Self-Crossing Leads to Weak Co-Variation of the Bacterial and Fungal Communities in the Rice Rhizosphere

 , , and
, , and 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
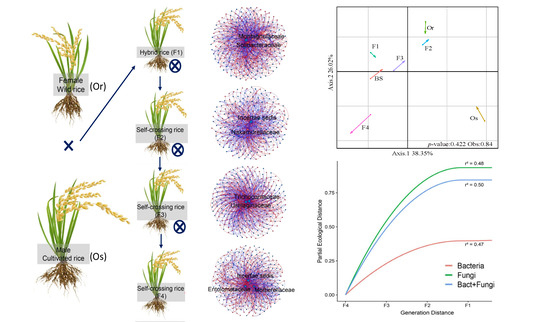
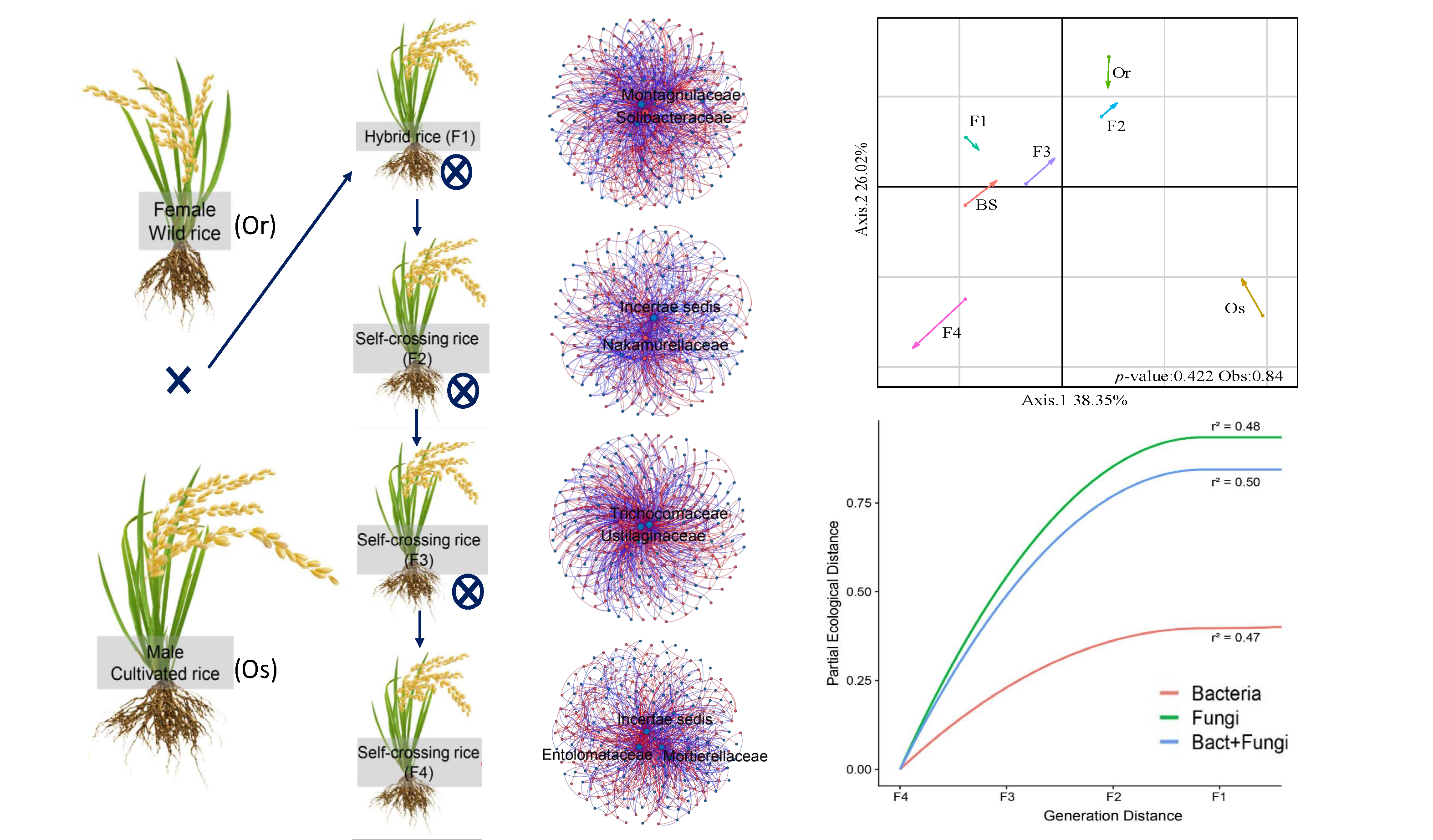
2.1. Plant Material and Rhizosphere Samples
