]. Moreover, studies of comparative genomics of Vibrio species have shown that genetic exchange among Vibrionaceae family species is a continuous process [6,9,10,11,12].
V. mimicus was first described as an atypical non-pathogenic strain of Vibrio cholerae; now, both are considered closely related species and share almost 64% of their genome [13]. Wang et al. [14] studied the genome mutations responsible for the biochemical metabolism differentiation between V. mimicus and V. cholerae. Those biochemical differences are due mainly by gene deletion. Additionally, V. mimicus has been used in comparative analyses of the core and pan-genome of V. cholerae, sharing approximately eight core genes from the superintegron (SI) [15,16]. V. mimicus and V. cholerae share many phenotypic characteristics but also virulence genes, such as cholera toxin (Ctx), toxin-coregulated pilus, and pathogenicity islands, among others [13]. This makes V. cholerae an appropriate outgroup when conducting evolutionary studies of V. mimicus [3,9,17]. Recently, the comparative genome analysis of Vibrio metoecus (RC341) and Vibrio parilis (RC586), previously characterized as environmental non-pathogenic variants of V. cholerae, demonstrated that V. metoecus evolved from V. cholera/V. mimicus lineages, while V. parilis from an ancestral V. mimicus lineage [16,18,19]. This type of information contributes to the understanding of Vibrio species diversity [10].
Several virulence genes typical of pathogenic Vibrio species have been found in the genome of V. mimicus strains. Nevertheless, no differences in the virulence potential have been found between environmental and clinical strains of V. mimicus [6,20,21]. It has been reported that these virulence genes could have an important role in V. mimicus adaptation to their natural environment [6], where pathogenic strains could emerge.
Several comparative genomic studies have attempted to explain the evolutionary history of Vibrio species. For instance, Lin et al. [22] studied twenty Vibrio genomes, suggesting a high variation as a response to its adaptation to the environment. Thompson et al. [9] analyzed 17 Vibrio species with Comparative Microbial Genomics (CMG) biotools [23] and obtained a pan-genome of approximately 26,504 genes. This study included 18 strains of V. cholerae (pan-genome = 6923 genes) and two strains of V. mimicus (pan-genome = 8306 genes). Lilburn et al. [10] also studied 11 Vibrionaceae family genomes, but the V. mimicus genome was not included. A pan-genome of 51,517 genes was reported, where 49,588 corresponded to CDS (coding DNA sequence).
Molecular phylogenetic has been used for decades to elucidate the relationships between species. Today, the wide availability of genomes facilitates the study of most pathogenic bacteria [24,25,26]. At the time of this study, there were about 4000 complete genomes of Vibrio species with 65% being of V. cholerae and Vibrio parahaemolyticus, while only 0.5% corresponded to V. mimicus (https://www.ncbi.nlm.nih.gov/genome/browse#!/prokaryotes/).
Despite the importance of V. mimicus as a human pathogen, little is known about its evolution and biology. The availability of genomic sequences of several V. mimicus strains makes it possible to study the evolution and pathogenic potential of this species. In this study, nine genomes of V. mimicus were analyzed. The pan-, core-, and accessory genomes were determined to characterize the virulence and biology of V. mimicus; in order to document their diversity and obtain the first outline of their pan-genome. In addition, a multilocus sequence analysis (MLSA) of eight housekeeping genes was performed to obtain information regarding their variability.
2. Materials and Methods
2.1. Bacterial Genomes
Nine genomes of V. mimicus were analyzed in this study. The genomes of V. mimicus CAIM-602T (Vm602, PRJNA179483) [7], V. mimicus CAIM-1882 (Vm1882, PRJNA219179), V. mimicus CAIM-1883 (Vm1883, PRJNA219181) [20], and V. mimicus ATCC-33654 (Vm 33654, PRJNA231624) were sequenced by our research group, as previously reported [7,20]. The remaining five genomes sequences were obtained from the database of the National Center for Biotechnology Information (NCBI): V. mimicus MB451 (Vm451, PRJNA40509), V. mimicus VM223 (Vm223, PRJNA40483), V. mimicus VM603 (Vm603, PRJNA40241), V. mimicus VM573 (Vm573, PRJNA40243), and V. mimicus SX-4 (Vm SX4, PRJNA47421) [6,10,14]. Additionally, two V. cholerae genomes were downloaded and used as outgroups, V. cholerae O1 biovar El Tor str. N16961 (Vc 16961, PRJNA36) [27] and V. cholerae O395 (Vc 0395, PRJNA32853) [28]. Original contigs were annotated by RAST (Rapid Annotations using Subsystems Technology) [29] (http://rast.nmpdr.org) and NCBI (http://www.ncbi.nlm.nih.gov).
2.2. Generation of a Coding Core Genome
A genome-wide assembly and contig synteny were constructed with Mauve Genome Alignment software ver. 2.3.1 (Sydney, Australia) [30] using V. mimicus MB451 as a reference strain [6]. The contigs were assembled with Geneious R6 ver. 6.0.3 (Biomatters Ltd. Auckland, New Zealand) to obtain the two chromosomes (ChI and ChII) of all V. mimicus strains [20]. The nine assembled sequences of each chromosome of V. mimicus strains and their corresponding counterparts of V. cholerae strains were aligned using progressiveMAUVE ver. 2.3.1 (Sydney Australia) [31], and the output was transformed into a plain FASTA format alignment using a Perl script [32]. The genome of V. mimicus strain MB451 was used as a reference [6], and all the positions from the alignment that were not shared with this strain were removed from the core genome analysis. Annotation files from NCBI were processed using R [33] to detect the coordinates of the genes in the reference genome, and to determine the core genes or those highly conserved genes that were present in all strains [26,34]. Core genes were defined as genes that shared significant homology on at least 80% with the corresponding reference gene. The core genomes of both chromosomes were built by concatenating the FASTA files of the corresponding aligned core genes [32].
