Multiplex Detection of Salmonella spp., E. coli O157 and L. monocytogenes by qPCR Melt Curve Analysis in Spiked Infant Formula



Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Stains
2.2. Enrichment Optimization
2.2.1. Media Formulation
2.2.2. Media Supplementation
2.3. Growth Kinetic Model and Statistical analysis
2.4. Food Spiking
2.5. DNA Extraction
2.5.1. Pure Culture
2.5.2. Food Samples
2.6. qPCR Reaction
2.7. Evaluation
2.7.1. Evaluation of qPCR Assay (Efficiency, Dynamic Range, Inclusivity/Exclusivity)
2.7.2. Evaluation of the Full Methodology
3. Results
3.1. Evaluation of mTA10 Medium Modifications and Supplements
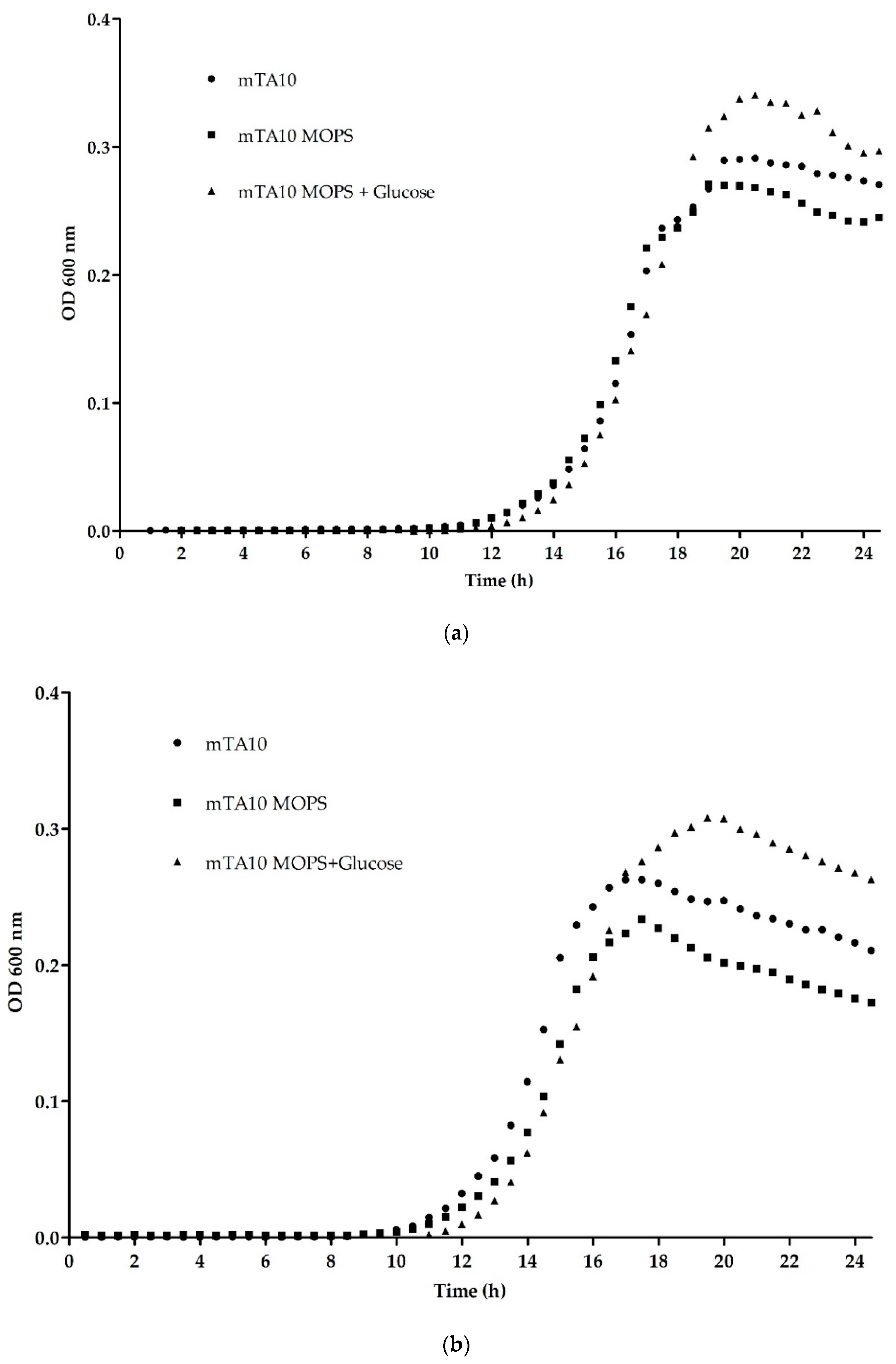
3.1.1. Optimization of Buffering Agent and Temperature
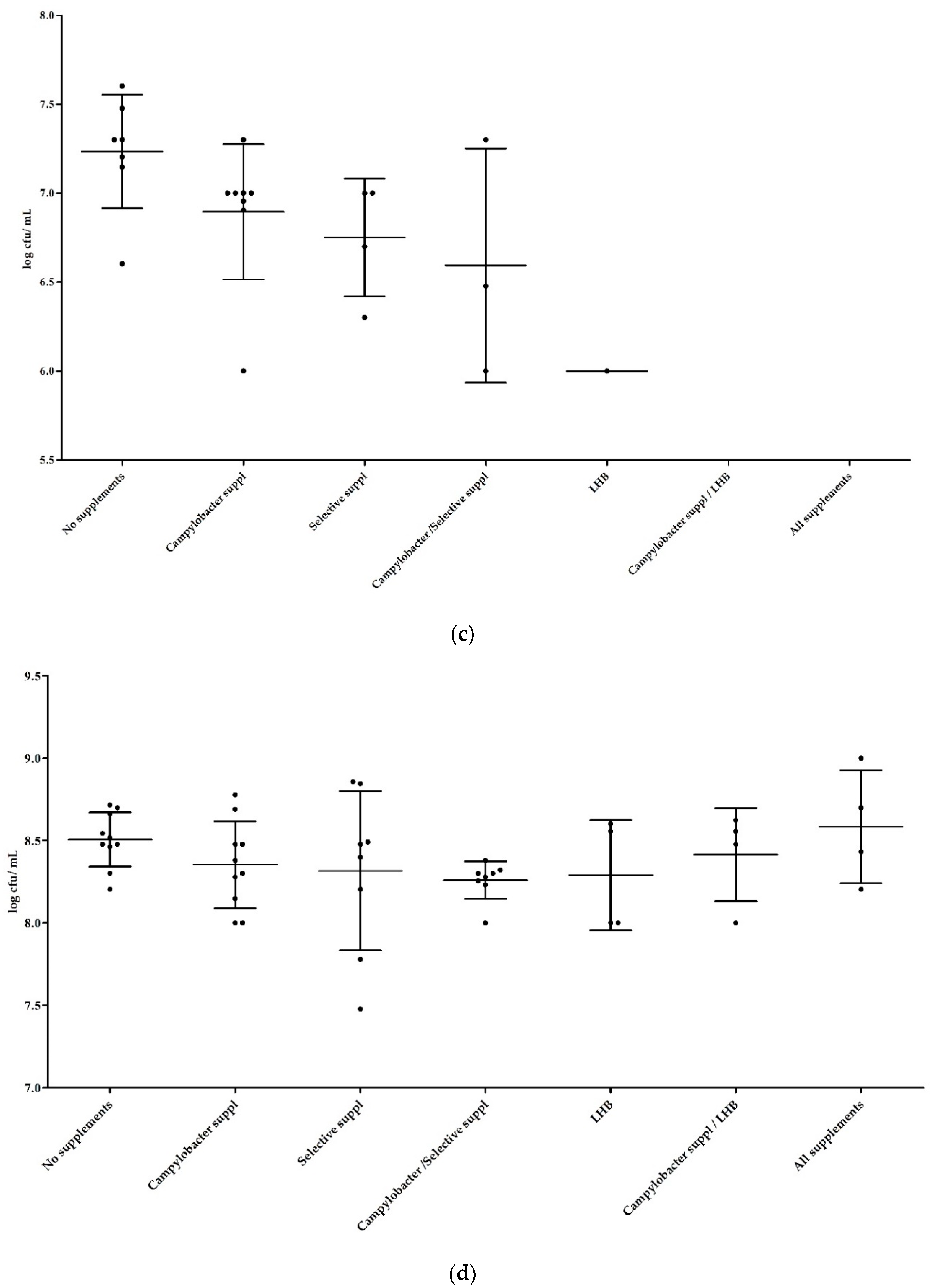
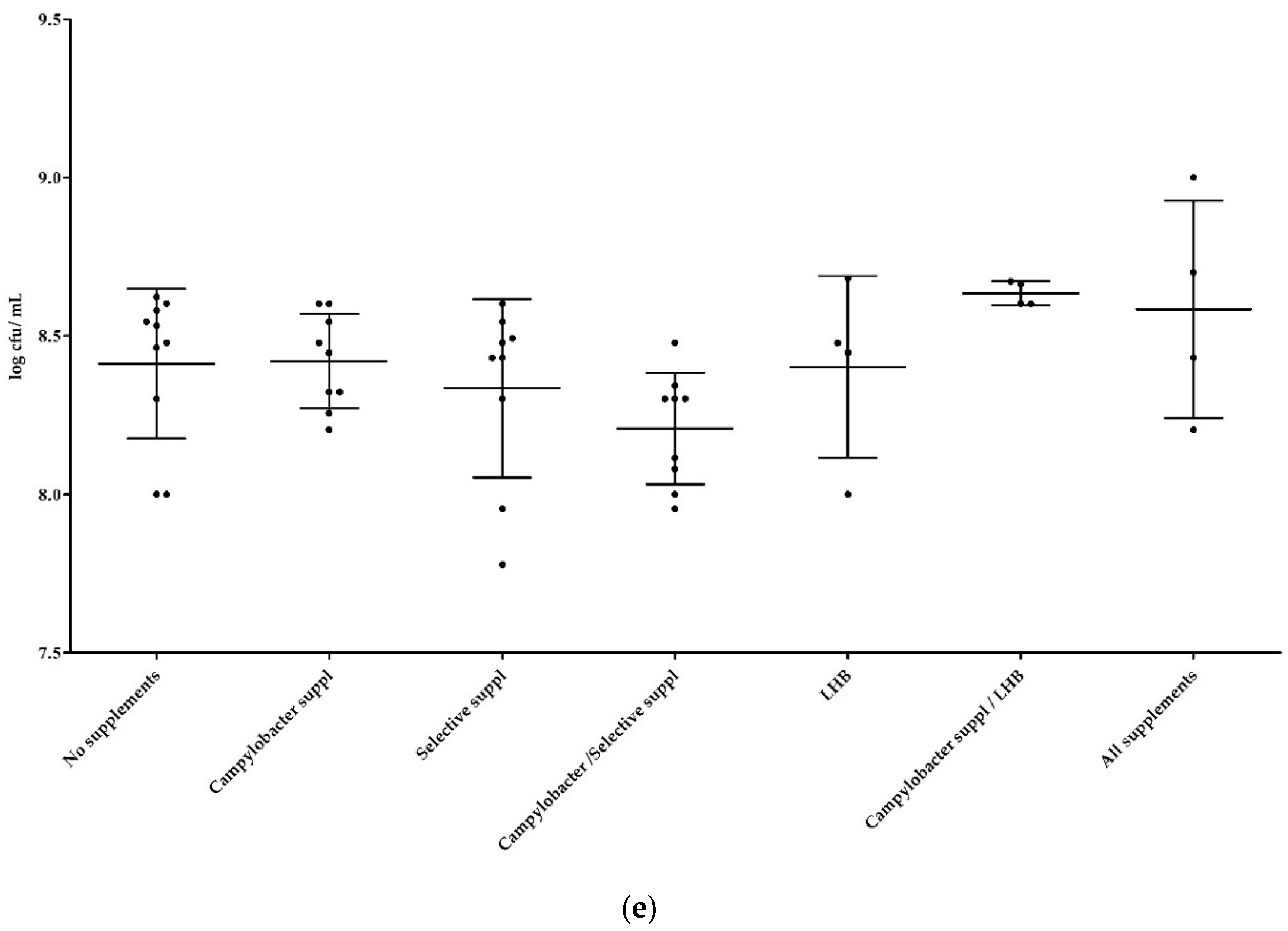
3.1.2. Influence of Supplements in Mixed Cultures
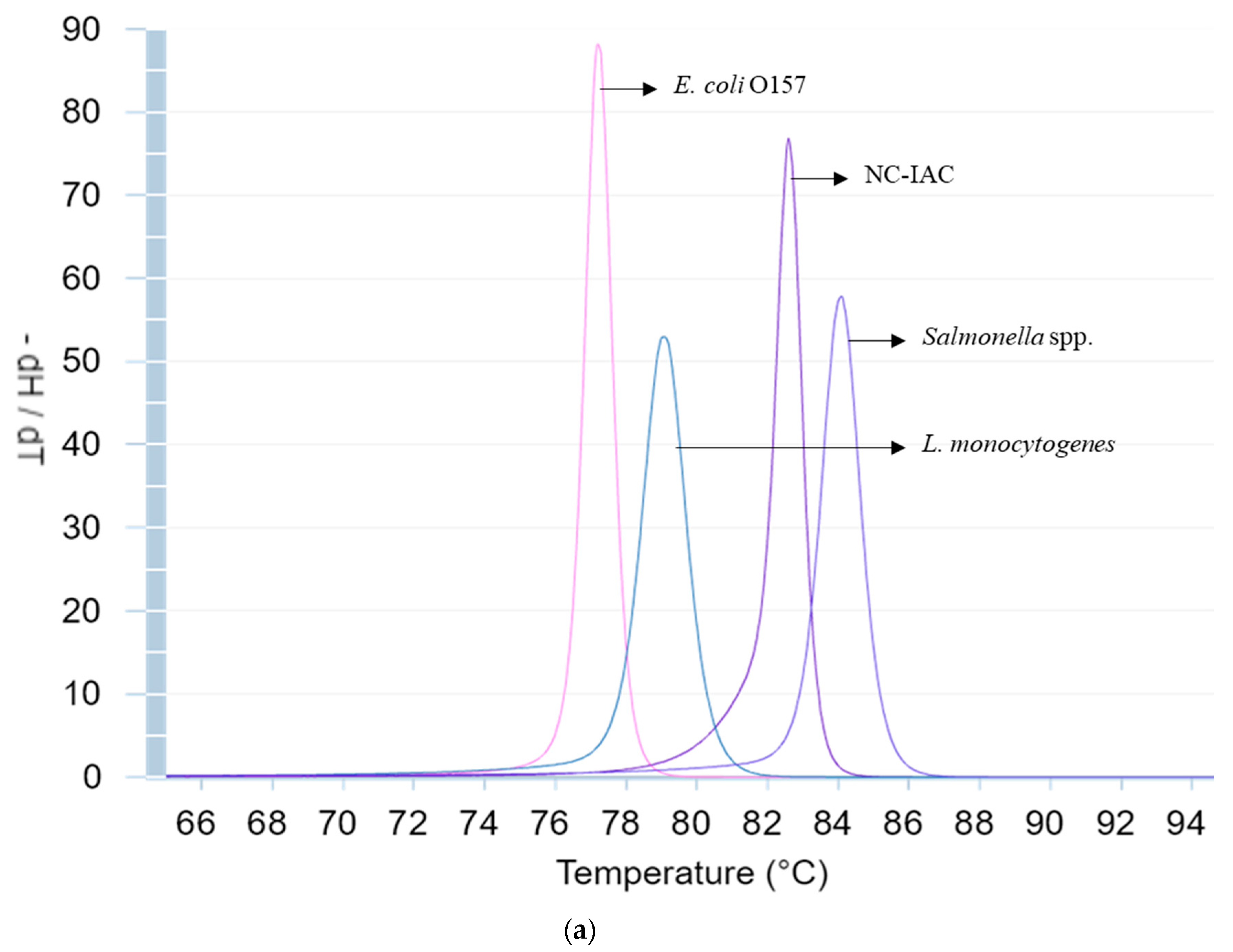
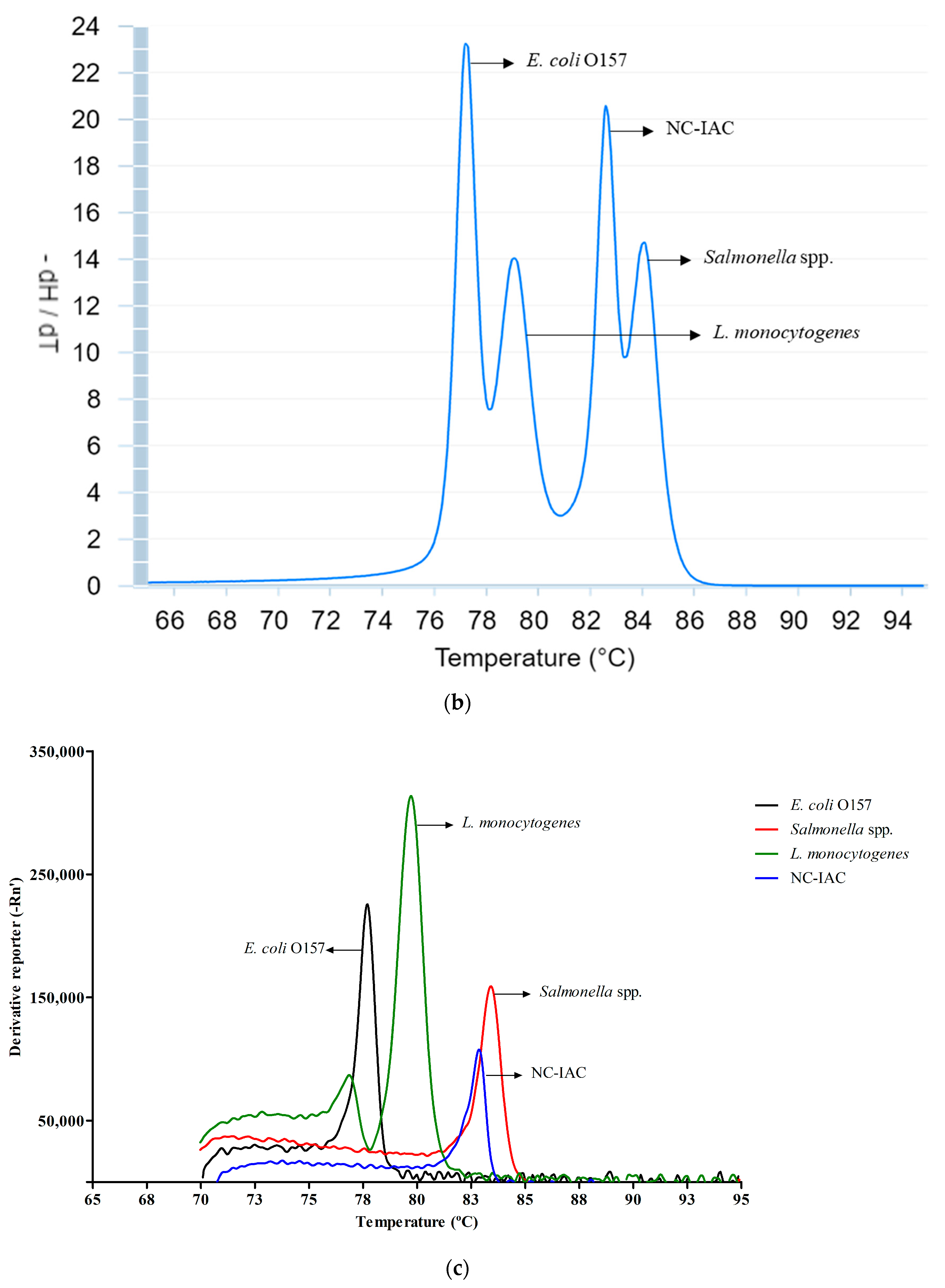
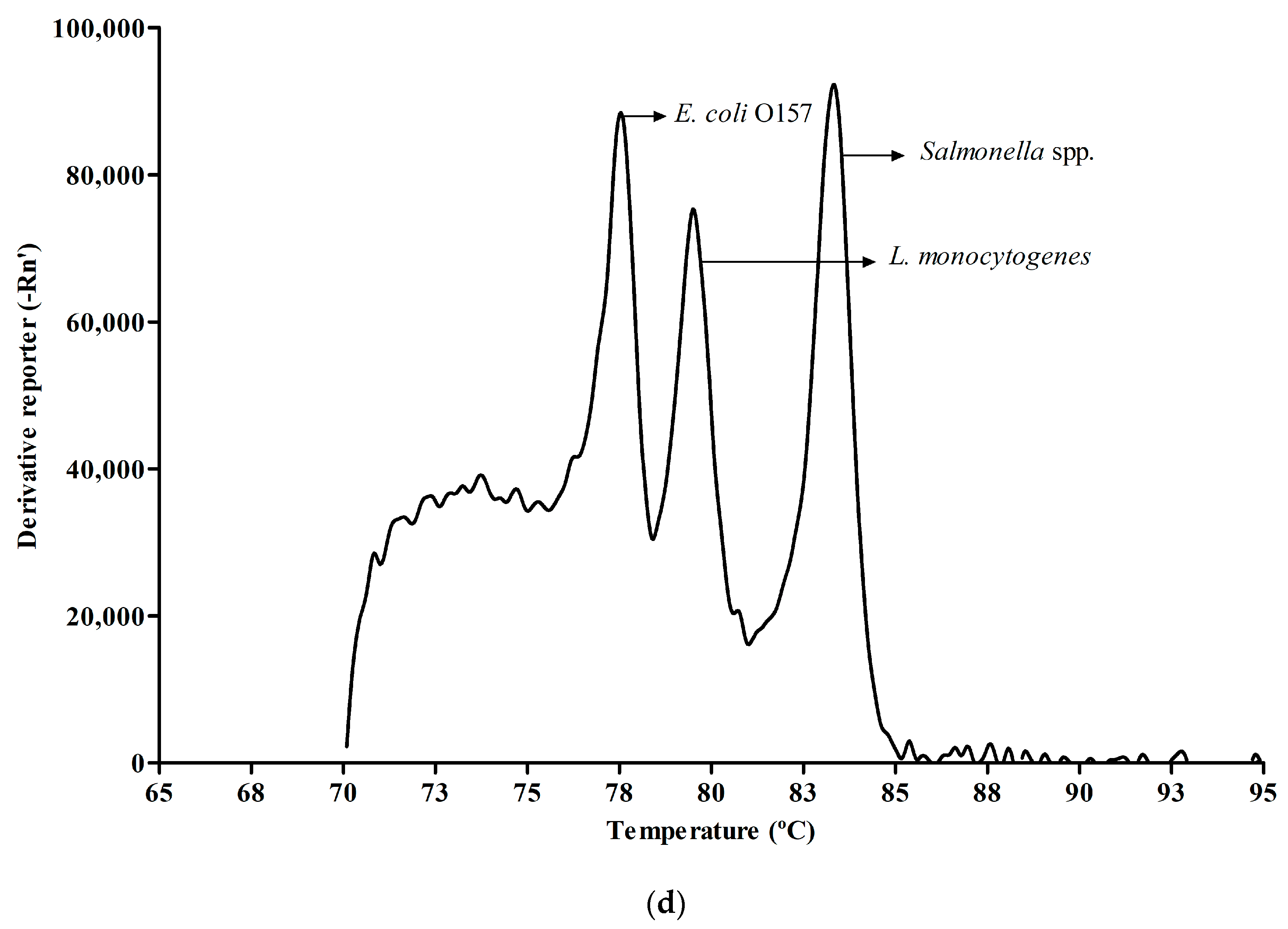
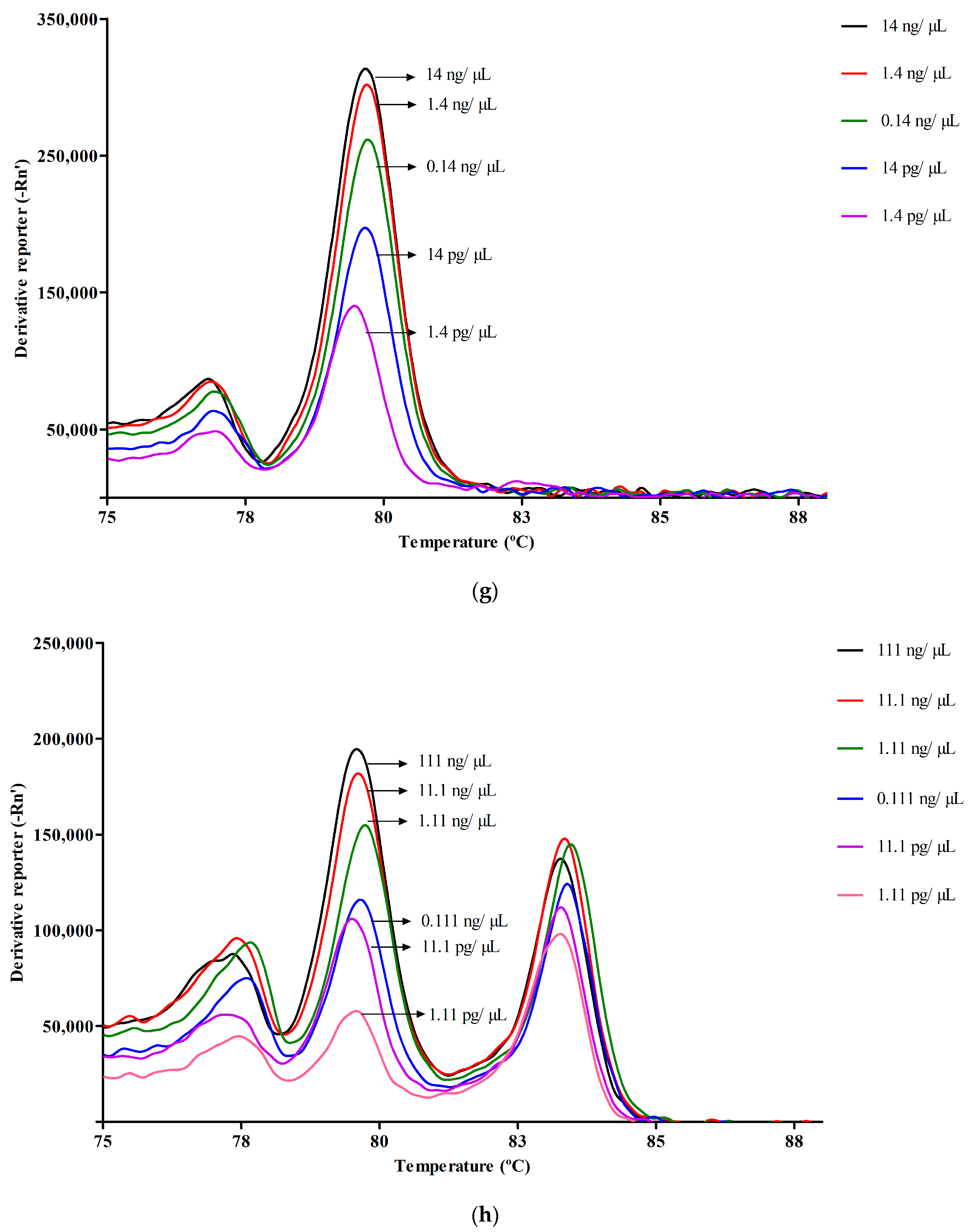
3.2. Melt Peak Analysis
3.3. Evaluation of qPCR Reaction
3.3.1. Inclusivity/Exclusivity
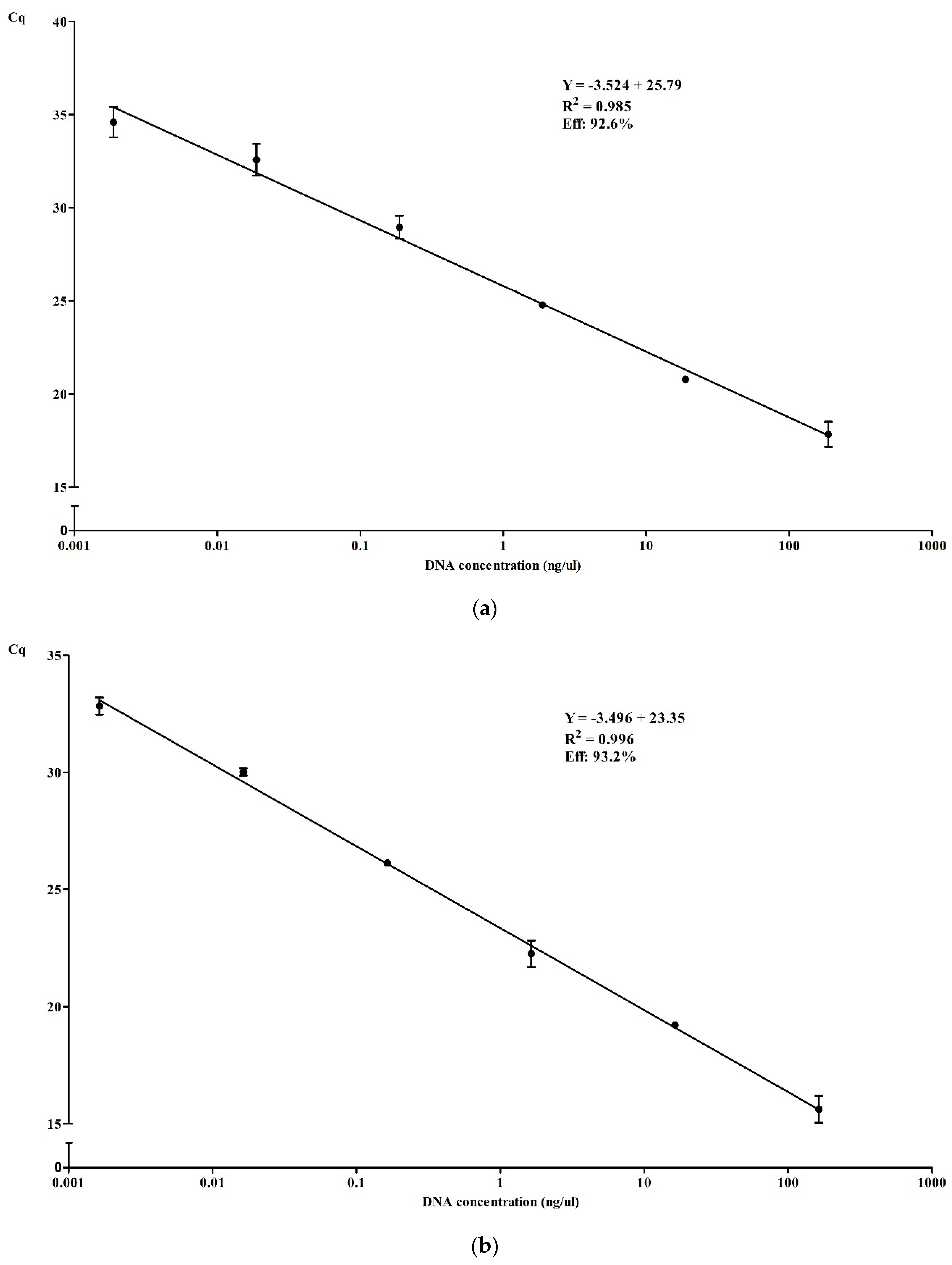
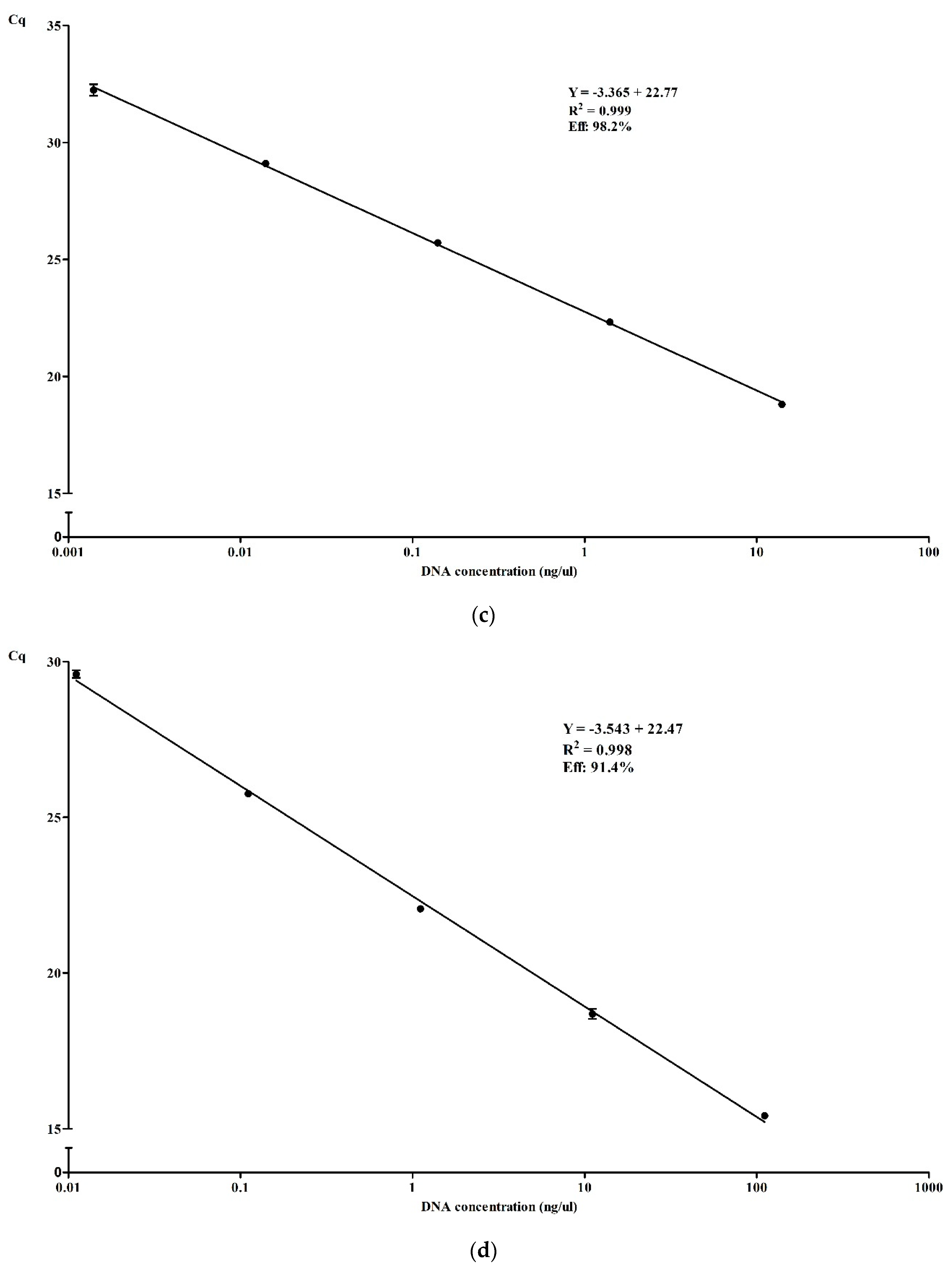
3.3.2. Efficiency and Dynamic Range of qPCR in Simplex and Multiplex
3.4. Methodology Evaluation
3.4.1. Comparison of Expected and Obtained Results
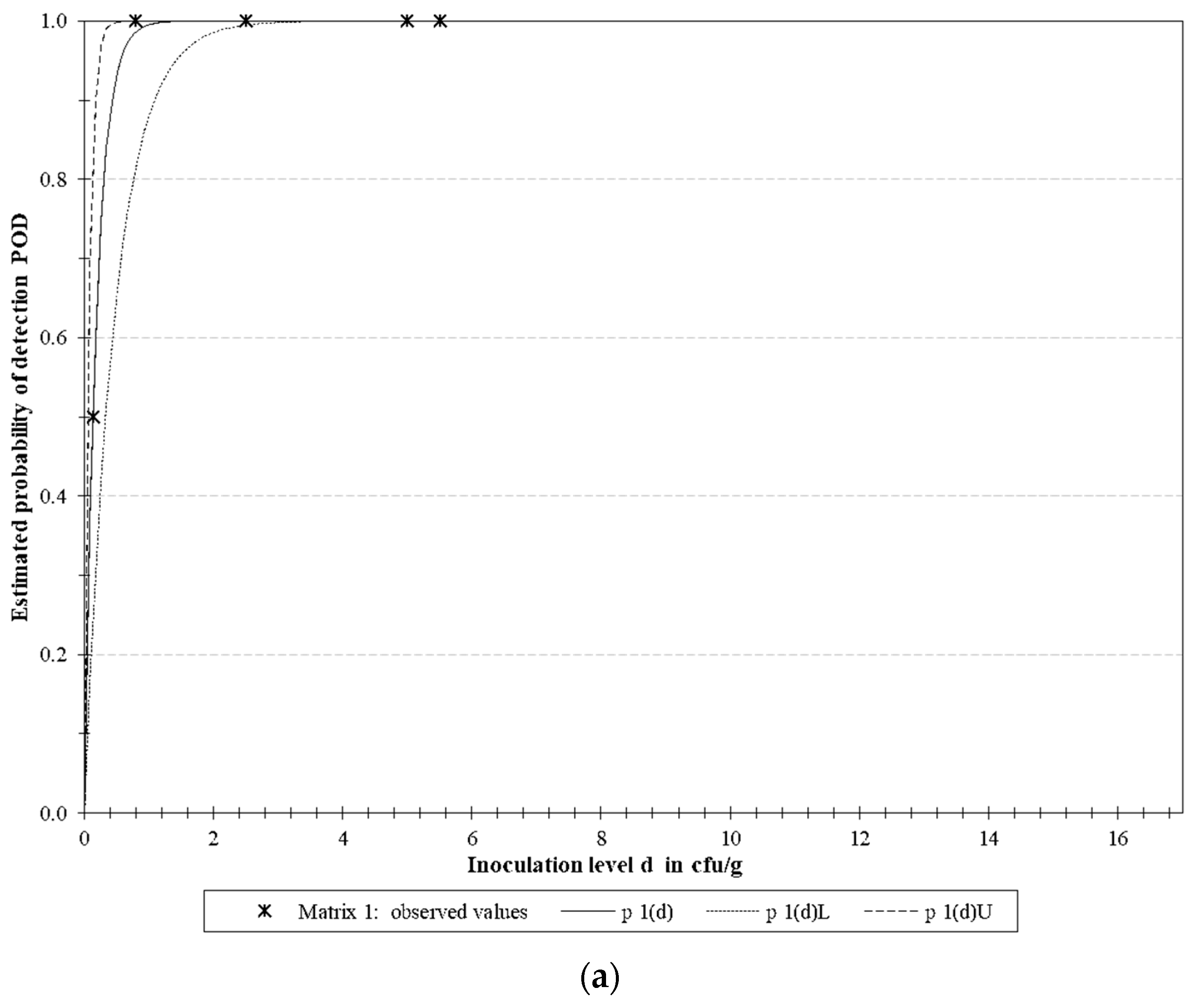
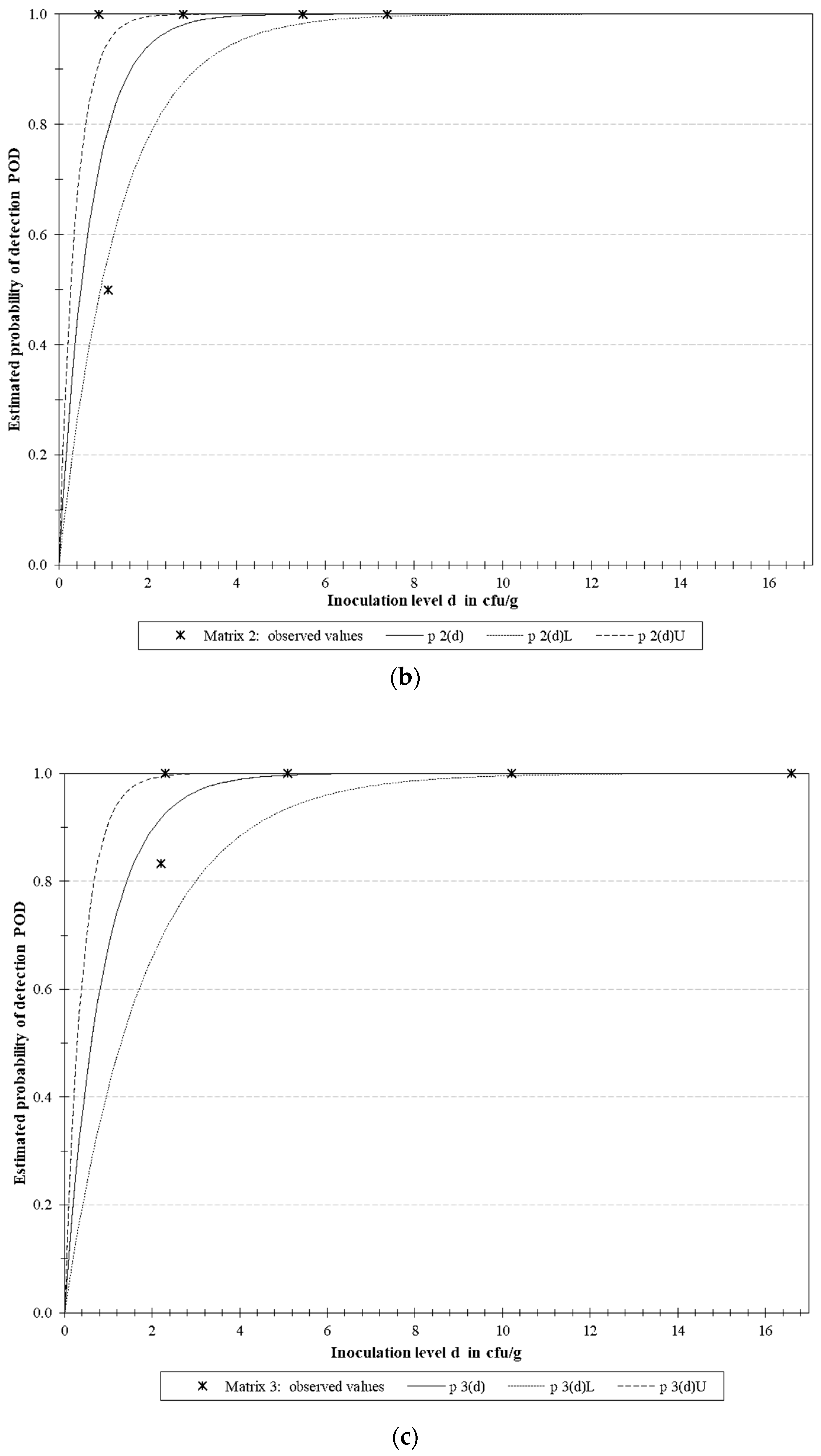
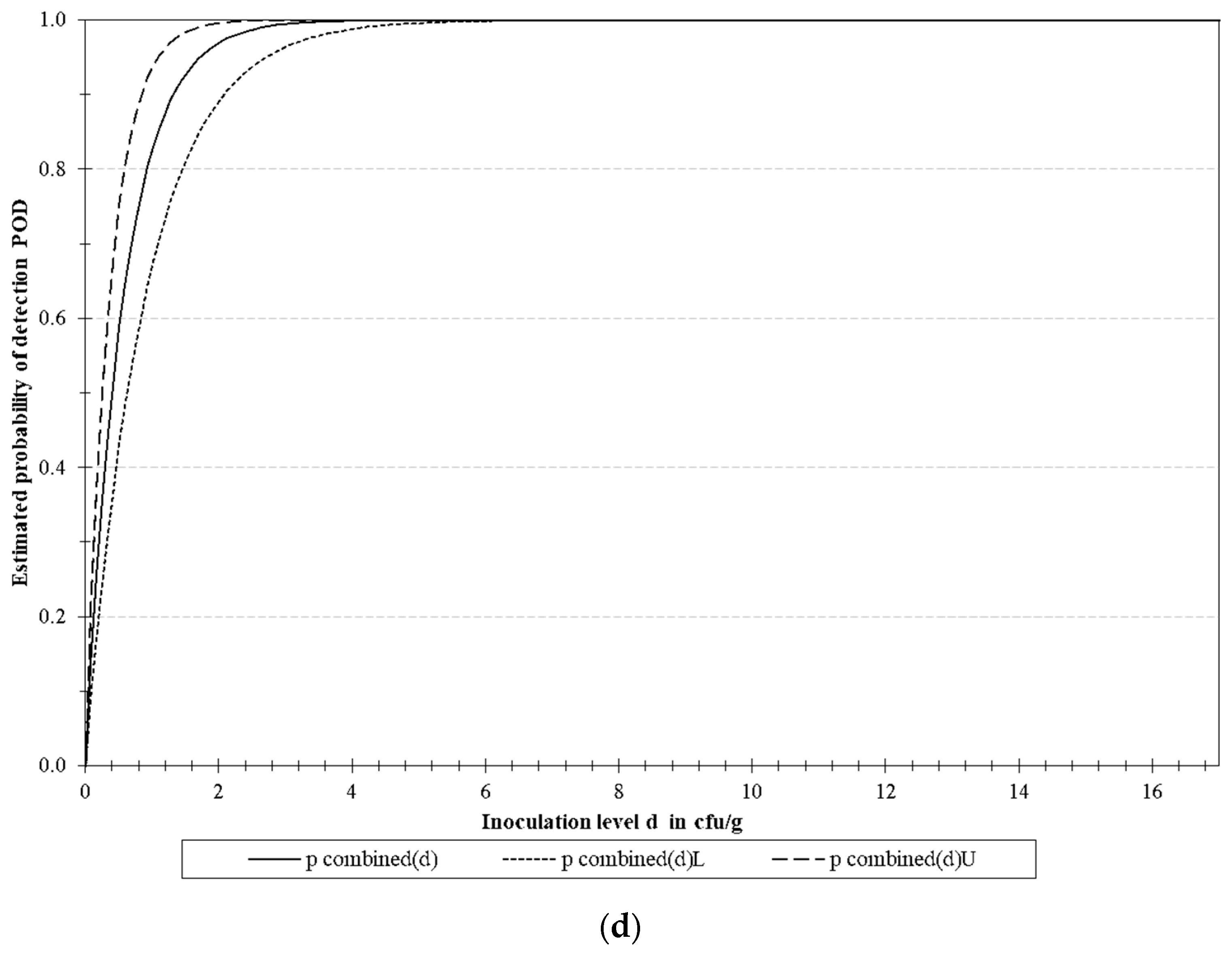
3.4.2. LoD Determination
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hoffmann, S.; Devleesschauwer, B.; Aspinall, W.; Cooke, R.; Corrigan, T.; Havelaar, A.; Angulo, F.; Gibb, H.; Kirk, M.; Lake, R.; et al. Attribution of global foodborne disease to specific foods: Findings from a World Health Organization structured expert elicitation. PLoS ONE 2017, 12, e0183641. [Google Scholar] [CrossRef]
- Liu, J.; Bai, L.; Li, W.; Han, H.; Fu, P.; Ma, X.; Bi, Z.; Yang, X.; Zhang, X.; Zhen, S.; et al. Trends of foodborne diseases in China: Lessons from laboratory-based surveillance since 2011. Front. Med. 2018, 12, 48–57. [Google Scholar] [CrossRef] [PubMed]
- EFSA; ECDC. The European Union summary report on trends and sources of zoonoses, zoonotic agents and food-borne outbreaks in 2016. EFSA J. 2017, 15, e05500. [Google Scholar] [CrossRef]
- Castro-Ibáñez, I.; Gil, M.I.; Allende, A. Ready-to-eat vegetables: Current problems and potential solutions to reduce microbial risk in the production chain. LWT Food Sci. Technol. 2017, 85, 284–292. [Google Scholar] [CrossRef]