2.2. Measurements of Soil Physical and Chemical Properties
2.3. DNA Extraction and Amplicon Sequencing
2.4. Bioinformatics and Statistical Analysis
3. Results
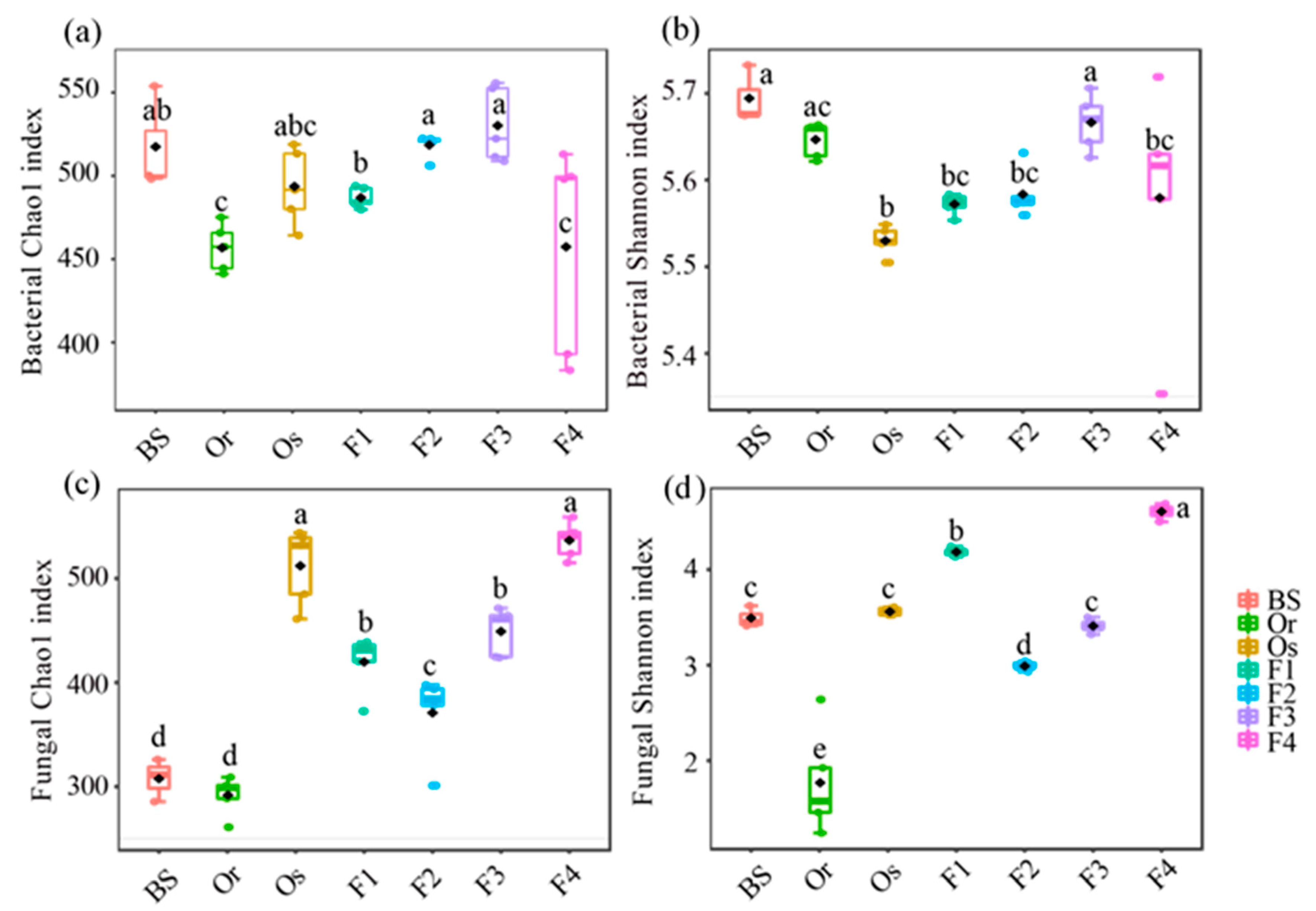
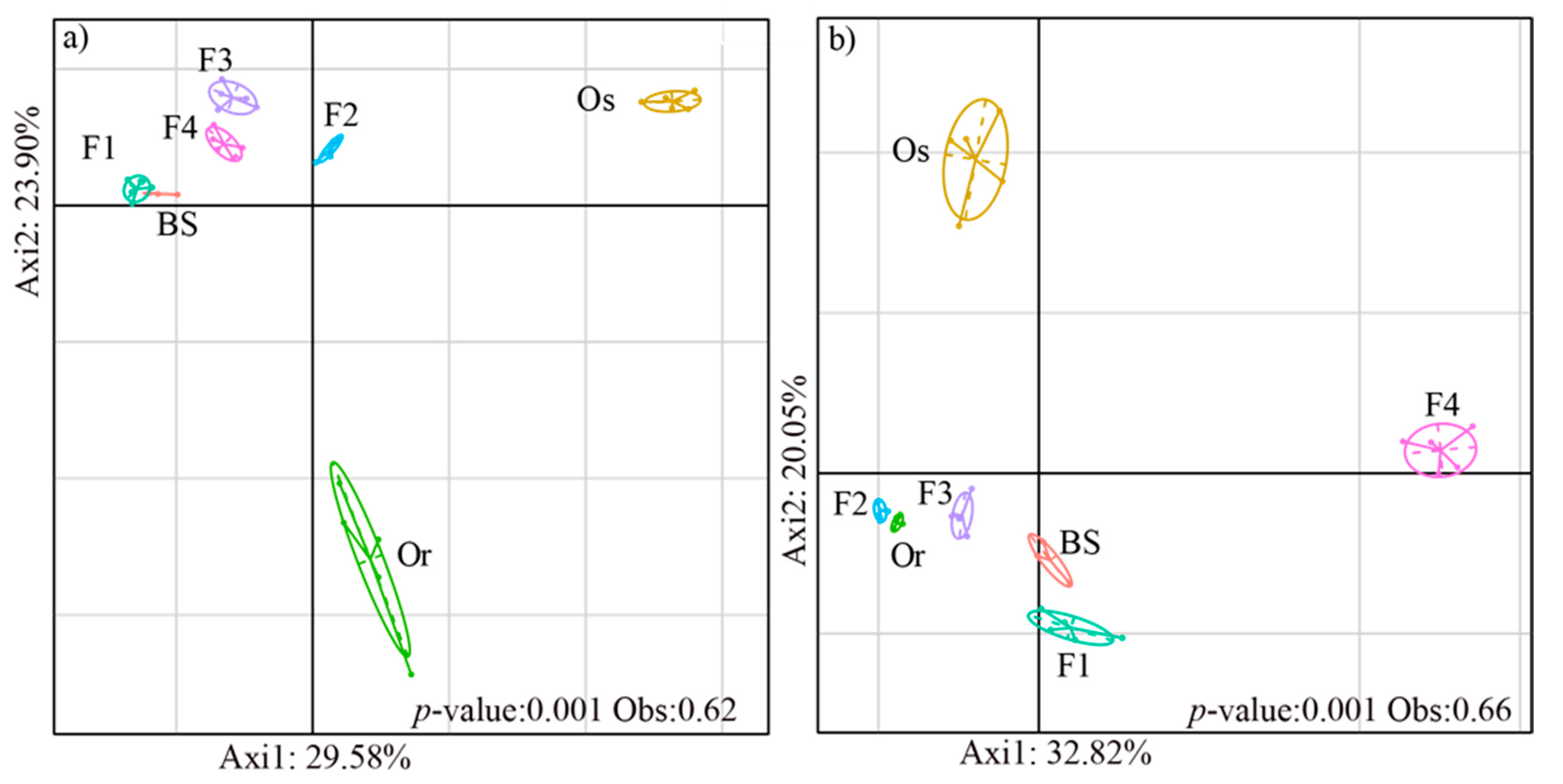
3.1. Structures of Bacterial and Fungal Communities