2.3. Phylogenetic Reconstruction
Phylogenetic reconstructions of the core genome of each chromosome were performed using Maximum Likelihood (ML). The ML phylogenetic tree was obtained using RAxML (Randomized Axelerated Maximum Likelihood v7.2.7) (Heidelberg, Germany) [35], applying the GTRGAMMA model of nucleotide substitution and 1000 bootstrap replicates. V. cholerae was used as an outgroup for this analysis.
2.4. Analysis of the Accessory Genome
A matrix of presence-absence genes of the accessory genome was created for each chromosome and clustered using the heatmap.2 function of the gplots R package (clustering method hclust) [36].
2.5. Comparative Microbial Genomics (CMG)
The nine genomes of V. mimicus were analyzed by the CMG-Biotools (Lyngby, Denmark) [23] to obtain a genome BLAST atlas of each chromosome using V. mimicus 451 as a reference genome.
2.6. Pan-genome Atlas
In addition, twenty-one genomes of V. mimicus uploaded until September 2019 were used to obtain the pan-genome atlas and the Average Nucleotide Identity (ANI) by Anvi’o ver.6.1 (http://merenlab.org/2016/11/08/pangenomics-v2/) [37]. The genomes analyzed were the nine genomes previously described plus twelve new genomes obtained from the NCBI: V. mimicus FDAARGOS113 (PRJNA231221), V. mimicus FDAARGOS112 (PRJNA231221), V. mimicus 523-80 (PRJNA242443), V. mimicus NCTC11435 (PRJEB6403), V. mimicus N2733 (PRJNA548872), V. mimicus N2763 (PRJNA548872), V. mimicus N2781 (PRJNA548872), V. mimicus N2789 (PRJNA548872), V. mimicus N2790 (PRJNA548872), V. mimicus N2810 (PRJNA548872), V. mimicus N2816 (PRJNA548872), and V. mimicus SCCF01 (PRJNA327733).
2.7. Multilocus Sequence Analysis (MLSA)
The sequences of eight housekeeping genes, ftsZ (1200 nt), gapA (991 nt), gyrB (2065 nt), topA (1788 nt), rpoA (993 nt), recA (1041 nt), mreB (1044 nt), and pyrH (732 nt), were obtained from the nine genomes of V. mimicus and the genomes of V. cholerae 16961, V. parilis (RC586), V. metoecus (RC341) [16,19] and Vibrio caribbeanicus ATCC-BAA-2122 [38], which were downloaded from NCBI. The nucleotide sequences were aligned with the MUSCLE program implemented in Geneious R6 ver. 4.8.5 (Biomatters Ltd.) with UPGMB (unweighted pair group method with arithmetic mean) as the clustering method (kmer4_6 as a distance measure for iteration 1 and pctid_kimura as a distance measure for iterations 2). An ML phylogenetic tree was created using RAxML by CIPRES Science Gateway [39], and the robustness of the topology was checked by 1000 bootstrap replicates. The consensus tree was elaborated by the 95% majority-rule of the replicates using DendroPy [40], and the edition of the phylogenetic tree was performed with the FigTree figure drawing tool (Ver 1.4.4), where V. caribbeanicus ATCC-BAA-2122 was used as an outgroup.
3. Results
3.1. Pan-Genome of V. mimicus
The general characteristics of the nine genomes of V. mimicus used in this study are shown in Table 1. The estimated size of Chromosome I (ChI) ranged between 2.82 and 3.05 Mbp with an average of 46.6% GC (guanine-cytosine content) and 2689 CDS were detected, whereas, in Chromosome II (ChII), the size ranged between 1.11 and 1.46 Mbp with an average of 46.0% GC, and 1164 CDS were identified. Additionally, according to the RAST server, the annotation was classified in several subsystems, reaching 436 in ChI and 132 in ChII (Table 1). The analysis of the pan-genome resulted in 3539 genes for ChI and 2355 genes for ChII.

Table 1.
General characteristics of the nine genomes of V. mimicus included in this study.
A BLAST atlas for the genome of both chromosomes (ChI and ChII) of nine V. mimicus and two V. cholerae strains were obtained using V. mimicus MB451 as the reference strain (Figure 1). The analysis revealed several variable regions in both chromosomes. In ChI, ten major variable regions were detected. Regions 1, 4, 5, 9, and 10 were only present in the reference strain V. mimicus MB451, whereas regions 2, 3, 6, 7, and 8 were in most of the V. mimicus strains, but were absent in the V. cholerae strains. For example, the CDS observed at region 1 were a phage transcriptional regulator AlpA and an integrase (1176 pb), while, in region 4, repeats-in-toxins (RTX) proteins were detected; region 5 consists of the f237 prophage with a zona occludens toxin, accessory cholera enterotoxin, and other CDS of the bacteriophage. Additionally, in region 9, a phage integrase was identified, and, in region 10, putative polysaccharide biosynthesis genes were detected. Differences between the gene content of V. mimicus and V. cholerae were observed in regions 2, 3, 6, and 8. Such genes as outer membrane proteins, type II/IV secretion system proteins, transcriptional regulators, and a bacteroid aerotolerance operon were identified in V. mimicus but not in V. cholerae. In region 7, genes, such as chemotaxis proteins, response, and transcriptional regulators, were not present in V. mimicus VM573 nor V. cholerae strains.
