Modulating Kinetics of the Amyloid-Like Aggregation of S. aureus Phenol-Soluble Modulins by Changes in pH


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
3. Results
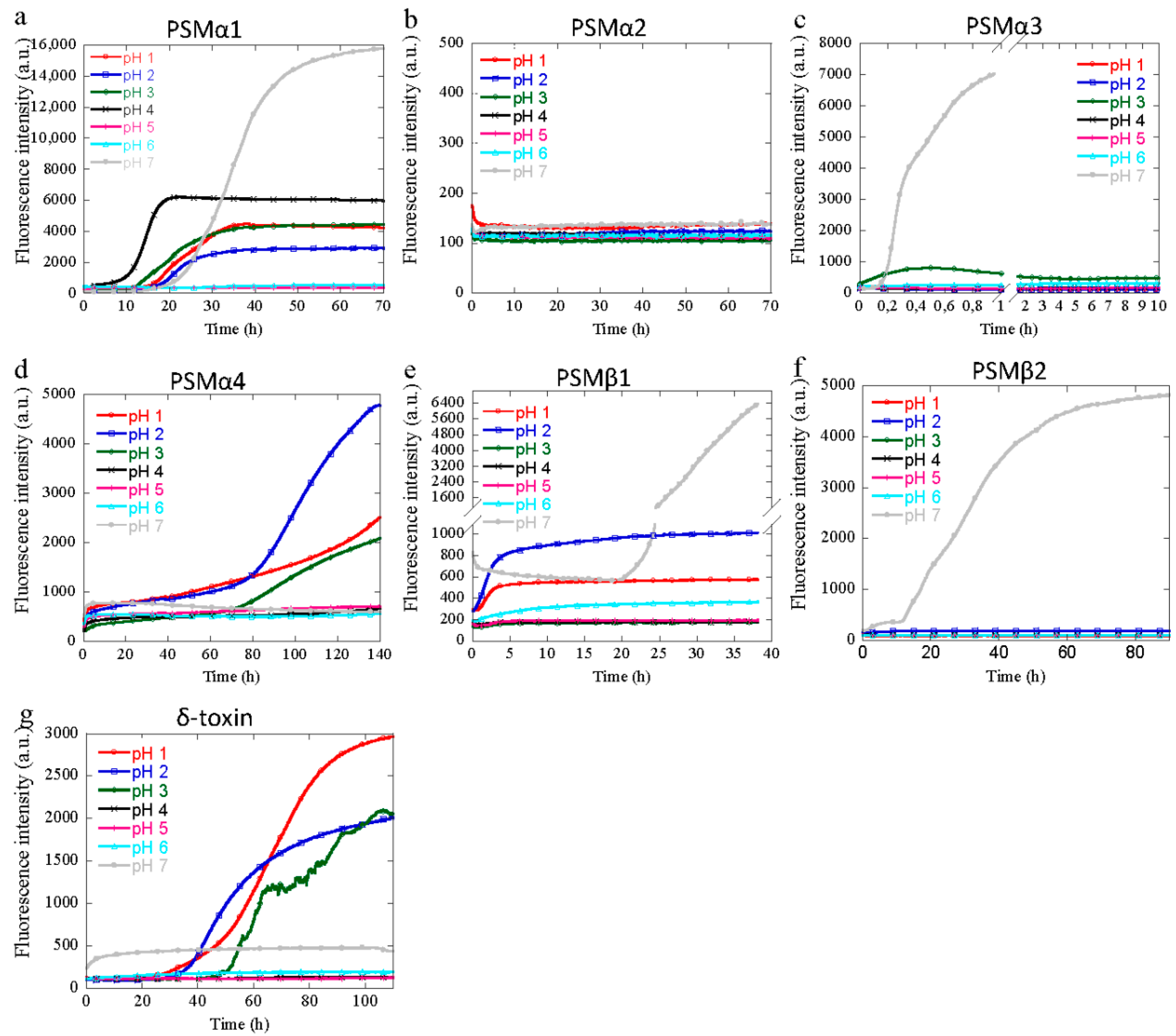
3.1. Aggregation of PSM Peptides at Acidic pH
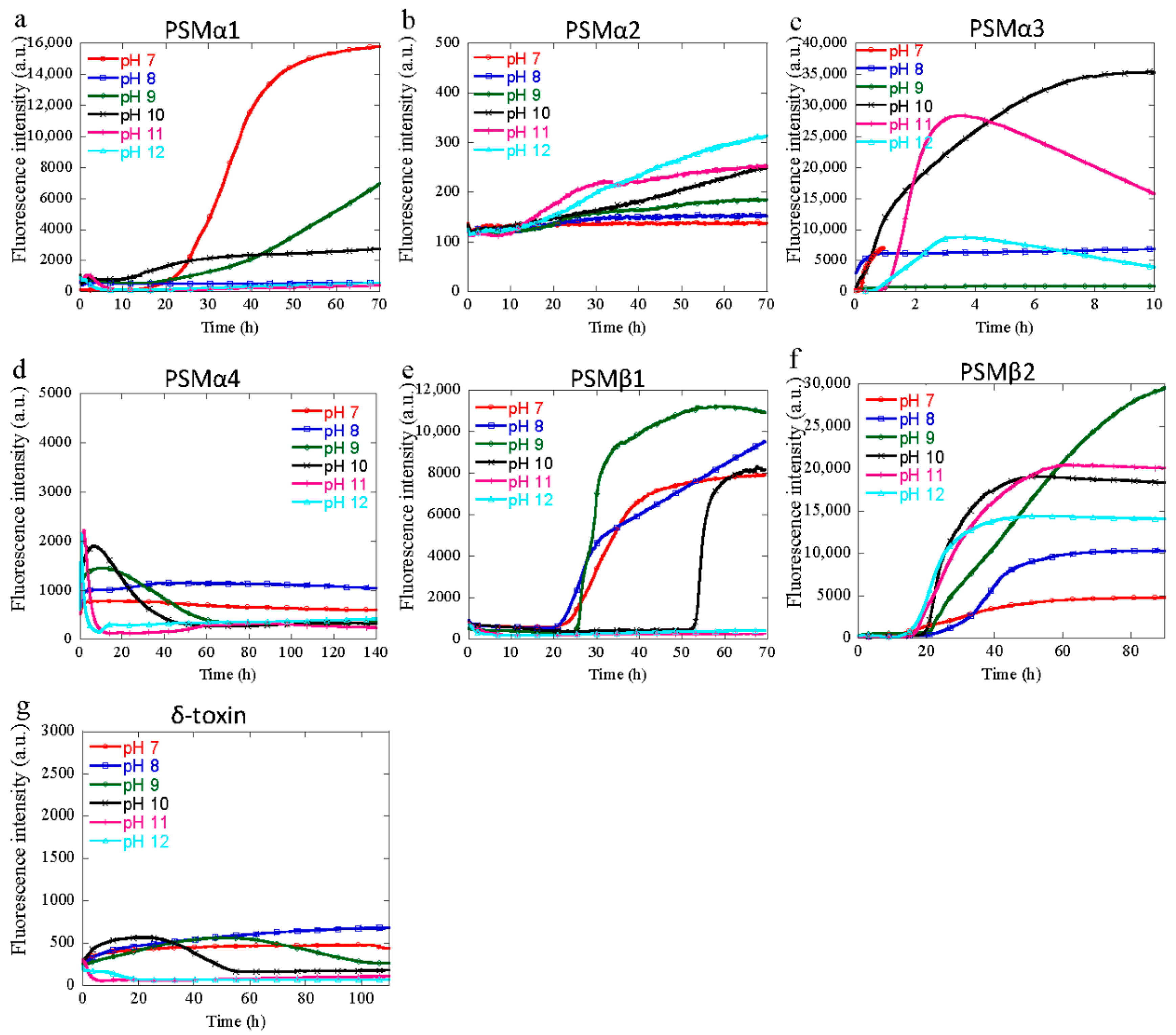
3.2. Aggregation of PSM Peptides at Basic pH
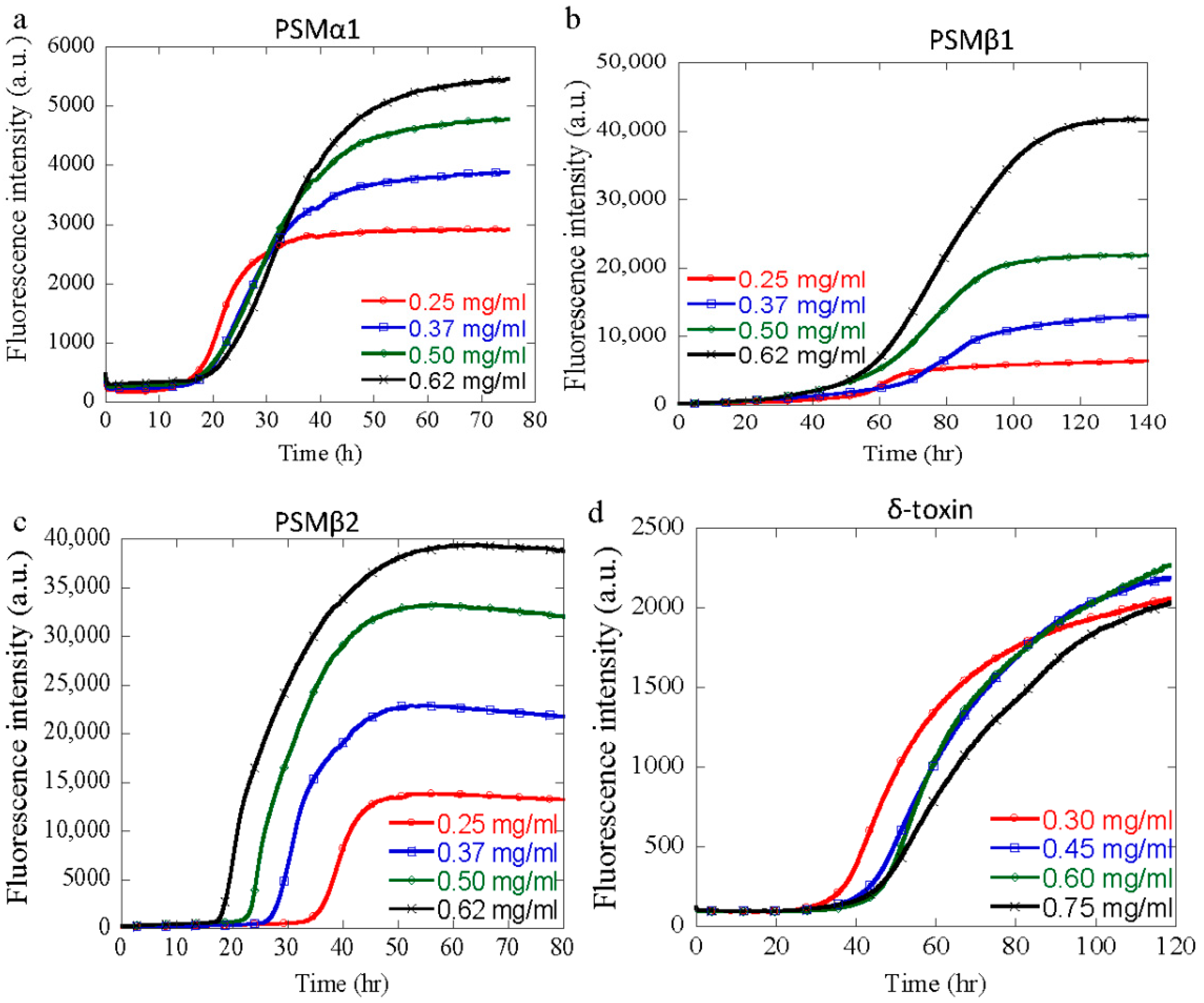
3.3. Effect of Peptide Concentration
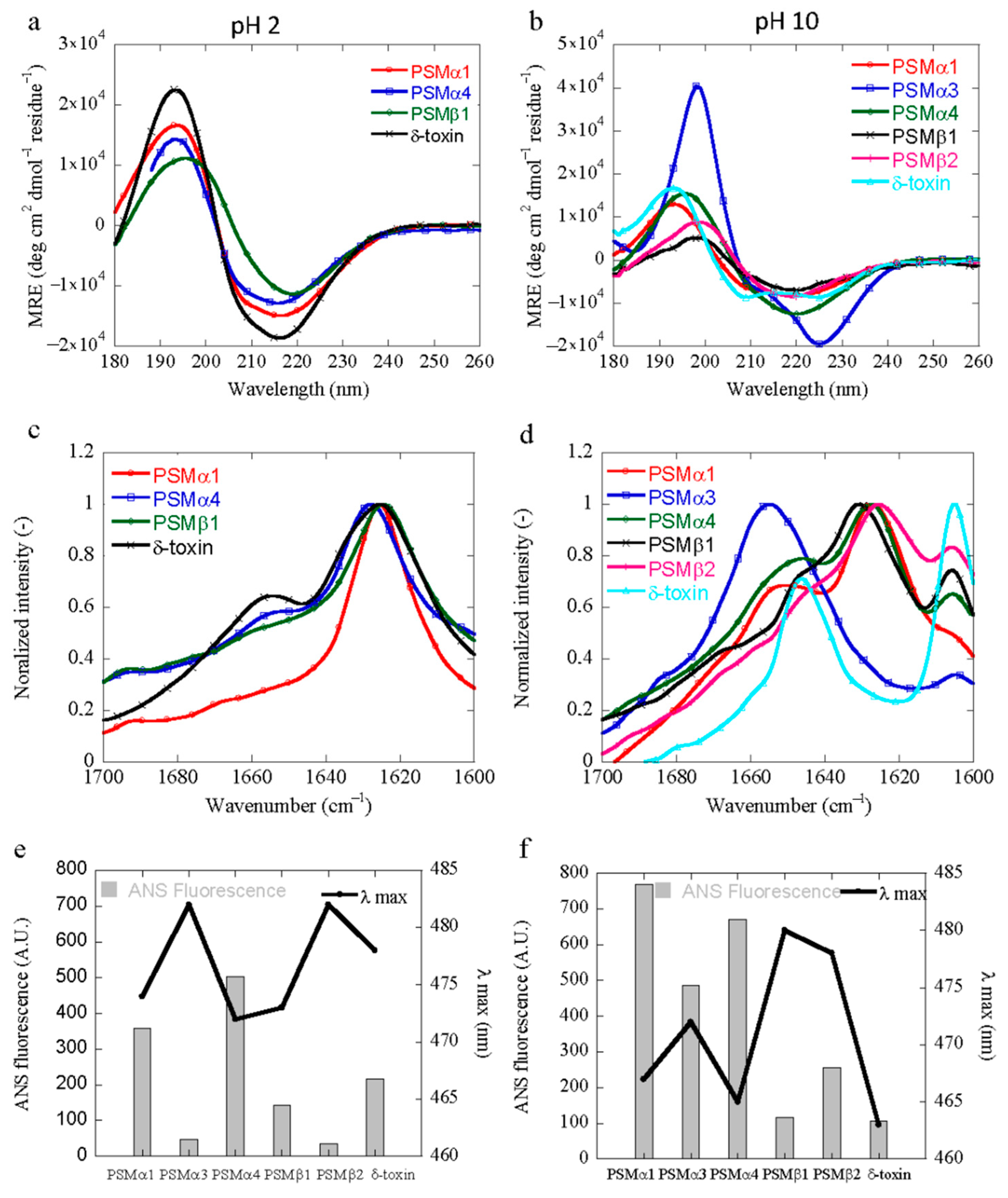
3.4. Structural Characterization of PSM Aggregation In Vitro
3.5. Characterization of the Hydrophobicity of the PSM Aggregates
3.6. Morphological Characterization of PSM Aggregates
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sugimoto, S.; Sato, F.; Miyakawa, R.; Chiba, A.; Onodera, S.; Hori, S.; Mizunoe, Y. Broad impact of extracellular DNA on biofilm formation by clinically isolated Methicillin-resistant and -sensitive strains of Staphylococcus aureus. Sci. Rep. 2018, 8, 2254. [Google Scholar] [CrossRef]
- Romero, D.; Aguilar, C.; Losick, R.; Kolter, R. Amyloid fibers provide structural integrity to Bacillus subtilis biofilms. Proc. Natl. Acad. Sci. USA 2010, 107, 2230–2234. [Google Scholar] [CrossRef]
- Flemming, H.C.; Wingender, J. The biofilm matrix. Nat. Rev. Microbiol. 2010, 8, 623–633. [Google Scholar] [CrossRef]
- Flemming, H.C.; Wingender, J.; Szewzyk, U.; Steinberg, P.; Rice, S.A.; Kjelleberg, S. Biofilms: An emergent form of bacterial life. Nat. Rev. Microbiol. 2016, 14, 563–575. [Google Scholar] [CrossRef]
- Kim, J.Y.; Sahu, S.; Yau, Y.H.; Wang, X.; Shochat, S.G.; Nielsen, P.H.; Dueholm, M.S.; Otzen, D.E.; Lee, J.; Delos Santos, M.M.; et al. Detection of Pathogenic Biofilms with Bacterial Amyloid Targeting Fluorescent Probe, CDy11. J. Am. Chem. Soc. 2016, 138, 402–407. [Google Scholar] [CrossRef] [PubMed]
- Mah, T.F.; O’Toole, G.A. Mechanisms of biofilm resistance to antimicrobial agents. Trends Microbiol. 2001, 9, 34–39. [Google Scholar] [CrossRef]
- Jones, E.M.; Cochrane, C.A.; Percival, S.L. The Effect of pH on the Extracellular Matrix and Biofilms. Adv. Wound Care 2015, 4, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Andreasen, M.; Meisl, G.; Taylor, J.D.; Michaels, T.C.T.; Levin, A.; Otzen, D.E.; Chapman, M.R.; Dobson, C.M.; Matthews, S.J.; Knowles, T.P.J. Physical Determinants of Amyloid Assembly in Biofilm Formation. mBio 2019, 10. [Google Scholar] [CrossRef]