in Different Generations of Rice Progenies Rhizosphere
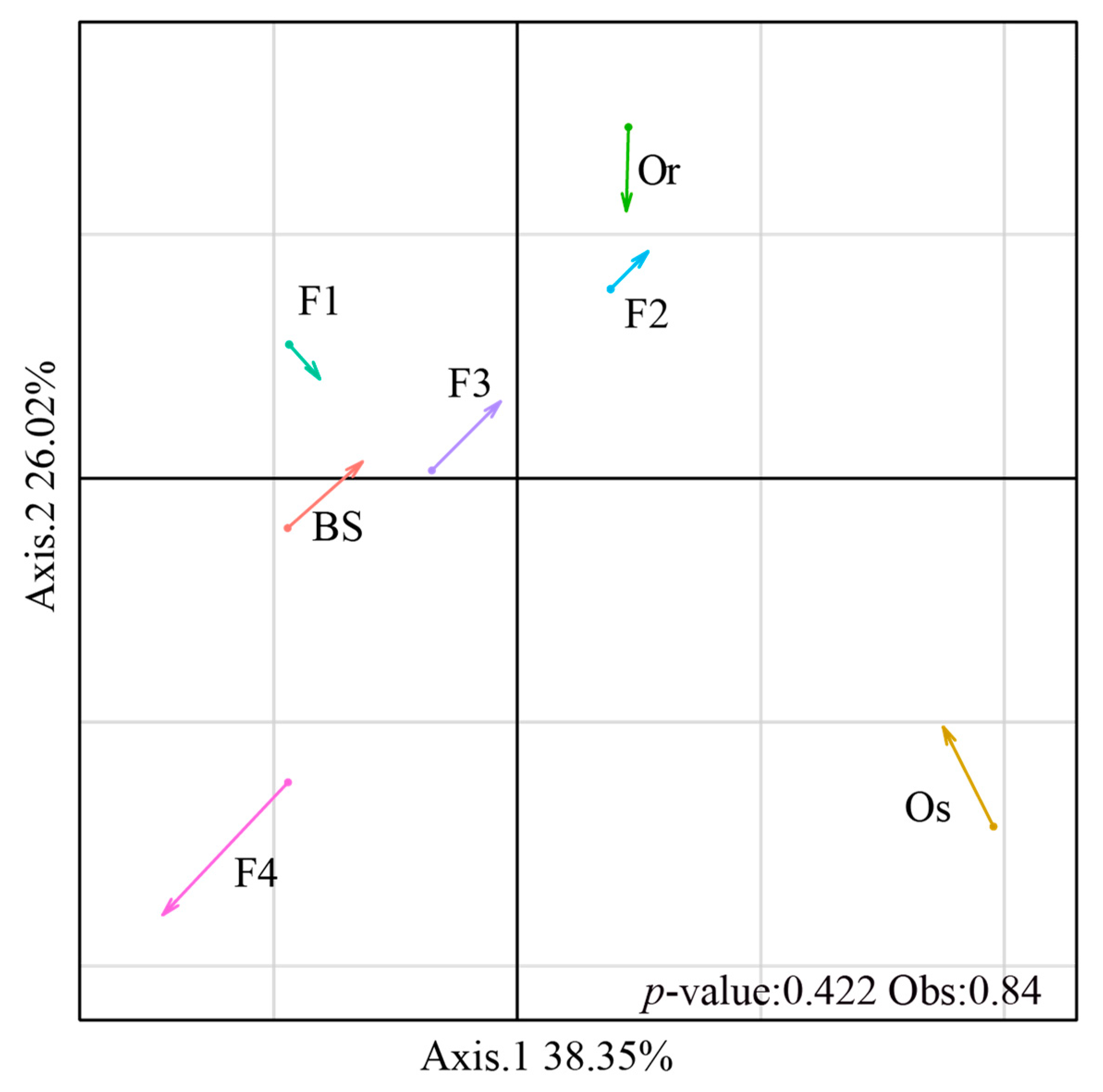
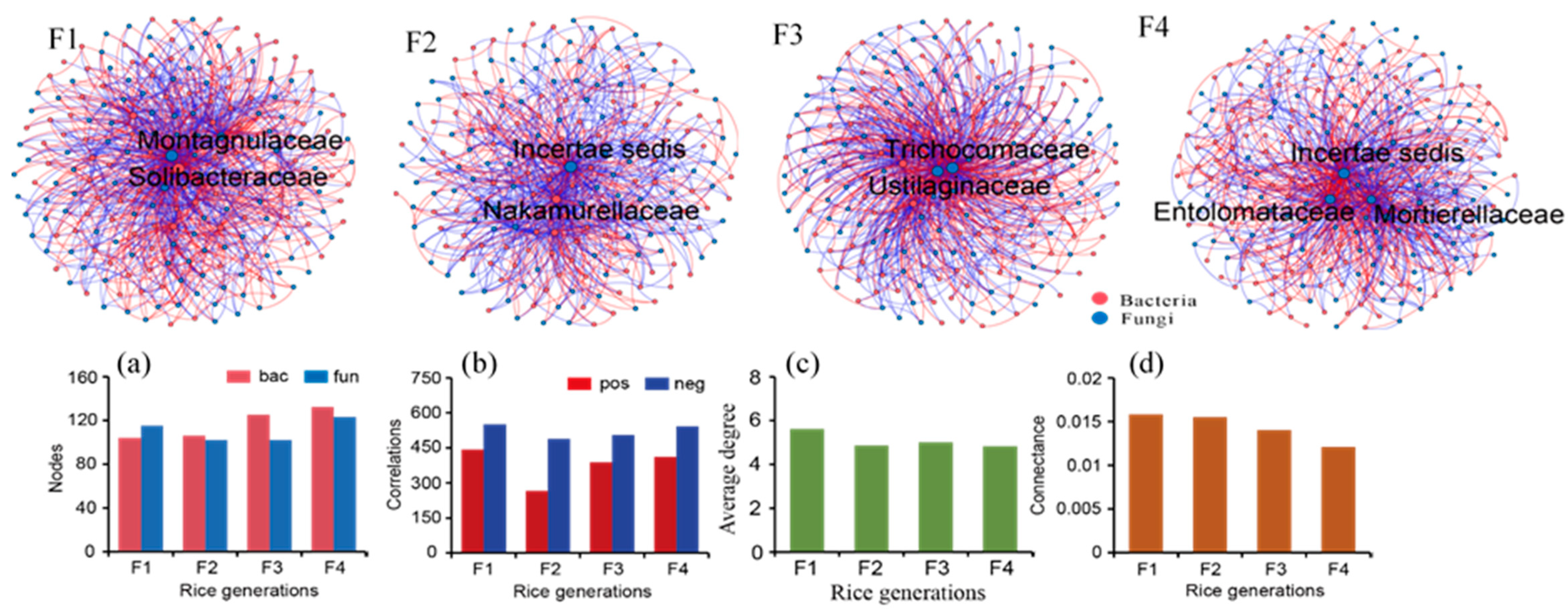
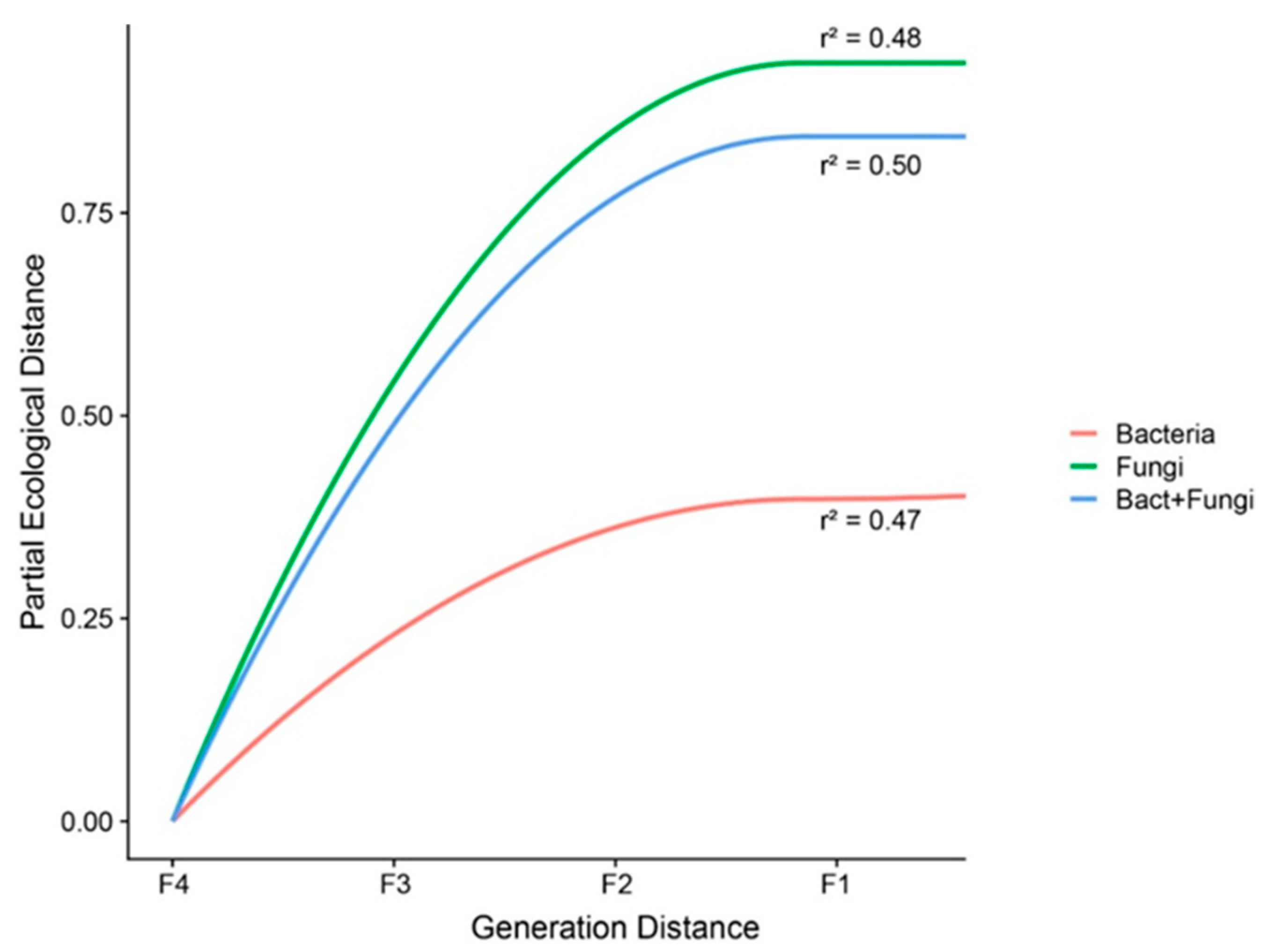
3.2. Co-Assembly of Bacterial and Fungal Communities in Different Generations of Rice Progenies Rhizosphere
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Elert, E. Rice by the numbers: A good grain. Nature 2014, 514, 50–51. [Google Scholar] [CrossRef] [PubMed]
- FAO. FAOSTAT: Food and Agriculture Data. 2020. Available online: http://www.fao.org/faostat/en/#data/QC (accessed on 20 September 2020).