- Faour-Klingbeil, D.; Todd, E.; Faour-Klingbeil, D.; Todd, E.C.D. A Review on the Rising Prevalence of International Standards: Threats or Opportunities for the Agri-Food Produce Sector in Developing Countries, with a Focus on Examples from the MENA Region. Foods 2018, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Chapela, M.; Garrido-Maestu, A.; Cabado, A.G. Detection of foodborne pathogens by qPCR: A practical approach for food industry applications. Cogent Food Agric. 2015, 1, 1–19. [Google Scholar] [CrossRef]
- Wang, L.; Li, Y.; Mustapha, A. Rapid and Simultaneous Quantitation of Escherichia coli O157:H7, Salmonella, and Shigella in Ground Beef by Multiplex Real-Time PCR and Immunomagnetic Separation. J. Food Prot. 2007, 70, 1366–1372. [Google Scholar] [CrossRef]
- Van, T.T.H.; Anwar, A.; Scott, P.C.; Moore, R.J. Rapid and Specific Methods to Differentiate Foodborne Pathogens, Campylobacter jejuni, Campylobacter coli, and the New Species Causing Spotty Liver Disease in Chickens, Campylobacter hepaticus. Foodborne Pathog. Dis. 2018, 15, 526–530. [Google Scholar] [CrossRef]
- Garrido-Maestu, A.; Fuciños, P.; Azinheiro, S.; Carvalho, J.; Prado, M. Systematic loop-mediated isothermal amplification assays for rapid detection and characterization of Salmonella spp., Enteritidis and Typhimurium in food samples. Food Control 2017, 80, 297–306. [Google Scholar] [CrossRef]
- Garrido, A.; Chapela, M.J.; Román, B.; Fajardo, P.; Lago, J.; Vieites, J.M.; Cabado, A.G. A new multiplex real-time PCR developed method for Salmonella spp. and Listeria monocytogenes detection in food and environmental samples. Food Control 2013, 30, 76–85. [Google Scholar] [CrossRef]
- Monis, P.T.; Giglio, S. Nucleic acid amplification-based techniques for pathogen detection and identification. Infect. Genet. Evol. 2006, 6, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Druml, B.; Cichna-Markl, M. High resolution melting (HRM) analysis of DNA—Its role and potential in food analysis. Food Chem. 2014, 158, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Singh, P.; Mustapha, A. Multiplex high resolution melt-curve real-time PCR assay for reliable detection of Salmonella. Food Control 2018, 91, 225–230. [Google Scholar] [CrossRef]
- Hoseinpour, F.; Foroughi, A.; Nomanpour, B.; Nasab, R.S. Identification and differentiation of Campylobacter species by high-resolution melting curve analysis. Microb. Pathog. 2017, 108, 109–113. [Google Scholar] [CrossRef]
- Forghani, F.; Wei, S.; Oh, D. A Rapid Multiplex Real-Time PCR High-Resolution Melt Curve Assay for the Simultaneous Detection of Bacillus cereus, Listeria monocytogenes, and Staphylococcus aureus in Food. J. Food Prot. 2016, 79, 810–815. [Google Scholar] [CrossRef] [PubMed]