- Fowler, D.M.; Koulov, A.V.; Balch, W.E.; Kelly, J.W. Functional amyloid—From bacteria to humans. Trends Biochem. Sci. 2007, 32, 217–224. [Google Scholar] [CrossRef]
- Otzen, D.; Riek, R. Functional Amyloids. Cold Spring Harb. Perspect. Biol. 2019. [Google Scholar] [CrossRef]
- Marinelli, P.; Pallares, I.; Navarro, S.; Ventura, S. Dissecting the contribution of Staphylococcus aureus alpha-phenol-soluble modulins to biofilm amyloid structure. Sci. Rep. 2016, 6, 34552. [Google Scholar] [CrossRef] [PubMed]
- Otto, M. Phenol-soluble modulins. Int. J. Med. Microbiol. 2014, 304, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Periasamy, S.; Joo, H.S.; Duong, A.C.; Bach, T.H.; Tan, V.Y.; Chatterjee, S.S.; Cheung, G.Y.; Otto, M. How Staphylococcus aureus biofilms develop their characteristic structure. Proc. Natl. Acad. Sci. USA 2012, 109, 1281–1286. [Google Scholar] [CrossRef] [PubMed]
- Peschel, A.; Otto, M. Phenol-soluble modulins and staphylococcal infection. Nat. Rev. Microbiol. 2013, 11, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Cheung, G.Y.; Joo, H.S.; Chatterjee, S.S.; Otto, M. Phenol-soluble modulins--critical determinants of staphylococcal virulence. FEMS Microbiol. Rev. 2014, 38, 698–719. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, K.; Syed, A.K.; Stephenson, R.E.; Rickard, A.H.; Boles, B.R. Functional amyloids composed of phenol soluble modulins stabilize Staphylococcus aureus biofilms. PLoS Pathog. 2012, 8, e1002744. [Google Scholar] [CrossRef]
- Salinas, N.; Colletier, J.P.; Moshe, A.; Landau, M. Extreme amyloid polymorphism in Staphylococcus aureus virulent PSMalpha peptides. Nat. Commun. 2018, 9, 3512. [Google Scholar] [CrossRef]
- Tayeb-Fligelman, E.; Tabachnikov, O.; Moshe, A.; Goldshmidt-Tran, O.; Sawaya, M.R.; Coquelle, N.; Colletier, J.P.; Landau, M. The cytotoxic Staphylococcus aureus PSMalpha3 reveals a cross-alpha amyloid-like fibril. Science 2017, 355, 831–833. [Google Scholar] [CrossRef]
- Zaman, M.; Andreasen, M. Cross-talk between individual phenol-soluble modulins in Staphylococcus aureus biofilm enables rapid and efficient amyloid formation. eLife 2020, 9. [Google Scholar] [CrossRef]