- Grant, V.; Oka, H.I. Origin of cultivated rice. Taxon 1988, 37, 935. [Google Scholar] [CrossRef]
- Bin Rahman, A.N.M.; Zhang, J. Flood and drought tolerance in rice: Opposite but may coexist. Food Energy Secur. 2016, 5, 76–88. [Google Scholar] [CrossRef]
- Ji, H.; Kim, S.R.; Kim, Y.H.; Sun, J.P.; Park, H.M.; Sreenivasulu, N.; Misra, G.; Kim, S.M.; Hechanova, S.L.; Kim, H.; et al. Map-based cloning and characterization of the BPH18 gene from wild rice conferring resistance to brown planthopper (BPH) insect pest. Sci. Rep.-UK 2016, 6, 34376. [Google Scholar] [CrossRef] [PubMed]
- Bulgarelli, D.; Schlaeppi, K.; Spaepen, S.; Themaat, E.V.L.V.; Schulze-Lefert, P. Structure and functions of the bacterial microbiota of plants. Annu. Rev. Plant. Biol. 2013, 64, 807–838. [Google Scholar] [CrossRef] [PubMed]
- Lareen, A.; Burton, F.; Schäfer, P. Plant root-microbe communication in shaping root microbiomes. Plant Mol. Biol. 2016, 90, 575–587. [Google Scholar] [CrossRef]
- Stringlis, I.A.; Yu, K.; Feussner, K.; de Jonge, R.; Van Bentum, S.; Van Verk, M.C.; Berendsen, R.L.; Bakker, P.; Feussner, I.; Pieterse, C.M.J. MYB72-dependent coumarin exudation shapes root microbiome assembly to promote plant health. Proc. Natl. Acad. Sci. USA 2018, 115, 5213–5222. [Google Scholar] [CrossRef]
- Tyc, O.; Song, C.; Dickschat, J.S.; Vos, M.; Garbeva, P. The ecological role of volatile and soluble secondary metabolites produced by soil bacteria. Trends Microbiol. 2017, 25, 280–292. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Y.X.; Zhang, N.; Hu, B.; Jin, T.; Xu, H.X.; Qin, Y.; Yan, P.X.; Zhang, X.N.; Guo, X.X.; et al. NRT1. 1B is associated with root microbiota composition and nitrogen use in field-grown rice. Nat. Biotechnol. 2019, 37, 676–684. [Google Scholar] [CrossRef]
- Li, X.G.; Jousset, A.; De-Boer, W.; Carrion, V.; Zhang, T.L.; Wang, X.X.; Kuramae, E.E. Legacy of land use history determines reprogramming of plant physiology by soil microbiome. ISME J. 2019, 13, 738–751. [Google Scholar] [CrossRef]
- Pascale, A.; Proietti, S.; Pantelides, I.S.; Stringlis, I. Modulation of the root microbiome by plant molecules: The basis for targeted disease suppression and plant growth promotion. Front. Plant. Sci. 2020, 10, 1741. [Google Scholar] [CrossRef] [PubMed]
- Schlemper, T.R.; Leite, M.F.A.; Lucheta, A.R.; Shimels, M.; Bouwmeester, H.J.; van Veen, J.A.; Kuramae, E.E. Rhizobacterial community structure differences among sorghum cultivars in different growth stages and soils. FEMS Microbiol. Ecol. 2017, 93, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Beckers, B.; De Beeck, M.O.; Weyens, N.; Van, A.R.; Van, M.M.; Boerjan, W.; Vangronsveld, J. Lignin engineering in field-grown poplar trees affects the endosphere bacterial microbiome. Proc. Natl. Acad. Sci. USA 2016, 113, 2312–2317. [Google Scholar] [CrossRef]
- Hu, L.; Robert, C.A.M.; Cadot, S.; Zhang, X.; Ye, M.; Li, B.; Manzo, D.; Chervet, N.; Steinger, T.; van-der-Heijden, M.G.A.; et al. Root exudate metabolites drive plant-soil feedbacks on growth and defense by shaping the rhizosphere microbiota. Nat. Commun. 2018, 9, 2738. [Google Scholar] [CrossRef] [PubMed]
- Mitter, B.; Brader, G.; Pfaffenbichler, N.; Sessitsch, A. Next generation microbiome applications for crop production-limitations and the need of knowledge-based solutions. Curr. Opin. Microbiol. 2019, 49, 59–65. [Google Scholar] [CrossRef]
- Rolfe, S.A.; Griffiths, J.; Ton, J. Crying out for help with root exudates: Adaptive mechanisms by which stressed plants assemble health-promoting soil microbiomes. Curr. Opin. Microbiol. 2019, 49, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Mendes, L.W.; Kuramae, E.E.; Navarrete, A.A.; Veen, J.A.V.; Tsai, S.M. Taxonomical and functional microbial community selection in soybean rhizosphere. ISME J. 2014, 8, 1577–1587. [Google Scholar] [CrossRef]
- Tian, L.; Zhou, X.; Ma, L.; Xu, S.Q.; Nasir, F.; Tian, C.J. Root-associated bacterial diversities of Oryza rufipogon and Oryza sativa and their influencing environmental factors. Arch. Microbiol. 2017, 199, 563–571. [Google Scholar] [CrossRef]
- Shenton, M.; Iwamoto, C.; Kurata, N.; Ikeo, K. Effect of wild and cultivated rice genotypes on rhizosphere bacterial community composition. Rice 2016, 9, 42. [Google Scholar] [CrossRef]
- Yu, Y.; Woo, M.O.; Rihua, P.; Koh, H.J. The drooping leaf (DR) gene encoding GDSL esterase is involved in silica deposition in rice (Oryza sativa L.). PLoS ONE 2020, 15, e0238887. [Google Scholar] [CrossRef]