- Zwietering, M.H.; Jongenburger, I.; Rombouts, F.M.; Riet, K. van’t Modeling of the Bacterial Growth Curve. Appl. Environ. Microbiol. 1990, 56, 1875. [Google Scholar] [CrossRef]
- Kawasaki, S.; Horikoshi, N.; Okada, Y.; Takeshita, K.; Sameshima, T.; Kawamoto, S. Multiplex PCR for Simultaneous Detection of Salmonella spp., Listeria monocytogenes, and Escherichia coli O157:H7 in Meat Samples. J. Food Prot. 2005, 68, 551–556. [Google Scholar] [CrossRef]
- Untergasser, A.; Nijveen, H.; Rao, X.; Bisseling, T.; Geurts, R.; Leunissen, J.A.M. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 2007, 35, 71–74. [Google Scholar] [CrossRef]
- Garrido-Maestu, A.; Azinheiro, S.; Carvalho, J.; Prado, M. Combination of Immunomagnetic separation and real-time Recombinase Polymerase Amplification (IMS-qRPA) for specific detection of Listeria monocytogenes in smoked salmon samples. J. Food Sci. 2019, 84, 1881–1887. [Google Scholar] [CrossRef]
- Azinheiro, S.; Kant, K.; Shahbazi, M.A.; Garrido-Maestu, A.; Prado, M.; Dieguez, L. A smart microfluidic platform for rapid multiplexed detection of foodborne pathogens. Food Control 2020, 114, 107242. [Google Scholar] [CrossRef]
- Garrido-Maestu, A.; Azinheiro, S.; Carvalho, J.; Fuciños, P.; Prado, M. Optimized sample treatment, combined with real-time PCR, for same-day detection of E. coli O157 in ground beef and leafy greens. Food Control 2019, 108, 106790. [Google Scholar] [CrossRef]
- Zhang, Z.; Xiao, L.; Lou, Y.; Jin, M.; Liao, C.; Malakar, P.K.; Pan, Y.; Zhao, Y. Development of a multiplex real-time PCR method for simultaneous detection of Vibrio parahaemolyticus, Listeria monocytogenes and Salmonella spp. in raw shrimp. Food Control 2015, 51, 31–36. [Google Scholar] [CrossRef]
- Wilrich, C.; Wilrich, P.T. Estimation of the pod function and the LOD of a qualitative microbiological measurement method. J. AOAC Int. 2009, 92, 1763–1772. [Google Scholar] [CrossRef] [PubMed]
- Schneebeli, R.; Egli, T. A Defined, Glucose-Limited Mineral Medium for the Cultivation of Listeria spp. Appl. Environ. Microbiol. 2013, 79, 2503–2511. [Google Scholar] [CrossRef]
- Tsai, H.-N.; Hodgson, D.A. Development of a Synthetic Minimal Medium for Listeria monocytogenes. Appl. Environ. Microbiol. 2003, 69, 6943–6945. [Google Scholar] [CrossRef]
- Garrido, A.; Chapela, M.-J.; Román, B.; Ferreira, M.; Lago, J.; Vieites, J.M.; Cabado, A.G. Development of a multiplex real-time PCR method for simultaneous detection of Salmonella enterica, Shigella flexneri and Listeria monocytogenes in processed food samples. Eur. Food Res. Technol. 2012, 234, 571–580. [Google Scholar] [CrossRef]