- Tipping, K.W.; Karamanos, T.K.; Jakhria, T.; Iadanza, M.G.; Goodchild, S.C.; Tuma, R.; Ranson, N.A.; Hewitt, E.W.; Radford, S.E. pH-induced molecular shedding drives the formation of amyloid fibril-derived oligomers. Proc. Natl. Acad. Sci. USA 2015, 112, 5691–5696. [Google Scholar] [CrossRef]
- Ramirez-Alvarado, M.; Merkel, J.S.; Regan, L. A systematic exploration of the influence of the protein stability on amyloid fibril formation in vitro. Proc. Natl. Acad. Sci. USA 2000, 97, 8979–8984. [Google Scholar] [CrossRef] [PubMed]
- Bieschke, J.; Herbst, M.; Wiglenda, T.; Friedrich, R.P.; Boeddrich, A.; Schiele, F.; Kleckers, D.; Lopez del Amo, J.M.; Gruning, B.A.; Wang, Q.; et al. Small-molecule conversion of toxic oligomers to nontoxic beta-sheet-rich amyloid fibrils. Nat. Chem. Biol. 2011, 8, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Liu, C.; Leibly, D.; Landau, M.; Zhao, M.; Hughes, M.P.; Eisenberg, D.S. Structure-based discovery of fiber-binding compounds that reduce the cytotoxicity of amyloid beta. eLife 2013, 2, e00857. [Google Scholar] [CrossRef] [PubMed]
- Marek, P.J.; Patsalo, V.; Green, D.F.; Raleigh, D.P. Ionic strength effects on amyloid formation by amylin are a complicated interplay among Debye screening, ion selectivity, and Hofmeister effects. Biochemistry 2012, 51, 8478–8490. [Google Scholar] [CrossRef]
- Nostro, A.; Cellini, L.; Di Giulio, M.; D’Arrigo, M.; Marino, A.; Blanco, A.R.; Favaloro, A.; Cutroneo, G.; Bisignano, G. Effect of alkaline pH on staphylococcal biofilm formation. Acta Pathol. Microbiol. Immunol. Scand. 2012, 120, 733–742. [Google Scholar] [CrossRef]
- Maji, S.K.; Perrin, M.H.; Sawaya, M.R.; Jessberger, S.; Vadodaria, K.; Rissman, R.A.; Singru, P.S.; Nilsson, K.P.; Simon, R.; Schubert, D.; et al. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science 2009, 325, 328–332. [Google Scholar] [CrossRef]
- Zmantar, T.; Kouidhi, B.; Miladi, H.; Mahdouani, K.; Bakhrouf, A. A Microtiter plate assay for Staphylococcus aureus biofilm quantification at various pH levels and hydrogen peroxide supplementation. New Microbiol. 2010, 33, 137–145. [Google Scholar]
- Whitmore, L.; Wallace, B.A. Protein secondary structure analyses from circular dichroism spectroscopy: Methods and reference databases. Biopolymers 2008, 89, 392–400. [Google Scholar] [CrossRef]