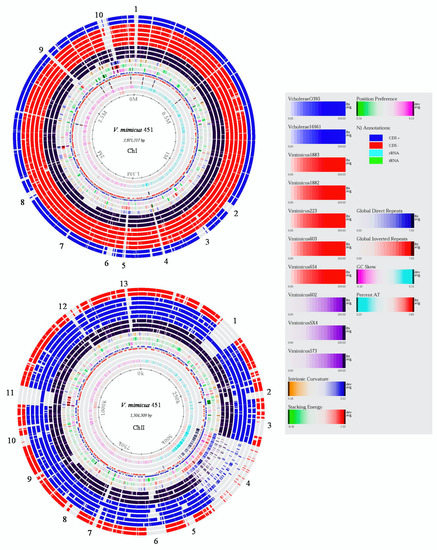
Figure 1.
Genome BLAST atlas of both chromosomes of V. mimicus using V. mimicus MB451 as a reference. Order of the genomes from the inner dark circle: V. mimicus VM573, V. mimicus SX-4, V. mimicus CAIM-602T, V. mimicus ATCC-33654, V. mimicus VM603, V. mimicus VM223, V. mimicus CAIM-1882, V. mimicus CAIM-1883, V. cholerae 16961, and V. cholerae O395. Genomic regions unique to the reference strain that are not present in the other strains are without color (no blast hit). The outside numbers (1 to 10 in Ch I and 1 to 13 in ChII), corresponds to the variable regions identified.
In ChII, thirteen major variable regions were detected. Region 13 and part of region 4 were only present in the reference strain V. mimicus MB451. Regions 3, 5, 8, 9, and 10 were present in most V. mimicus strains and were absent in the V. cholerae strains. Moreover, regions 1, 2, 6, 7, 11, and 12 were absent in most V. mimicus strains and in both V. cholerae strains. Some of the genes detected only in the reference strain were in region 4, consisting of integral and inner membrane protein genes, DNA damage-inducible genes, Doc toxins, mobile elements, and transcriptional regulators. In region 13, valine glycine repeat G (VgrG) protein genes were observed in the reference strain. In regions 3, 5, 8, 9, and 10, some CDS genes were present in V. mimicus, but absent in V. cholerae, e.g., outer membrane porin, pilus assembly proteins, type II/IV protein secretion system, and transcriptional regulators. In regions 1, 2, 6, 7, 11, and 12, various genes, such as transcriptional regulators, RTX toxins, and enterotoxins, were absent for most V. mimicus strains and V. cholerae strains. In addition, a second copy of various flagellar proteins were detected in ChII of V. mimicus MB451, VM573, VM603, and SX-4.
As supplementary material, we also included the ANI data of the 21 V. mimicus genomes available since the conception of this manuscript to generate the pan-genome atlas (Figure S1), although some of the strains are the same but sequenced by different research groups.
3.2. Core and Accessory Genome of V. mimicus
The core genome of V. mimicus was obtained by selecting homologous nucleotide sequences with >80% of similarity with the reference genome (V. mimicus MB451). Thus, all the positions from the alignment that were not shared with this strain were removed from the core genome analysis. This alignment resulted in 2,972,217 bp for ChI and 1,304,309 for ChII. Next, the core genomes for both chromosomes were built by concatenating the FASTA files of the corresponding aligned core genes, obtaining up to 2,378,529 bp for ChI (2424 core genes) and 826,416 bp for ChII (822 core genes).
To determine the phylogenetic relationships of the core genome (CDS) of V. mimicus strains, an ML phylogenetic tree was obtained for each chromosome (Figure 2). A clear differentiation of the clades formed by V. mimicus and V. cholerae was observed. In the phylogenetic trees, a different clustering between V. mimicus strains in ChI and ChII was observed, primarily due to the V. mimicus MB451 strain. For instance, the environmental V. mimicus strains VM603, CAIM-1882, and CAIM-1883 were grouped in ChI but not in ChII, whereas the environmental strains CAIM-1882 and CAIM-1883 clustered with the clinical strain V. mimicus MB451. Nonetheless, it can be observed that within the clade of V. mimicus the strain ATCC-33654 was the closest to the outgroup in both chromosomes. In addition, species diversity and similar cluster formation were observed (Figure S1).
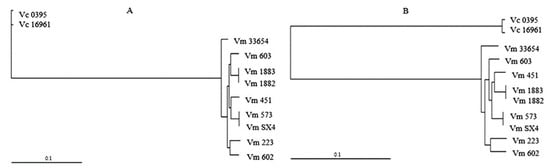
Figure 2.
Phylogenetic tree of the core genes of V. mimicus MB451, V. mimicus VM573, V. mimicus SX4, V. mimicus CAIM-602T, V. mimicus ATCC-33654, V. mimicus VM603, V. mimicus VM223, V. mimicus CAIM-1882, V. mimicus CAIM-1883, and two V. cholerae (Vc O395 and Vc 16961). (A): Maximum likelihood (ML) tree of ChI. (B): ML tree of ChII.
Furthermore, we obtained genes from the core genome with phylogenetic signal by a likelihood mapping approach. We found 301 genes out of 2196 genes in ChI and 99 genes out of 651 genes in ChII (data not shown). Those genes are considered to have a signal of external origin and could be considered to be the result of recombination and horizontal gene transfer events [22,32]. Virulence genes and transcriptional regulators could be taken as examples of this phylogenetic signal.