- Suo, B.; Wang, Y. Evaluation of a multiplex selective enrichment broth SEL for simultaneous detection of injured Salmonella, Escherichia coli O157:H7 and Listeria monocytogenes. Braz. J. Microbiol. 2013, 44, 737–742. [Google Scholar] [CrossRef]
- Kim, H.; Bhunia, A.K. SEL, a Selective Enrichment Broth for Simultaneous Growth of Salmonella enterica, Escherichia coli O157:H7, and Listeria monocytogenes. Appl. Environ. Microbiol. 2008, 74, 4853–4866. [Google Scholar] [CrossRef]
- Cai, S.; Kabuki, D.Y.; Kuaye, A.Y.; Cargioli, T.G.; Chung, M.S.; Nielsen, R.; Wiedmann, M. Rational design of DNA sequence-based strategies for subtyping Listeria monocytogenes. J. Clin. Microbiol. 2002, 40, 3319–3325. [Google Scholar] [CrossRef]
- Oravcova, K.; Kaclikova, E.; Krascsenicsova, K.; Pangallo, D.; Brezna, B.; Siekel, P.; Kuchta, T. Detection and quantification of Listeria monocytogenes by 5’-nuclease polymerase chain reaction targeting the actA gene. Lett. Appl. Microbiol. 2006, 42, 15–18. [Google Scholar] [CrossRef]
- Zhou, X.; Jiao, X. Polymerase chain reaction detection of Listeria monocytogenes using oligonucleotide primers targeting actA gene. Food Control 2005, 16, 125–130. [Google Scholar] [CrossRef]
- Mann, E.; Hein, I.; Mester, P.; Stessl, B.; Rossmanith, P.; Wagner, M.; Dzieciol, M. A Robust and Poisson Validated Quantitative 5′ Nuclease TaqMan® Real-Time PCR Assay Targeting fimA for the Rapid Detection of Salmonella spp. in Food. Food Anal. Methods 2013, 6, 991–995. [Google Scholar] [CrossRef]
- Csordas, A.T.; Barak, J.D.; Delwiche, M.J. Comparison of primers for the detection of Salmonella enterica serovars using real-time PCR. Lett. Appl. Microbiol. 2004, 39, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Giglio, S.; Monis, P.T.; Saint, C.P. Demonstration of preferential binding of SYBR Green I to specific DNA fragments in real-time multiplex PCR. Nucleic Acids Res. 2003, 31, 136. [Google Scholar] [CrossRef]
- Raymaekers, M.; Smets, R.; Maes, B.; Cartuyvels, R. Checklist for optimization and validation of real-time PCR assays. J. Clin. Lab. Anal. 2009, 23, 145–151. [Google Scholar] [CrossRef]
- Ruiz-Rueda, O.; Soler, M.; Calvó, L.; García-Gil, J.L. Multiplex Real-time PCR for the Simultaneous Detection of Salmonella spp. and Listeria monocytogenes in Food Samples. Food Anal. Methods 2011, 4, 131–138. [Google Scholar] [CrossRef]
- Garrido, A.; Chapela, M.-J.; Román, B.; Fajardo, P.; Vieites, J.M.; Cabado, A.G. In-house validation of a multiplex real-time PCR method for simultaneous detection of Salmonella spp., Escherichia coli O157 and Listeria monocytogenes. Int. J. Food Microbiol. 2013, 164, 92–98. [Google Scholar] [CrossRef]