- Whitmore, L.; Wallace, B.A. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 2004, 32, W668–W673. [Google Scholar] [CrossRef]
- Lees, J.G.; Miles, A.J.; Wien, F.; Wallace, B.A. A reference database for circular dichroism spectroscopy covering fold and secondary structure space. Bioinformatics 2006, 22, 1955–1962. [Google Scholar] [CrossRef]
- Kirk, W.R.; Kurian, E.; Prendergast, F.G. Characterization of the sources of protein-ligand affinity: 1-sulfonato-8-(1′)anilinonaphthalene binding to intestinal fatty acid binding protein. Biophys. J. 1996, 70, 69–83. [Google Scholar] [CrossRef]
- Bhak, G.; Choe, Y.J.; Paik, S.R. Mechanism of amyloidogenesis: Nucleation-dependent fibrillation versus double-concerted fibrillation. BMB Rep. 2009, 42, 541–551. [Google Scholar] [CrossRef] [PubMed]
- French, K.C.; Makhatadze, G.I. Core sequence of PAPf39 amyloid fibrils and mechanism of pH-dependent fibril formation: The role of monomer conformation. Biochemistry 2012, 51, 10127–10136. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.J. Therapeutic protein aggregation: Mechanisms, design, and control. Trends Biotechnol. 2014, 32, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Zapadka, K.L.; Becher, F.J.; Uddin, S.; Varley, P.G.; Bishop, S.; Gomes Dos Santos, A.L.; Jackson, S.E. A pH-Induced Switch in Human Glucagon-like Peptide-1 Aggregation Kinetics. J. Am. Chem. Soc. 2016, 138, 16259–16265. [Google Scholar] [CrossRef]
- Pedersen, J.S.; Andersen, C.B.; Otzen, D.E. Amyloid structure--one but not the same: The many levels of fibrillar polymorphism. FEBS J. 2010, 277, 4591–4601. [Google Scholar] [CrossRef]
- Zaman, M.; Ehtram, A.; Chaturvedi, S.K.; Nusrat, S.; Khan, R.H. Amyloidogenic behavior of different intermediate state of stem bromelain: A biophysical insight. Int. J. Biol. Macromol. 2016, 91, 477–485. [Google Scholar] [CrossRef]
- Nielsen, L.; Khurana, R.; Coats, A.; Frokjaer, S.; Brange, J.; Vyas, S.; Uversky, V.N.; Fink, A.L. Effect of environmental factors on the kinetics of insulin fibril formation: Elucidation of the molecular mechanism. Biochemistry 2001, 40, 6036–6046. [Google Scholar] [CrossRef]
- Deva, T.; Lorenzen, N.; Vad, B.S.; Petersen, S.V.; Thorgersen, I.; Enghild, J.J.; Kristensen, T.; Otzen, D.E. Off-pathway aggregation can inhibit fibrillation at high protein concentrations. Biochim. Biophys. Acta 2013, 1834, 677–687. [Google Scholar] [CrossRef]