- Nasir, F.; Tian, L.; Shi, S.; Bahadur, A.; Batool, A.; Ma, L.N. Asian cultivated rice domestication suppresses the expression of abiotic stress- and reactive oxygen species scavening-related genes in roots. Pak. J. Bot. 2019, 51, 49–54. [Google Scholar] [CrossRef]
- Tian, L.; Shi, S.; Ma, L.; Nasir, F.; Li, X.; Phan Tran, L.S. Co-evolutionary associations between root-associated microbiomes and root transcriptomes in wild and cultivated rice varieties. Plant Physiol. Biochem. 2018, 128, 134–141. [Google Scholar] [CrossRef]
- Shi, S.; Chang, J.; Tian, L.; Nasir, F.; Ji, L.; Li, X.; Tian, C. Comparative analysis of the rhizomicrobiome of the wild versus cultivated crop: Insights from rice and soybean. Arch. Microbiol. 2019, 201, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Raju, B.R.; Narayanaswamy, B.R.; Mohankumar, M.V.; Sumanth, K.K.; Rajanna, M.P.; Mohanraju, B.; Udayakumar, M. Root traits and cellular level tolerance hold the key in maintaining higher spikelet fertility of rice under water limited conditions. Funct. Plant Biol. 2014, 41, 930–939. [Google Scholar] [CrossRef]
- Miyamoto, K.; Fujita, M.; Shenton, M.R.; Akashi, S.; Sugawara, C.; Sakai, A.; Horie, K.; Hasegawa, M.; Kawaide, H.; Mitsuhashi, W.; et al. Evolutionary trajectory of phytoalexin biosynthetic gene clusters in rice. Plant J. 2016, 87, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Tian, L.; Chang, C.; Li, X.J.; Tian, C.J. Cultivated rice rhizomicrobiome is more sensitive to environmental shifts than that of wild rice in natural environments. Appl. Soil Ecol. 2019, 140, 68–77. [Google Scholar] [CrossRef]
- Effmert, U.; Kalderás, J.; Warnke, R.; Piechulla, B. Volatile mediated interactions between bacteria and fungi in the soil. J. Chem. Ecol. 2012, 38, 665–703. [Google Scholar] [CrossRef]
- Zhang, S.X.; Huang, D.X.; Yi, X.Y.; Zhang, S.; Yao, R.; Li, C.G.; Liang, A.; Zhang, X.P. Rice yield corresponding to the seedling growth under supplemental green light in mixed light-emitting diodes. Plant Soil Environ. 2016, 62, 222–229. [Google Scholar] [CrossRef]
- Chang, C.; Chen, W.; Luo, S.S.; Ma, L.N.; Li, X.J.; Tian, C.J. Rhizosphere microbiota assemblage associated with wild and cultivated soybeans grown in three types of soil suspensions. Arch. Agrono. Soil Sci. 2019, 65, 74–87. [Google Scholar] [CrossRef]
- Luo, S.; Tian, L.; Chang, C.; Wang, S.; Zhang, J.; Zhou, X.; Li, X.; Tran, L.S.; Tian, C. Grass and maize vegetation systems restore saline-sodic soils in the Songnen Plain of Northeast China. Land Degrad. Dev. 2018, 29, 1107–1119. [Google Scholar] [CrossRef]
- Van Bemmelen, J. Über die Bestimmung des Wassers, des Humus, des Schwefels, der in den colloïdalen Silikaten gebundenen Kieselsäure, des Mangans usw im Ackerboden. Landwirthschaftlichen Vers-Stn. 1890, 37, e290. [Google Scholar]
- Pribyl, D.W. A critical review of the conventional SOC to SOM conversion factor. Geoderma 2010, 156, 75–83. [Google Scholar] [CrossRef]
- Lao, J.C. Handbook of Soil Chemical Analysis; Agriculture Press: Beijing, China, 1988; pp. 120–124. [Google Scholar]
- Yadav, R.L.; Dwivedi, B.S.; Prasad, K.; Tomar, O.K.; Shurpali, N.J.; Pandey, P.S. Yield trends, and changes in soil organic-C and available NPK in a long-term rice-wheat system under integrated use of manures and fertilisers. Field Crop Res. 2000, 68, 219–246. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; BergLyons, D.; Huntley, J.; Fierer, N.; Owens, S.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [PubMed]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Kõljalg, U.; Nilsson, R.H.; Abarenkov, K. Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 2013, 22, 5271–5277. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Gregory Caporaso, J. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.L.; Agellon, L.B.; Xia, J.G. MicrobiomeAnalyst: A web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017, 45, 180–188. [Google Scholar] [CrossRef]
- Chao, A.; Shen, T.J. Nonparametric prediction in species sampling. J. Agric. Biol. Environ. Stat. 2004, 9, 253–269. [Google Scholar] [CrossRef]
- Chang, J.J.; Zhu, J.X.; Xu, L.; Su, H.X.; Gao, Y.; Cai, X.L.; Peng, T.; Wen, X.F.; Zhang, J.J.; He, N.P. Rational land-use types in the karst regions of China: Insights from soil organic matter composition and stability. Catena 2018, 160, 345–353. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, 60–78. [Google Scholar] [CrossRef] [PubMed]
- Legendre, P.; Gallagher, E. Ecologically meaningful transformations for ordination of species data. Oecologia 2001, 129, 271–280. [Google Scholar] [CrossRef]