- Suo, B.; He, Y.; Tu, S.I.; Shi, X. A multiplex real-time polymerase chain reaction for simultaneous detection of Salmonella spp., Escherichia coli o157, and Listeria monocytogenes in meat products. Foodborne Pathog. Dis. 2010, 7, 619–628. [Google Scholar] [CrossRef]
- Hyeon, J.Y.; Park, C.; Choi, I.S.; Holt, P.S.; Seo, K.H. Development of multiplex real-time PCR with Internal amplification control for simultaneous detection of Salmonella and Cronobacter in powdered infant formula. Int. J. Food Microbiol. 2010, 144, 177–181. [Google Scholar] [CrossRef]
- Brusa, V.; Galli, L.; Linares, L.H.; Ortega, E.E.; Lirón, J.P.; Leotta, G.A. Development and validation of two SYBR green PCR assays and a multiplex real-time PCR for the detection of Shiga toxin-producing Escherichia coli in meat. J. Microbiol. Methods 2015, 119, 10–17. [Google Scholar] [CrossRef]
- Agrimonti, C.; Sanangelantoni, A.M.; Marmiroli, N. Simultaneous enumeration of Campylobacter jejuni and Salmonella enterica genome equivalents by melting curve analysis following duplex real time PCR in the presence of SYBR Green. LWT 2018, 93, 542–548. [Google Scholar] [CrossRef]
- Bundidamorn, D.; Supawasit, W.; Trevanich, S. A new single-tube platform of melting temperature curve analysis based on multiplex real-time PCR using EvaGreen for simultaneous screening detection of Shiga toxin-producing Escherichia coli, Salmonella spp. and Listeria monocytogenes in food. Food Control 2018, 94, 195–204. [Google Scholar] [CrossRef]
- Bhagwat, A.A. Simultaneous detection of Escherichia coli O157:H7, Listeria monocytogenes and Salmonella strains by real-time PCR. Int. J. Food Microbiol. 2003, 84, 217–224. [Google Scholar] [CrossRef]
- ISO. Microbiology of Food and Animal Feeding Stuffs—Polymerase Chain Reaction (PCR) for the Detection of Food-Borne Pathogens—Requirements for Sample Preparation for Qualitative Detection; ISO: Geneve, Switzerland, 2006. [Google Scholar]
- Anderson, A.; Pietsch, K.; Zucker, R.; Mayr, A.; Müller-Hohe, E.; Messelhäusser, U.; Sing, A.; Busch, U.; Huber, I. Validation of a Duplex Real-Time PCR for the Detection of Salmonella spp. in Different Food Products. Food Anal. Methods 2011, 4, 259–267. [Google Scholar] [CrossRef]
- Gianfranceschi, M.V.; Rodriguez-Lazaro, D.; Hernandez, M.; Gonzalez-Garcia, P.; Comin, D.; Gattuso, A.; Delibato, E.; Sonnessa, M.; Pasquali, F.; Prencipe, V. European validation of a Real-Time PCR-based method for detection of Listeria monocytogenes in soft cheese. Int. J. Food Microbiol. 2014, 184, 128–133. [Google Scholar] [CrossRef]