- Souillac, P.O.; Uversky, V.N.; Fink, A.L. Structural transformations of oligomeric intermediates in the fibrillation of the immunoglobulin light chain LEN. Biochemistry 2003, 42, 8094–8104. [Google Scholar] [CrossRef]
- Dueholm, M.S.; Petersen, S.V.; Sonderkaer, M.; Larsen, P.; Christiansen, G.; Hein, K.L.; Enghild, J.J.; Nielsen, J.L.; Nielsen, K.L.; Nielsen, P.H.; et al. Functional amyloid in Pseudomonas. Mol. Microbiol. 2010, 77, 1009–1020. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Yang, S.; Kong, J.; Dong, A.; Yu, S. Obtaining information about protein secondary structures in aqueous solution using Fourier transform IR spectroscopy. Nat. Protoc. 2015, 10, 382–396. [Google Scholar] [CrossRef] [PubMed]
- Natalello, A.; Doglia, S.M. Insoluble protein assemblies characterized by fourier transform infrared spectroscopy. Methods Mol. Biol. 2015, 1258, 347–369. [Google Scholar] [CrossRef]
- Fischer, G.; Cao, X.; Cox, N.; Francis, M. The FT-IR spectra of glycine and glycylglycine zwitterions isolated in alkali halide matrices. Chem. Phys. 2005, 313, 39–49. [Google Scholar] [CrossRef]
- Lindgren, M.; Sorgjerd, K.; Hammarstrom, P. Detection and characterization of aggregates, prefibrillar amyloidogenic oligomers, and protofibrils using fluorescence spectroscopy. Biophys. J. 2005, 88, 4200–4212. [Google Scholar] [CrossRef]
- Hawe, A.; Sutter, M.; Jiskoot, W. Extrinsic fluorescent dyes as tools for protein characterization. Pharm. Res. 2008, 25, 1487–1499. [Google Scholar] [CrossRef]
- Wang, R.; Braughton, K.R.; Kretschmer, D.; Bach, T.H.; Queck, S.Y.; Li, M.; Kennedy, A.D.; Dorward, D.W.; Klebanoff, S.J.; Peschel, A.; et al. Identification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nat. Med. 2007, 13, 1510–1514. [Google Scholar] [CrossRef]
- Cheung, G.Y.; Duong, A.C.; Otto, M. Direct and synergistic hemolysis caused by Staphylococcus phenol-soluble modulins: Implications for diagnosis and pathogenesis. Microbes Infect. 2012, 14, 380–386. [Google Scholar] [CrossRef]
- Otto, M. Staphylococcal infections: Mechanisms of biofilm maturation and detachment as critical determinants of pathogenicity. Annu. Rev. Med. 2013, 64, 175–188. [Google Scholar] [CrossRef]