For the accessory genome of V. mimicus, between 218 and 378 genes were detected in ChI, and for ChII, between 214 and 490 genes were identified (Figure 3), which represents on average 10.9% in ChI and 29% in ChII from the complete genome. The accessory genome was analyzed in terms of the number of genes shared among V. mimicus strains. It was observed that most of the genes that were not shared were present only in a single strain or in several. Some genes were present in the nine strains but showed less than 80% of homology; hence, they do not fulfill the criteria for the core genome (Figure S2).
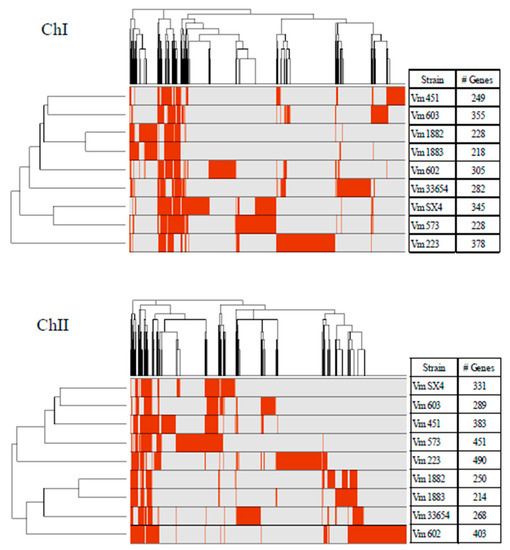
Figure 3.
Presence-absence matrix of the genes of the accessory genome of the nine V. mimicus strains (V. mimicus MB451, V. mimicus VM573, V. mimicus SX-4, V. mimicus CAIM-602T, V. mimicus ATCC-33654, V. mimicus VM603, V. mimicus VM223, V. mimicus CAIM-1882, and V. mimicus CAIM-1883) for each chromosome. Where red color indicates gene presence
3.3. Virulence Classification
Virulence genes of the core and accessory genome were identified and classified for each chromosome according to Kimes et al. [41] (Figure 4). In the core genome, 512 virulence genes were identified in ChI and 200 in ChII. The main categories detected in ChI were type I protein secretion system (139 genes), extracellular components (52 genes), flagellar proteins (52 genes), and potential regulators (41 genes). In ChII, the main categories were type I protein secretion system (56 genes), chemotaxis proteins (28 genes), and antibiotic resistance proteins (19 genes). In the accessory genome, 233 virulence genes were identified in ChI and 221 in ChII. The main categories detected in ChI were mobile elements (94 genes), extracellular components (48 genes), and toxins (28 genes). In ChII, the main categories were mobile elements (63 genes), toxins (38 genes), and flagellar proteins (37 genes).
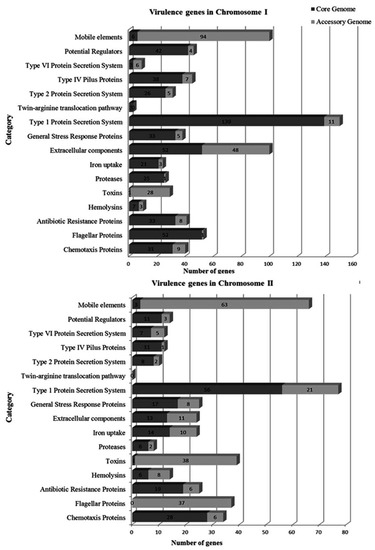
Figure 4.
Classification of virulence genes in the core and accessory genome of V. mimicus by category.
Moreover, virulence genes with phylogenetic signals of the core genome were identified in both chromosomes resulting in 56 genes in ChI and 29 genes in ChII. Phylogenetic trees of some of the virulence genes of ChI (protease, ompU, mshQ, toxR, luxR, and luxO) and ChII (tonB, zinc metalloprotease, chitinase, lolC, methyl-accepting chemotaxis protein, and acriflavine resistance protein) are presented in Figures S3 and S4, respectively. Each phylogenetic tree shows different clusters where V. mimicus MB451, V. mimicus CAIM-602, V. mimicus ATCC-33654, V. mimicus VM603, and V. mimicus VM223 displayed more variability in each tree of both chromosomes.
3.4. MLSA
A phylogenetic tree based on MLSA of eight housekeeping genes (ftsZ, gapA, gyrB, topA, rpoA, recA, mreB, and pyrH) was performed in conjunction with V. cholerae 16961, V. parilis (RC586), V. metoecus (RC341), and V. caribbeanicus ATCC-BAA-2122 (Figure 5). The phylogenetic trees assigned the analyzed species to a different clade, where V. mimicus forms a clearly separated clade from the others. The clusters observed provide evidence of the differentiation of V. mimicus strains with other Vibrio species and its variability.
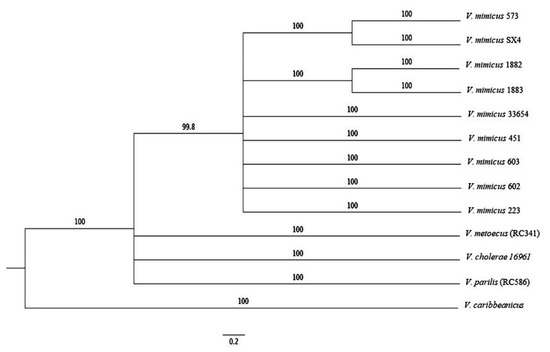
Figure 5.
ML phylogenetic tree of eight housekeeping genes (ftsZ, gapA, gyrB, topA, rpoA, recA, mreB, and pyrH) of the nine strains of V. mimicus, V. cholerae 16961, V. parilis (RC586), V. metoecus (RC341), and V. caribbeanicus ATCC BAA-2122.