- Skerniškytė, J.; Armalytė, J.; Kvietkauskaitė, R.; Šeputienė, V.; Povilonis, J.; Sužiedėlienė, E. Detection of Salmonella spp., Yersinia enterocolitica, Listeria monocytogenes and Campylobacter spp. by real-time multiplex PCR using amplicon DNA melting analysis and probe-based assay. Int. J. Food Sci. Technol. 2016, 51, 519–529. [Google Scholar] [CrossRef]
- Singh, J.; Batish, V.K.; Grover, S. Simultaneous detection of Listeria monocytogenes and Salmonella spp. in dairy products using real time PCR-melt curve analysis. J. Food Sci. Technol. 2012, 49, 234–239. [Google Scholar] [CrossRef][Green Version]
- Kaynak, A.; Şakalar, E. A Rapid and Cost-Efficient Technique for Simultaneous/Duplex Detection of Listeria monocytogenes and Escherichia coli O157:H7 Using Real Time PCR. J. Food Saf. 2016, 36, 375–382. [Google Scholar] [CrossRef]
- Xiao, X.-l.; Zhang, L.; Wu, H.; Yu, Y.-g.; Tang, Y.-q.; Liu, D.-m.; Li, X.-f. Simultaneous Detection of Salmonella, Listeria monocytogenes, and Staphylococcus aureus by Multiplex Real-Time PCR Assays Using High-Resolution Melting. Food Anal. Methods 2014, 7, 1960–1972. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacterial Species | Source | N | fimA | actA | rfbE | IAC |
|---|---|---|---|---|---|---|
| Salmonella spp. | Mollusk (AMC 90, 238, 253) | 3 | + | - | - | -* |
| Salmonella spp. | Unknown (AMC 260, 261) | 2 | + | - | - | -* |
| Salmonella Oranienburg | Mollusk (AMC 28) | 1 | + | - | - | -* |
| Salmonella Veneziana | Mollusk (AMC 200) | 1 | + | - | - | -* |
| Salmonella Anatum | Proficiency test (AMC 60) | 1 | + | - | - | -* |
| Salmonella Enteritidis | Proficiency test (AMC 82) | 1 | + | - | - | -* |
| Salmonella Enteritidis | University of Bristol | 1 | + | - | - | -* |
| Salmonella Wentworth | Proficiency test (AMC 84) | 1 | + | - | - | -* |
| Salmonella Liverpool | Proficiency test (AMC 198) | 1 | + | - | - | -* |
| Salmonella Typhimurium | Proficiency test (AMC 96) | 1 | + | - | - | -* |
| Salmonella Typhimurium | Unknown (WDCM 00031) | 1 | + | - | - | -* |
| Listeria monocytogenes | Unknown (WDCM 00021) | 1 | - | + | - | -* |
| Listeria monocytogenes | Mollusk | 8 | - | + | - | -* |
| Listeria monocytogenes | Chestnut | 6 | - | + | - | -* |
| Listeria monocytogenes | Chicken | 3 | - | + | - | -* |
| Listeria ivanovii | WDCM 00018 | 1 | - | - | - | + |
| Listeria innocua | WDCM 00017, CECT 5376, 4030, 1325, 1141, 2110 | 6 | - | - | - | + |
| Staphylococcus aureus | WDCM 00034, 00033 | 2 | - | - | - | + |
| Staphylococcus coagulase + | Proficiency test | 1 | - | - | - | + |
| Campylobacter coli | University of Minho | 1 | - | - | - | + |
| Escherichia coli | WDCM 00013, 00012 | 2 | - | - | - | + |