- Le, K.Y.; Dastgheyb, S.; Ho, T.V.; Otto, M. Molecular determinants of staphylococcal biofilm dispersal and structuring. Front. Cell. Infect. Microbiol. 2014, 4, 167. [Google Scholar] [CrossRef]
- Wu, K.P.; Weinstock, D.S.; Narayanan, C.; Levy, R.M.; Baum, J. Structural reorganization of alpha-synuclein at low pH observed by NMR and REMD simulations. J. Mol. Biol. 2009, 391, 784–796. [Google Scholar] [CrossRef]
- Otzen, D. Functional amyloid: Turning swords into plowshares. Prion 2010, 4, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Pfefferkorn, C.M.; McGlinchey, P.R.; Lee, J.C. Effects of pH on aggregation kinetics of the repeat domain of a functional amyloid, Pmel17. Proc. Natl. Acad. Sci. USA 2010, 107, 2147–21452. [Google Scholar] [CrossRef] [PubMed]
- Vuong, C.; Gotz, F.; Otto, M. Construction and characterization of an agr deletion mutant of Staphylococcus epidermidis. Infect. Immun. 2000, 68, 1048–1053. [Google Scholar] [CrossRef] [PubMed]
- Kong, K.F.; Vuong, C.; Otto, M. Staphylococcus quorum sensing in biofilm formation and infection. Int. J. Med. Microbiol. 2006, 296, 133–139. [Google Scholar] [CrossRef]
- Cogen, A.L.; Yamasaki, K.; Sanchez, K.M.; Dorschner, R.A.; Lai, Y.; MacLeod, D.T.; Torpey, J.W.; Otto, M.; Nizet, V.; Kim, J.E.; et al. Selective antimicrobial action is provided by phenol-soluble modulins derived from Staphylococcus epidermidis, a normal resident of the skin. J. Investig. Dermatol. 2010, 130, 192–200. [Google Scholar] [CrossRef]
- Cogen, A.L.; Yamasaki, K.; Muto, J.; Sanchez, K.M.; Crotty Alexander, L.; Tanios, J.; Lai, Y.; Kim, J.E.; Nizet, V.; Gallo, R.L. Staphylococcus epidermidis antimicrobial delta-toxin (phenol-soluble modulin-gamma) cooperates with host antimicrobial peptides to kill group A Streptococcus. PLoS ONE 2010, 5, e8557. [Google Scholar] [CrossRef]
- Marchand, A.; Verdon, J.; Lacombe, C.; Crapart, S.; Hechard, Y.; Berjeaud, J.M. Anti-Legionella activity of staphylococcal hemolytic peptides. Peptides 2011, 32, 845–851. [Google Scholar] [CrossRef]
- Diep, B.A.; Otto, M. The role of virulence determinants in community-associated MRSA pathogenesis. Trends Microbiol. 2008, 16, 361–369. [Google Scholar] [CrossRef]
- Kaito, C.; Saito, Y.; Nagano, G.; Ikuo, M.; Omae, Y.; Hanada, Y.; Han, X.; Kuwahara-Arai, K.; Hishinuma, T.; Baba, T.; et al. Transcription and translation products of the cytolysin gene psm-mec on the mobile genetic element SCCmec regulate Staphylococcus aureus virulence. PLoS Pathog. 2011, 7, e1001267. [Google Scholar] [CrossRef]