- Schlemper, T.R.; van Veen, J.A.; Kuramae, E.E. Co-variation of bacterial and fungal communities in different sorghum cultivars and growth stages is soil dependent. Microb. Ecol. 2018, 76, 205–214. [Google Scholar] [CrossRef]
- Culhane, A.C.; Perrière, G.; Higgins, D.G. Cross-platform comparison and visualisation of gene expression data using co-inertia analysis. BMC Bioinform. 2003, 4, 59–74. [Google Scholar] [CrossRef]
- Friedman, J.; Alm, E.J. Inferring correlation networks from genomic survey data. PLoS Comput. Biol. 2012, 8, e1002687. [Google Scholar] [CrossRef]
- Warton, D.I.; Wright, S.T.; Wang, Y. Distance-based multivariate analyses confound location and dispersion effects. Methods Ecol. Evol. 2012, 3, 89–101. [Google Scholar] [CrossRef]
- Ferrier, S.; Manion, G.; Elith, J.; Richardson, K. Using generalized dissimilarity modelling to analyse and predict patterns of beta diversity in regional biodiversity assessment. Divers. Distrib. 2007, 13, 252–264. [Google Scholar] [CrossRef]
- Reiner, A.; Yekutieli, D.; Benjamini, Y. Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics 2003, 19, 368–375. [Google Scholar] [CrossRef]
- Peiffer, J.A.; Spor, A.; Koren, O.; Zhao, J. Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc. Natl. Acad. Sci. USA 2013, 110, 6548–6553. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Liu, X.; Zheng, J.; Zhang, B.; Lu, H.F.; Chi, Z.Z.; Pan, G.X.; Li, L.Q.; Zheng, J.F.; Zhang, X.H.; et al. Biochar soil amendment increased bacterial but decreased fungal gene abundance with shifts in community structure in a slightly acid rice paddy from Southwest China. Appl. Soil Ecol. 2013, 71, 33–44. [Google Scholar] [CrossRef]
- Guo, M.; Zhou, Q.; Zhou, Y.; Yang, L.; Liu, T.; Yang, J.; Chen, Y.; Su, L.; Xu, J.; Chen, J. Genomic evolution of 11 type strains within family Planctomycetaceae. PLoS ONE 2014, 9, e86752. [Google Scholar] [CrossRef]
- Rheims, H.; Schumann, P.; Rohde, M.; Stackbrandt, E. Verrucosispora gifhornensis gen. nov., a new member of the actinobacterial family Micromonosporaceae. Int. J. Syst. Bacteriol. 1998, 48, 1119–1127. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.Y.; Zhang, W.X. Analysis of microbial community structure in pit mud from two Chinese Luzhou-flavor liquor producing areas. Microbiol. China 2014, 41, 1498–1506. [Google Scholar]
- Normand, P.; Benson, D.R.; Berry, A.M.; Tisa, L.S. The family Frankiaceae. Prokaryotes 2014, 339–356. [Google Scholar] [CrossRef]
- Huhndorf, S.M.; Miller, A.N.; Fernández, F.A. Molecular systematics of the Sordariales: The order and the family Lasiosphaeriaceae redefined. Mycologia 2004, 96, 368–387. [Google Scholar] [CrossRef] [PubMed]
- Kruys, Å.; Huhndorf, S.M.; Miller, A.N. Coprophilous contributions to the phylogeny of Lasiosphaeriaceae and allied taxa within Sordariales (Ascomycota, Fungi). Fungal Divers. 2015, 70, 101–113. [Google Scholar] [CrossRef]
- Valenzuela-Lopez, N.; Cano-Lira, J.F.; Guarro, J.; Sutton, D.A.; Wiederhold, N.; Crous, P.W.; Stchigel, A.M. Coelomycetous Dothideomycetes with emphasis on the families Cucurbitariaceae and Didymellaceae. Stud. Mycol. 2018, 90, 1–69. [Google Scholar] [CrossRef]
- Kurihara, Y.; Degawa, Y. Pinnaticoemansia, a new genus of Kickxellales, with a revised key to the genera of Kickxellales. Mycoscience 2006, 47, 205–211. [Google Scholar] [CrossRef]
- Shawkat Ali, J.D.L.; Linning, R.; Cervantes-Chávez, J.A.; Denis, G.; Guus, B. An immunity-triggering effector from the barley smut fungus Ustilago hordei resides in an Ustilaginaceae-specific cluster bearing signs of transposable element-assisted evolution. PLoS Pathog. 2014, 10, e1004223. [Google Scholar] [CrossRef]
- Taylor, J.E.; Groenewald, J.Z.E.; Crous, P.W. A phylogenetic analysis of Mycosphaerellaceae leaf spot pathogens of Proteaceae. Mycol. Res. 2003, 107, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Harrell, E.A.; Miller, E.S. Genome sequence of Aeromicrobium erythreum NRRL B-3381, an erythromycin-producing bacterium of the Nocardioidaceae. Genome Announc. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Labeda, D.P.; Dunlap, C.A.; Rong, X.; Huang, Y.; Doroghazi, J.R.; Ju, K.S.; Metcalf, W.W. Phylogenetic relationships in the family Streptomycetaceae using multi-locus sequence analysis. Antonie Van Leeuwenhoek 2017, 110, 563–583. [Google Scholar] [CrossRef]
- Wüst, P.K.; Horn, M.A.; Drake, H.L. Clostridiaceae and Enterobacteriaceae as active fermenters in earthworm gut content. ISME J. 2011, 5, 92. [Google Scholar]