| Escherichia coli | WDCM 00014 | 1 | - | - | + | - |
| Target Microorganism | Target Gene | Primers | Sequence 5′–3′ | Concentration Used | Reference |
|---|---|---|---|---|---|
| L. monocytogenes | actA | actA F | TTAAGACTTGCTTTGCCAGAGAC | 900 nM | [20] |
| actA R | GGTGGTGGAAATTCGAATGAGC | ||||
| Salmonella spp. | fimA | fimA F | CACTAAATCCGCCGATCAAACG | 100 nM | In this study |
| fimA R | TTCAGGACGATGGAGAAAGGC | ||||
| E. coli O157 | rfbE | rfbE F | TCAACAGTCTTGTACAAGTCCAC | 100 nM | [21] |
| rfbE R | ACTGGCCTTGTTTCGATGAG | ||||
| NC-IAC * | - | NC-IAC F | TTAAGACTTGCTTTGCCAGAGAC | 100 nM | [19] |
| NC-IAC R | GGTGGTGGAAATTCGAATGAGC |
| 30 °C | 35 °C | |||||
|---|---|---|---|---|---|---|
| mTA10 | mTA10-MOPS | mTA10-MOPS + Glucose | mTA10 | mTA10-MOPS | mTA10-MOPS + Glucose | |
| OD600 max | 0.288 ± 0.004 | 0.261 ± 0.013 | 0.328 ± 0.010 | 0.242 ± 0.003 | 0.202 ± 0.006 | 0.292 ± 0.002 |
| µmax | 0.074 ± 0.004 | 0.078 ± 0.003 | 0.088 ± 0.001 | 0.089 ± 0.017 | 0.095 ± 0.003 | 0.085 ± 0.004 |
| λ | 14.166 ± 0.166 | 14.002 ± 0.157 | 14.770 ± 0.085 | 12.403 ± 0.143 | 13.090 ± 0.184 | 13.370 ± 0.079 |
| Type of Sample | Contamination Level (CFU/25 g of Sample) * | N | ||||||
|---|---|---|---|---|---|---|---|---|
| L. monocytogenes | S. Typhimurium | E. coli O157 | actA | fimA | rfbE | IAC | ||
| Infant Milk LoD | 16.6 | 7 | 6 | 6 | + | + | + | - |
| 10.2 | 6 | 5 | 6 | + | + | + | - | |
| 5 | 3 | 2 | 6 | + | + | + | - | |
| 2 | 1 | 1 | 6 | + (2ND) | + (3ND) | + | - | |
| 2 | <1 | <1 | 6 | + | + | +(3ND) | - | |
| Infant Milk | - | - | - | 4 | - | - | - | + |
| 9 | 6 | 4 | 2 | + | + | + | - | |
| 9.1 | 13 | 8 | 1 | + | + | + | - | |
| 9 | 6 | 1 | + | + | - | - | ||
| 6 | 4 | 1 | - | + | + | - | ||
| 9 | 4 | 1 | + | - | + | - | ||
| 91 | 1.3 × 103 | 1 | + | + | - | - | ||
| 9 | 8.0 × 102 | 1 | + | - | + | - | ||
| 1.3 × 104 | 8.0 × 103 | 1 | - | + | + | - | ||
| 9.1 × 103 | 1.3 × 104 | 8.0 × 103 | 1 | + | + | + | - | |
| Microorganism | Target | N | PA | PD | NA | ND | SE | SP | AC | PPV | NPV | k |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| E. coli O157 | rfbE | 44 | 32 | 0 | 12 | 0 | 100 | 100 | 100 | 100 | 100 | 1.00 |
| S. Typhimurium | fimA | 44 | 26 | 0 | 18 | 0 | 100 | 100 | 100 | 100 | 100 | 1.00 |
| L. monocytogenes | actA | 44 | 26 | 0 | 18 | 0 | 100 | 100 | 100 | 100 | 100 | 1.00 |
| Microorganism | Target | LoD50 * | LoD95 * | ||||
|---|---|---|---|---|---|---|---|
| LoD | Lower Conf. Limit | Upper Conf. Limit | LoD | Lower Conf. Limit | Upper Conf. Limit | ||
| E. coli O157 | rfbE | 0.129 | 0.051 | 0.327 | 0.558 | 0.220 | 1.414 |
| S. Typhimurium | fimA | 0.488 | 0.257 | 0.930 | 2.111 | 1.109 | 4.018 |