4. Discussion
Comparative genomics analyses have been performed for years, primarily with partial genomes. Due to advances in technology and accessibility, increasingly complete bacterial genomes are now available. Hence, it is possible to identify all the genes present in a specific genome [42]. However, at the time of this study, only nine genomes sequences of V. mimicus were available, including one obtained by our research group that was uploaded in 2019 (Accession: PRJNAV.231624). By September 2019, twenty-one assembled genomes were available at NBCI, and this number is still increasing. A pan-genome atlas, including all 21 V. mimicus genomes, was constructed, although part of the strain’s information is missing. In addition, some of the available genomes correspond to the same strain but sequenced by different research groups (e.g., CAIM602 and NCTC11435; FDAARGOS_112 and ATCC 33654), and there were slight differences and a different number of unique genes observed. Probably, those differences could be due to bacterial subculturing along time by each research group, which can lead to the selection of different variants carrying a distinct set of mutations [43]. Differences in storage and preservation methods, and general laboratory practices, may also contribute to this phenomenon. Moreover, some strains were isolated from the same region, year, and clustered together (Figure S1). Therefore, a reduced number of strains, including all the genome and metadata, were selected for genome comparison. Despite the low number of genomes of V. mimicus included, this work presents the first comparative genome analysis of V. mimicus, obtaining a first outline of the pan-genome, core genome, and accessory genome.
Quantification of genome relatedness can be accomplished by different methods [44]. The ML phylogenetic trees were obtained for the core genome of V. mimicus. These phylogenetic trees showed no correlations among strains relatedness and its source (clinical or environmental), isolation year, or geographical origin. Surprisingly, only a few strains clustered together based on its source, and two clinical (573 and SX4) and two environmental (1882 and 1883) strains showed consistency between them in the phylogenetic trees. Nevertheless, it is important to note the major lapse between strains and the distant geographical areas from which they were isolated, which may help to explain the lack of correlation.
Eight housekeeping genes were used to study the relationships between V. mimicus and three closely related species, V. cholerae, V. parilis (RC586), and V. metoecus (RC341) using V. caribbeanicus as an outgroup. A separate clade formed by V. mimicus strains was observed, corroborating the differences with V. cholerae and the others. Surprisingly, V. cholerae was more closely related to the two other species than V. mimicus. Sawabe et al. [45] realized MLSA with the same eight housekeeping genes in several Vibrio species. These researchers identified a cholerae clade that includes V. cincinnatiensis, V. cholerae, V. furnissii, V. fluvialis, V. metschnikovii, V. cholerae, and V. mimicus, and the similarity of the MLSA concatenation was between 85.4% and 94.7% within this clade. Recently, an update of the Vibrio clades was published, and V. parilis was added to the cholera clade with almost the same similarity values (83.4–94.4%) [46]. The MLSA of V. mimicus species shows in the ML tree consistency in their clustering. Thus, this approach is useful for studying phylogenetic relationships, and MLSA offers a good option because fewer data and less time are required for the analysis.
Phylogenetic signals within the core genome genes were observed, although there was a small number of positive genes (301 genes in ChI and 99 genes in ChII). As the core genome includes genes sharing >80% homology in the sequence, it is unsurprising to detect few differences between them, which may help to explain the low number of genes showing phylogenetic signals. However, these genes with phylogenetic signals could be considered to have a different phylogenetic history and therefore could be potentially recombinant (either by recombination or horizontal gene transfer) [32]. In the accessory genome analysis, strong variability was observed between the strains; different numbers of genes were reported in each strain, and an aggrupation between strains was made depending on the number of strains that shared the same gene. These results help us to elucidate the variability of the species.
The development of molecular and genomic techniques allows the identification of genes that encode virulence factors [47]. Although V. mimicus and V. cholerae are closely related and share some phenotypic and genomic characteristics, differences in the genome Blast atlas have been reported [3,6,9]. The main differences found in this study were on virulence genes, hypothetical proteins, and transcriptional regulators. These differences in virulence content could explain the distinctiveness of the pathogenic potential of each species [48]. Several virulence genes were identified and classified in this study for V. mimicus strains. More virulence genes were found in the core-genome, with a higher number in ChI (511 genes) than in ChII (200 genes), the same pattern, but with lower values were observed in the accessory genome (233 genes in ChI and 221 genes in ChII). It has been reported that ChI has more plasticity [8] and more virulence genes than ChII. The genes present in ChI are responsible for growth and viability, while the genes in ChII are responsible for adaptation to environmental changes [1,6]. However, in V. cholerae, it has been proposed that the genes on both chromosomes function differently depending on the environment [49]. Thus, it would be important for future research to study how the gene content of V. mimicus could depend or be affected by the environment.
5. Conclusions
With the advances and development of biological technologies, information concerning bacterial evolution and pathogenesis has become increasingly available. The results obtained in this study regarding the pan-genome (core genome, accessory genome, and virulence genes), as well as phylogenetic analysis of V. mimicus, will help us to elucidate the variability of this species. These results, even in cases when they came from the same source, have provided a perspective on the gene content and virulence potential of V. mimicus. A great diversity of genes associated with virulence factors, previously linked to other pathogenic Vibrio species, were found in the core and accessory genomes of V. mimicus. This finding emphasizes the pathogenic potential of Vibrio mimicus, as well as the importance of continuing with the genomic study of this species. Specifically, further research is warranted to elucidate the pathogenic potential and its possible mechanisms, as well as to establish the behavior of V. mimicus in the environment and evaluate its real potential as a human pathogen.