- Wang, R.; Khan, B.A.; Cheung, G.Y.; Bach, T.H.; Jameson-Lee, M.; Kong, K.F.; Queck, S.Y.; Otto, M. Staphylococcus epidermidis surfactant peptides promote biofilm maturation and dissemination of biofilm-associated infection in mice. J. Clin. Investig. 2011, 121, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Jang, K.S.; Park, M.; Lee, J.Y.; Kim, J.S. Mass spectrometric identification of phenol-soluble modulins in the ATCC(R) 43300 standard strain of methicillin-resistant Staphylococcus aureus harboring two distinct phenotypes. European journal of clinical microbiology & infectious diseases. Off. Publ. Eur. Soc. Clin. Microbiol. 2017, 36, 1151–1157. [Google Scholar] [CrossRef]
- Gonzalez, D.J.; Okumura, C.Y.; Hollands, A.; Kersten, R.; Akong-Moore, K.; Pence, M.A.; Malone, C.L.; Derieux, J.; Moore, B.S.; Horswill, A.R.; et al. Novel phenol-soluble modulin derivatives in community-associated methicillin-resistant Staphylococcus aureus identified through imaging mass spectrometry. J. Biol. Chem. 2012, 287, 13889–13898. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, D.J.; Vuong, L.; Gonzalez, I.S.; Keller, N.; McGrosso, D.; Hwang, J.H.; Hung, J.; Zinkernagel, A.; Dixon, J.E.; Dorrestein, P.C.; et al. Phenol soluble modulin (PSM) variants of community-associated methicillin-resistant Staphylococcus aureus (MRSA) captured using mass spectrometry-based molecular networking. Mol. Cell. Proteom. 2014, 13, 1262–1272. [Google Scholar] [CrossRef] [PubMed]
- Joo, H.S.; Cheung, G.Y.; Otto, M. Antimicrobial activity of community-associated methicillin-resistant Staphylococcus aureus is caused by phenol-soluble modulin derivatives. J. Biol. Chem. 2011, 286, 8933–8940. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.S.; Joo, H.S.; Duong, A.C.; Dieringer, T.D.; Tan, V.Y.; Song, Y.; Fischer, E.R.; Cheung, G.Y.; Li, M.; Otto, M. Essential Staphylococcus aureus toxin export system. Nat. Med. 2013, 19, 364–367. [Google Scholar] [CrossRef] [PubMed]
- Laabei, M.; Jamieson, W.D.; Yang, Y.; van den Elsen, J.; Jenkins, A.T. Investigating the lytic activity and structural properties of Staphylococcus aureus phenol soluble modulin (PSM) peptide toxins. Biochim. Biophys. Acta 2014, 1838, 3153–3161. [Google Scholar] [CrossRef]