| L. monocytogenes | actA | 0.613 | 0.292 | 1.288 | 2.649 | 1.261 | 5.567 |
| Multiplex | 0.398 | 0.253 | 0.627 | 1.721 | 1.093 | 2.710 | |
| Reference | Pathogens Detected | Target | Fluorescent Dye | IAC | LoD and Matrix | Enrichment Time |
|---|---|---|---|---|---|---|
| Skerniškytė 2016 et al. [47] | Salmonella spp., Y. ente- rocolitica, L. monocytogenes and Campyl obacter spp. Salmonella spp. Yersinia enterocolitica Listeria monocytogenes Campylobacter spp. | invA ystA hly 16S_rrna | SYTO 9 | Yes | 3.01 log CFU/mL 2.67 log CFU/mL 4.49 log CFU/mL 2.51 log CFU/mL Chicken rinses | - |
| Singh 2012 et al. [48] | Listeria monocytogenes Salmonella spp. | hly invA | SYBR Green | No | 3 log CFU per mL 1 log CFU per mL Dried milk | 6 h |
| Bundidamorn 2018 et al. [42] | Shiga toxin-producing Escherichia coli Salmonella spp. Listeria monocytogenes | stx1,2 invA hly | EvaGreen | No | 1 CFU/25 g raw chicken meat, raw pork, raw beef and fresh mung bean sprout | 18 h |
| Kaynak 2016 et al. [49] | Listeria monocytogenes Escherichia coli O157:H7 | hly uid | EvaGreen | No | <10 cells/mL Bovine and poultry meat | 20 h |
| Xiao 2014 et al. [50] | Salmonella spp. Listeria monocytogenes Staphylococcus aureus | hly, invA, Sa442 | EvaGreen | No | 5 CFU/25 g Milk, chicken, beef, pork, egg | 24 h |
| This study | Listeria monocytogenes Escherichia coli O157 Salmonella spp. | actA rfbE fimA | SYBR Green | Yes | 2 CFU/25 g <1 CFU/25 g 2 CFU/25 g Infant formula | 24 h |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azinheiro, S.; Carvalho, J.; Prado, M.; Garrido-Maestu, A. Multiplex Detection of Salmonella spp., E. coli O157 and L. monocytogenes by qPCR Melt Curve Analysis in Spiked Infant Formula. Microorganisms 2020, 8, 1359. https://doi.org/10.3390/microorganisms8091359
Azinheiro S, Carvalho J, Prado M, Garrido-Maestu A. Multiplex Detection of Salmonella spp., E. coli O157 and L. monocytogenes by qPCR Melt Curve Analysis in Spiked Infant Formula. Microorganisms. 2020; 8(9):1359. https://doi.org/10.3390/microorganisms8091359
Chicago/Turabian StyleAzinheiro, Sarah, Joana Carvalho, Marta Prado, and Alejandro Garrido-Maestu. 2020. "Multiplex Detection of Salmonella spp., E. coli O157 and L. monocytogenes by qPCR Melt Curve Analysis in Spiked Infant Formula" Microorganisms 8, no. 9: 1359. https://doi.org/10.3390/microorganisms8091359
APA StyleAzinheiro, S., Carvalho, J., Prado, M., & Garrido-Maestu, A. (2020). Multiplex Detection of Salmonella spp., E. coli O157 and L. monocytogenes by qPCR Melt Curve Analysis in Spiked Infant Formula. Microorganisms, 8(9), 1359. https://doi.org/10.3390/microorganisms8091359