Supplementary Materials
The following are available online at https://www.mdpi.com/2076-2607/9/1/191/s1, Figure S1. Pan-genome atlas of 21 V. mimicus strains: V. mimicus MB451 V. mimicus VM573, V. mimicus SX-4, V. mimicus CAIM-602T, V. mimicus ATCC-33654, V. mimicus VM603, V. mimicus VM223, V. mimicus CAIM-1882, V. mimicus CAIM-1883, V. mimicus FDAARGOS113, V. mimicus FDAARGOS112, V. mimicus 523-80, V. mimicus NCTC11435, V. mimicus N2733, V. mimicus N2763, V. mimicus N2781, V. mimicus N2789, V. mimicus N2790, V. mimicus N2810, V. mimicus N2816, and V. mimicus SCCF01. Figure S2. Classification of the accessory genome of V. mimicus according to the number of shared genes between strains. Figure S3. Phylogenetic tree of six virulence genes with phylogenetic signal (protease, ompU, mshQ, toxR, luxR, and luxO) in ChI of V. mimicus MB451, V. mimicus VM573, V. mimicus VM603, V. mimicus SX4, V. mimicus CAIM 602T, V. mimicus ATCC 33654, V. mimicus VM223, V. mimicus CAIM 1882 and V. mimicus CAIM 1883; using V. cholerae 16961 and V. cholerae O395 as outgroups. The phylogenetic tree was obtained by an independent maximum likelihood (ML) tree reconstruction using RAxML v7.2.7 [25] and the topology testing methodology implemented in TreePuzzle v5.2 [32]. Figure S4. Phylogenetic tree of six virulence genes with phylogenetic signal (tonB, zinc metalloprotease, chitinase, lolC, methyl.accepting chemotaxis protein, and acriflavine resistance protein) in ChII of V. mimicus MB451, V. mimicus VM573, V. mimicus VM603, V. mimicus SX4, V. mimicus CAIM 602T, V. mimicus ATCC 33654, V. mimicus VM223, V. mimicus CAIM 1882 and V. mimicus CAIM 1883; using V. cholerae 16961 and V. cholerae O395 as outgroups. The phylogenetic tree was obtained by an independent maximum likelihood (ML) tree reconstruction using RAxML v7.2.7 [25] and the topology testing methodology implemented in TreePuzzle v5.2 [32].
Author Contributions
Conceptualization, E.A.-F. and L.N.-O.; methodology, B.G.-G. and E.A.-F.; software, L.S.-B.; validation, B.G.-G., F.G.-C., and M.Z.-C.; formal analysis, I.G.-A. and L.S.-B.; investigation, I.G.-A.; resources, E.A.-F. and L.N.-O.; data curation, I.G.-A.; writing—original draft preparation, I.G.-A.; writing—review and editing, E.A.-F., M.Z.-C., F.G.-C., and L.N.-O.; visualization, E.A.-F. and L.N.-O.; supervision, E.A.-F., B.G.-G., and L.N.-O.; project administration, E.A.-F. and L.N.-O.; funding acquisition, E.A.-F. and L.N.-O. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by CONACYT (National Council for Science and Technology), who provided a student scholarship (Grant No. 98245) and internal institutional funds from CIAD Guaymas and CIAD Hermosillo.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study will be openly available at CIAD thesis repository (https://ciad.repositorioinstitucional.mx/jspui/) from 2021.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Thompson, F.L.; Iida, T.; Swings, J. Biodiversity of vibrios. Microbiol. Mol. Biol. Rev. 2004, 68, 403–431. [Google Scholar] [CrossRef] [PubMed]
- Farmer, J., III. The family vibrionaceae. In The Prokaryotes; Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K.-H., Stackebrandt, E., Eds.; Springer: New York, NY, USA; Singapore, 2006; Volume 6, pp. 495–507. ISBN 978-0-387-25496-8. [Google Scholar]
- Davis, B.R.; Fanning, R.; Madden, J.M.; Steigerwalt, A.G.; Bradford, H.B., Jr.; Smith, H.L., Jr.; Brenner, D.J. Characterization of biochemically atypical vibrio cholerae strains and designation of a new pathogenic species, Vibrio mimicus. J. Clin. Microbiol. 1981, 14, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Gil, B.; Thompson, C.C.; Matsumura, Y.; Sawabe, T.; Iida, T.; Christen, R.; Thompson, F.; Sawabe, T. The famlily Vibrionaceae. In The Prokaryotes; Springer: Berlin/Heidelberg, Germany, 2014; pp. 659–747. [Google Scholar]
- Tercero-Alburo, J.J.; González-Márquez, H.; Bonilla-González, E.; Quiñones-Ramírez, E.I.; Vázquez-Salinas, C. Identification of capsule, biofilm, lateral flagellum, and type IV pili in Vibrio mimicus strains. Microb. Pathog. 2014, 76, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Hasan, N.A.; Grim, C.J.; Haley, B.J.; Chun, J.; Alam, M.; Taviani, E.; Hoq, M.; Munk, A.C.; Saunders, E.; Brettin, T.S.; et al. Comparative genomics of clinical and environmental Vibrio mimicus. Proc. Natl. Acad. Sci. USA 2010, 107, 21134–21139. [Google Scholar] [CrossRef]
- Guardiola-Avila, I.; Acedo Félix, E.; Noriega-Orozco, L.; Yepiz-Plascencia, G.; Sifuentes-Romero, I.; Gomez-Gil, B. Draft genome sequence of Vibrio mimicus strain CAIM 602T. Genome Announc. 2013, 1, 3–4. [Google Scholar] [CrossRef]
- Yu, Z.; Wang, E.; Geng, Y.; Wang, K.; Chen, D.; Huang, X.; Ouyang, P.; Zuo, Z.; Huang, C.; Fang, J.; et al. Complete genome analysis of Vibrio mimicus strain SCCF01, a highly virulent isolate from the freshwater catfish. Virulence 2020, 11, 23–31. [Google Scholar] [CrossRef]
- Thompson, C.C.; Vicente, A.C.P.; Souza, R.C.; Vasconcelos, A.T.R.; Vesth, T.; Alves, N.; Ussery, D.W.; Iida, T.; Thompson, F.L. Genomic taxonomy of vibrios. BMC Evol. Biol. 2009, 9, 258. [Google Scholar] [CrossRef]