- Pérez-Jaramillo, J.E.; Carrión, V.J.; de-Hollander, M.; Raaijmakers, J.M. The wild side of plant microbiomes. Microbiome 2018, 6, 143–149. [Google Scholar] [CrossRef]
- Pérez-Jaramillo, J.E.; Carrión, V.J.; Bosse, M.; Ferrao, L.F.V.; de-Hollander, M.; Garcia, A.A.F.; Ramirez, C.A.; Mendes, R.; Raaijmakers, J.M. Linking rhizosphere microbiome composition of wild and domesticated Phaseolus vulgaris to genotypic and root phenotypic traits. ISME J. 2017, 11, 2244–2257. [Google Scholar] [CrossRef]
- Pérez-Jaramillo, J.E.; de-Hollander, M.; Ramirez, C.A.; Mendes, R.; Raaijmakers, J.M. Deciphering rhizosphere microbiome assembly of wild and modern common bean (Phaseolus vulgaris) in native and agricultural soils from Colombia. Microbiome 2019, 7, 114–130. [Google Scholar] [CrossRef]
- Cardinale, M.; Grube, M.; Erlacher, A.; Quehenberger, J.; Berg, G. Bacterial networks and co-occurrence relationships in the lettuce root microbiota. Environ. Microbiol. 2015, 17, 239–252. [Google Scholar] [CrossRef]
- Leff, J.W.; Lynch, R.C.; Kane, N.C.; Fierer, N. Plant domestication and the assembly of bacterial and fungal communities associated with strains of the common sunflower, Helianthus annuus. New Phytol. 2017, 214, 412–423. [Google Scholar] [CrossRef]
- Edwards, J.; Johnson, C.; Santos-Medellín, C.; Lurie, E.; Podishetty, N.K.; Bhatnagar, S.; Eisen, J.A.; Sundaresan, V. Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. USA 2015, 112, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.Q.; Tang, Y.L.; Lu, W.; Zhang, R.X. Comparison on some aspects of photosynthetic declination of wild rice and cultivated rices. Acta Agron. Sin. 2001, 4, 453–459. [Google Scholar]
- Mohammadkhani, N.; Heidari, R. Drought-induced accumulation of soluble sugars and proline in two maize varieties. World Appl. Sci. J. 2008, 3, 448–453. [Google Scholar]
- Chen, D.Z.; Xiao, Y.Q.; Zhao, S.X.; Pi, Y.H.; Xiong, H.J.; Luo, L.J. Genetic study on the cold tolerance of Dongxiang wild rice at the seedling stage. Acta Agric. Jiangxi 1997, 19, 56–59. [Google Scholar]
- Xiang, Y.; He, H.H.; Liu, Y.B.; Fu, J.R. Advances in research on cold tolerance in Dongxiang wild rice. Acta Agric. Univ. Jiangxiensis 2003, 4, 482–486. [Google Scholar]
- Grigulis, K.; Clément, J.C. Relative contributions of plant traits and soil microbial properties to mountain grassland ecosystem services. J. Ecol. 2013, 101, 47–57. [Google Scholar] [CrossRef]
- Faust, K.; Raes, J. Microbial interactions: From networks to models. Nat. Rev. Microbiol. 2012, 10, 538–550. [Google Scholar] [CrossRef]
- Jiang, Y.J.; Li, S.Z.; Li, R.P.; Zhang, J.; Liu, Y.H.; Lv, L.F.; Zhu, H.; Wu, W.l.; Li, W.L. Plant cultivars imprint the rhizosphere bacterial community composition and association networks. Soil Biol. Biochem. 2017, 109, 145–155. [Google Scholar] [CrossRef]
- Dray, S.; Thioulouse, C.J. Co-inertia analysis and the linking of ecological tables. Ecology 2003, 84, 3078–3089. [Google Scholar] [CrossRef]
- Rahman, A.U.; Shah, S.M.A.; Rahman, H.U.; Khalil, I.H.; Ismaeel, M.; Raza, M.A.; Khan, I.A. Genetic variability for morphological parameters in F2 segregating populations of rice. Pak. J. Bot. 2015, 47, 1759–1764. [Google Scholar]
- Khan, A.S.; Imran, M.; Ashfaq, M. Estimation of genetic variability and correlation for grain yield components in rice (Oryza sativa L.). Am.-Eurasian J. Agric. Environ. Sci. 2009, 6, 585–590. [Google Scholar]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, J.; Shi, S.; Tian, L.; Leite, M.F.A.; Chang, C.; Ji, L.; Ma, L.; Tian, C.; Kuramae, E.E. Self-Crossing Leads to Weak Co-Variation of the Bacterial and Fungal Communities in the Rice Rhizosphere. Microorganisms 2021, 9, 175. https://doi.org/10.3390/microorganisms9010175
Chang J, Shi S, Tian L, Leite MFA, Chang C, Ji L, Ma L, Tian C, Kuramae EE. Self-Crossing Leads to Weak Co-Variation of the Bacterial and Fungal Communities in the Rice Rhizosphere. Microorganisms. 2021; 9(1):175. https://doi.org/10.3390/microorganisms9010175
Chicago/Turabian StyleChang, Jingjing, Shaohua Shi, Lei Tian, Marcio F. A. Leite, Chunling Chang, Li Ji, Lina Ma, Chunjie Tian, and Eiko E. Kuramae. 2021. "Self-Crossing Leads to Weak Co-Variation of the Bacterial and Fungal Communities in the Rice Rhizosphere" Microorganisms 9, no. 1: 175. https://doi.org/10.3390/microorganisms9010175
APA StyleChang, J., Shi, S., Tian, L., Leite, M. F. A., Chang, C., Ji, L., Ma, L., Tian, C., & Kuramae, E. E. (2021). Self-Crossing Leads to Weak Co-Variation of the Bacterial and Fungal Communities in the Rice Rhizosphere. Microorganisms, 9(1), 175. https://doi.org/10.3390/microorganisms9010175