- Cheung, G.Y.; Kretschmer, D.; Queck, S.Y.; Joo, H.S.; Wang, R.; Duong, A.C.; Nguyen, T.H.; Bach, T.H.; Porter, A.R.; DeLeo, F.R.; et al. Insight into structure-function relationship in phenol-soluble modulins using an alanine screen of the phenol-soluble modulin (PSM) alpha3 peptide. Off. Publ. Fed. Am. Soc. Exp. Biol. 2014, 28, 153–161. [Google Scholar] [CrossRef]
- Kim, W.; Hecht, M.H. Generic hydrophobic residues are sufficient to promote aggregation of the Alzheimer’s Abeta42 peptide. Proc. Natl. Acad. Sci. USA 2006, 103, 15824–15829. [Google Scholar] [CrossRef]
- Giangaspero, A.; Sandri, L.; Tossi, A. Amphipathic alpha helical antimicrobial peptides. Eur. J. Biochem. 2001, 268, 5589–5600. [Google Scholar] [CrossRef]
- Dean, D.N.; Lee, J.C. pH-Dependent fibril maturation of a Pmel17 repeat domain isoform revealed by tryptophan fluorescence. Biochim. Biophys. Acta Proteins Proteom. 2019, 10, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Elgersma, R.C.; Kroon-Batenburg, L.M.J.; Posthuma, G.J.D.; Meeldijk, J.D.; Rijkers, D.T.S.; Liskamp, R.M.J. pH-controlled aggregation polymorphism of amyloidogenic Aβ(16–22): Insights for obtaining peptide tapes and peptide nanotubes, as function of the N-terminal capping moiety. Eur. J. Med. Chem. 2014, 88, 55–65. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaman, M.; Andreasen, M. Modulating Kinetics of the Amyloid-Like Aggregation of S. aureus Phenol-Soluble Modulins by Changes in pH. Microorganisms 2021, 9, 117. https://doi.org/10.3390/microorganisms9010117
Zaman M, Andreasen M. Modulating Kinetics of the Amyloid-Like Aggregation of S. aureus Phenol-Soluble Modulins by Changes in pH. Microorganisms. 2021; 9(1):117. https://doi.org/10.3390/microorganisms9010117
Chicago/Turabian StyleZaman, Masihuz, and Maria Andreasen. 2021. "Modulating Kinetics of the Amyloid-Like Aggregation of S. aureus Phenol-Soluble Modulins by Changes in pH" Microorganisms 9, no. 1: 117. https://doi.org/10.3390/microorganisms9010117
APA StyleZaman, M., & Andreasen, M. (2021). Modulating Kinetics of the Amyloid-Like Aggregation of S. aureus Phenol-Soluble Modulins by Changes in pH. Microorganisms, 9(1), 117. https://doi.org/10.3390/microorganisms9010117