- Lilburn, T.G.; Gu, J.; Cai, H.; Wang, Y. Comparative genomics of the family Vibrionaceae reveals the wide distribution of genes encoding virulence-associated proteins. BMC Genomics 2010, 11. [Google Scholar] [CrossRef]
- Kahlke, T.; Goesmann, A.; Hjerde, E.; Willassen, N.P.; Haugen, P. Unique core genomes of the bacterial family vibrionaceae: Insights into niche adaptation and speciation. BMC Genomics 2012, 13. [Google Scholar] [CrossRef]
- Busschaert, P.; Frans, I.; Crauwels, S.; Zhu, B.; Willems, K.; Bossier, P.; Michiels, C.; Verstrepen, K.; Lievens, B.; Rediers, H. Comparative genome sequencing to assess the genetic diversity and virulence attributes of 15 Vibrio anguillarum isolates. J. Fish. Dis. 2015, 38, 795–807. [Google Scholar] [CrossRef]
- Ceccarelli, D.; Amaro, C.; Romalde, J.L.; Suffredini, E.; Vezzulli, L. Vibrio Species; Wiley: Hoboken, NJ, USA, 2019; ISBN 9781555819972. [Google Scholar]
- Wang, D.; Wang, H.; Zhou, Y.; Zhang, Q.; Zhang, F.; Du, P.; Wang, S.; Chen, C.; Kan, B. Genome sequencing reveals unique mutations in characteristic metabolic pathways and the transfer of virulence genes between V. mimicus and V. cholerae. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Marin, M.A.; Vicente, A.C.P. Architecture of the superintegron in Vibrio cholerae: Identification of core and unique genes. F1000 Res. 2013, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Haley, B.J.; Grim, C.J.; Hasan, N.A.; Choi, S.-Y.; Chun, J.; Brettin, T.S.; Bruce, D.C.; Challacombe, J.F.; Detter, J.C.; Han, C.S.; et al. Comparative genomic analysis reveals evidence of two novel Vibrio species closely related to V. cholerae. BMC Microbiol. 2010, 10, 154. [Google Scholar] [CrossRef] [PubMed]
- Vieira, V.V.; Teixeira, L.F.M.; Vicente, A.C.P.; Momen, H.; Salles, C.A. Differentiation of environmental and clinical isolates of Vibrio mimicus from Vibrio cholerae by multilocus enzyme electrophoresis. Appl. Environ. Microbiol. 2001, 67, 2360–2364. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Orata, F.D.; Kirchberger, P.C.; Méheust, R.; Barlow, E.J.; Tarr, C.L.; Boucher, Y. The dynamics of genetic interactions between Vibrio metoecus and Vibrio cholerae, two close relatives co-occurring in the environment. Genome Biol. Evol. 2015, 7, 2941–2954. [Google Scholar] [CrossRef]
- Kirchberger, P.C.; Turnsek, M.; Hunt, D.E.; Haley, B.J.; Colwell, R.R.; Polz, M.F.; Tarr, C.L.; Boucher, Y. Vibrio metoecus sp. nov., A close relative of Vibrio cholerae isolated from coastal brackish ponds and clinical specimens. Int. J. Syst. Evol. Microbiol. 2014, 64, 3208–3214. [Google Scholar] [CrossRef]
- Guardiola-Avila, I.; Acedo-Felix, E.; Sifuentes-Romero, I.; Yepiz-Plascencia, G.; Gomez-Gil, B.; Noriega-Orozco, L. Molecular and genomic characterization of Vibrio mimicus isolated from a frozen shrimp processing facility in Mexico. PLoS ONE 2016, 11, e0144885. [Google Scholar] [CrossRef]
- Guardiola-Avila, I.; Noriega-Orozco, L.; Gomez-Gil, B.; Acedo-Felix, E. Factores de virulencia de Vibrio mimicus—Vibrio mimicus Virulence factors. Biotecnia 2015, 17, 38–49. [Google Scholar] [CrossRef]
- Lin, H.; Yu, M.; Wang, X.; Zhang, X.H. Comparative genomic analysis reveals the evolution and environmental adaptation strategies of vibrios. BMC Genom. 2018, 19. [Google Scholar] [CrossRef]
- Vesth, T.; Lagesen, K.; Acar, Ö.; Ussery, D. CMG-biotools, a free workbench for basic comparative microbial genomics. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Sjölander, K. Phylogenomic inference of protein molecular function: Advances and challenges. Bioinformatics 2004, 20, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Prakash, O.; Verma, M.; Sharma, P.; Kumar, M.; Kumari, K.; Singh, A.; Kumari, H.; Jit, S.; Gupta, S.K.; Khanna, M.; et al. Polyphasic approach of bacterial classification—An overview of recent advances. Ind. J. Microbiol. 2007, 47, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Zeb, S.; Gulfam, S.M.; Bokhari, H. Comparative core/pan genome analysis of Vibrio cholerae isolates from Pakistan. Infect. Genet. Evol. 2020, 82, 104316. [Google Scholar] [CrossRef] [PubMed]
- Heidelberg, J.F.; Eisen, J.A.; Nelson, W.C.; Clayton, R.A.; Gwinn, M.L.; Dodson, R.J.; Haft, D.H.; Hickey, E.K.; Peterson, J.D.; Umayam, L.; et al. DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nat. Cell Biol. 2000, 406, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Reeves, P.R.; Lan, R.; Ren, Y.; Gao, C.; Zhou, Z.; Ren, Y.; Cheng, J.; Wang, W.; Wang, J.; et al. A recalibrated molecular clock and independent origins for the cholera pandemic clones. PLoS ONE 2008, 3. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef]
- Darling, A.E.; Mau, B.; Perna, N.T. Progressivemauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5. [Google Scholar] [CrossRef]
- Sánchez-Busó, L.; Comas, I.; Jorques, G.; González-Candelas, F. Recombination drives genome evolution in outbreak-related Legionella pneumophila isolates. Nat. Genet. 2014, 46, 1205–1211. [Google Scholar] [CrossRef]
- R Team. CR Core Team R: A Language and Environment for Statistical Computing. Available online: http://www.R-project.org/.
- Pant, A.; Bag, S.; Saha, B.; Verma, J.; Kumar, P.; Banerjee, S.; Kumar, B.; Kumar, Y.; Desigamani, A.; Maiti, S.; et al. Molecular insights into the genome dynamics and interactions between core and acquired genomes of Vibrio cholerae. Proc. Natl. Acad. Sci. USA 2020, 117, 23762–23773. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef] [PubMed]
- Warnes, G.R.; Bolker, B.; Bonebakker, L.; Gentleman, R.; Huber, W. gplots: Various R Programming Tools for Plotting Data. 2013. Available online: https://cran.r-project.org/web/packages/gplots/index.html (accessed on 19 November 2018).
- Eren, A.M.; Esen, O.C.; Quince, C.; Vineis, J.H.; Morrison, H.G.; Sogin, M.L.; Delmont, T.O. Anvi’o: An advanced analysis and visualization platformfor ’omics data. Peer J. 2015, 2015, e1319. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Monday, S.R.; Allard, M.W.; Strain, E.A.; Whittaker, P.; Naum, M.; Mccarthy, P.J.; Lopez, J.V.; Fischer, M.; Brown, E.W. Vibrio caribbeanicus sp. nov., isolated from the marine sponge Scleritoderma cyanea. Int. J. Syst. Evol. Microbiol. 2012, 62, 1736–1743. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the Gateway Computing Environments Workshop, New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar]
- Sukumaran, J.; Holder, M.T. DendroPy: A Python library for phylogenetic computing. Bioinformatics 2010, 26, 1569–1571. [Google Scholar] [CrossRef] [PubMed]
- Kimes, N.E.; Grim, C.J.; Johnson, W.R.; Hasan, N.A.; Tall, B.D.; Kothary, M.H.; Kiss, H.; Munk, A.C.; Tapia, R.; Green, L.; et al. Temperature regulation of virulence factors in the pathogen Vibrio coralliilyticus. ISME J. 2011, 6, 835–846. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V. Comparative genomics, minimal gene-sets and the last universal common ancestor. Nat. Rev. Microbiol. 2003, 1, 127–136. [Google Scholar] [CrossRef]
- Petronella, N.; Kundra, P.; Auclair, O.; Hébert, K.; Rao, M.; Kingsley, K.; De Bruyne, K.; Banerjee, S.; Gill, A.; Pagotto, F.; et al. Changes detected in the genome sequences of Escherichia coli, Listeria monocytogenes, Vibrio parahaemolyticus, and Salmonella enterica after serial subculturing. Can. J. Microbiol. 2019, 65, 842–850. [Google Scholar] [CrossRef]
- Zeigler, D.R. Gene sequences useful for predicting relatedness of whole genomes in bacteria. Int. J. Syst. Evol. Microbiol. 2003, 53, 1893–1900. [Google Scholar] [CrossRef]
- Sawabe, T.; Kita-Tsukamoto, K.; Thompson, F.L. Inferring the evolutionary history of vibrios by means of multilocus sequence analysis. J. Bacteriol. 2007, 189, 7932–7936. [Google Scholar] [CrossRef]
- Sawabe, T.; Ogura, Y.; Matsumura, Y.; Feng, G.; Rohul Amin, A.K.M.; Mino, S.; Nakagawa, S.; Sawabe, T.; Kumar, R.; Fukui, Y.; et al. Updating the Vibrio clades defined by multilocus sequence phylogeny: Proposal of eight new clades, and the description of Vibrio tritonius sp. nov. Front. Microbiol. 2013, 4, 1–14. [Google Scholar] [CrossRef]
- Wassenaar, T.M.; Gaastra, W. Bacterial virulence: Can we draw the line? FEMS Microbiol. Lett. 2001, 201, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Neogi, S.B.; Chowdhury, N.; Awasthi, S.P.; Asakura, M.; Okuno, K.; Mahmud, Z.H.; Islam, M.S.; Hinenoya, A.; Nair, G.B.; Yamasaki, S. Novel cholera toxin variant and ToxT regulon in environmental Vibrio mimicus isolates: Potential resources for the evolution of Vibrio cholerae hybrid strains. Appl. Environ. Microbiol. 2019, 85, 1–20. [Google Scholar] [CrossRef]
- Tagomori, K.; Iida, T.; Honda, T. Comparison of genome structures of vibrios, bacteria possessing two chromosomes. J. Bacteriol. 2002, 184, 4351–4358. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).