Comparative Genome Analysis of Bacillus sporothermodurans with Its Closest Phylogenetic Neighbor, Bacillus oleronius, and Bacillus cereus and Bacillus subtilis Groups


 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Isolation and Identification
2.2. Genome Sequencing and Analysis
3. Results
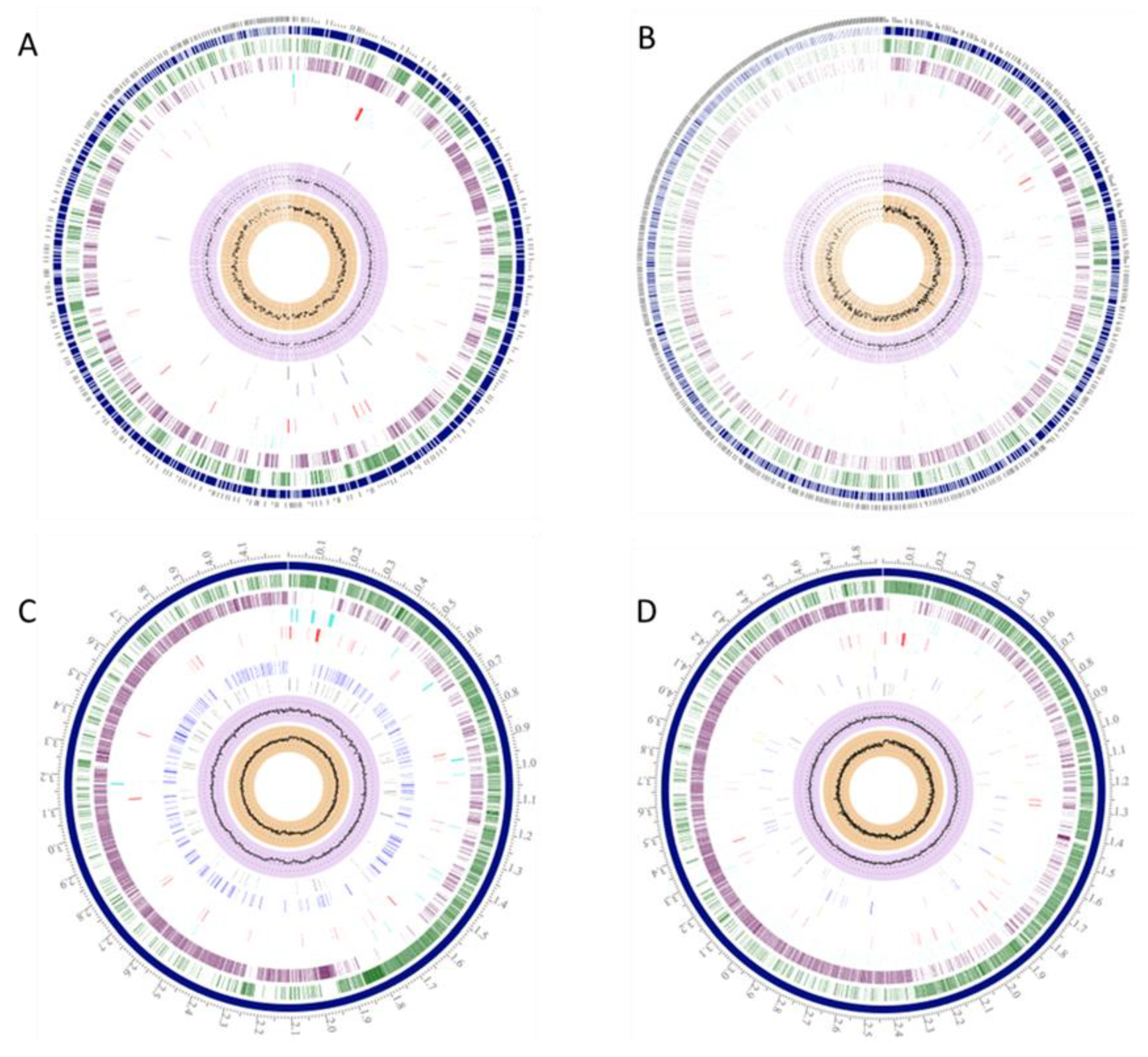
3.1. Wide Variations in Bacillus Species
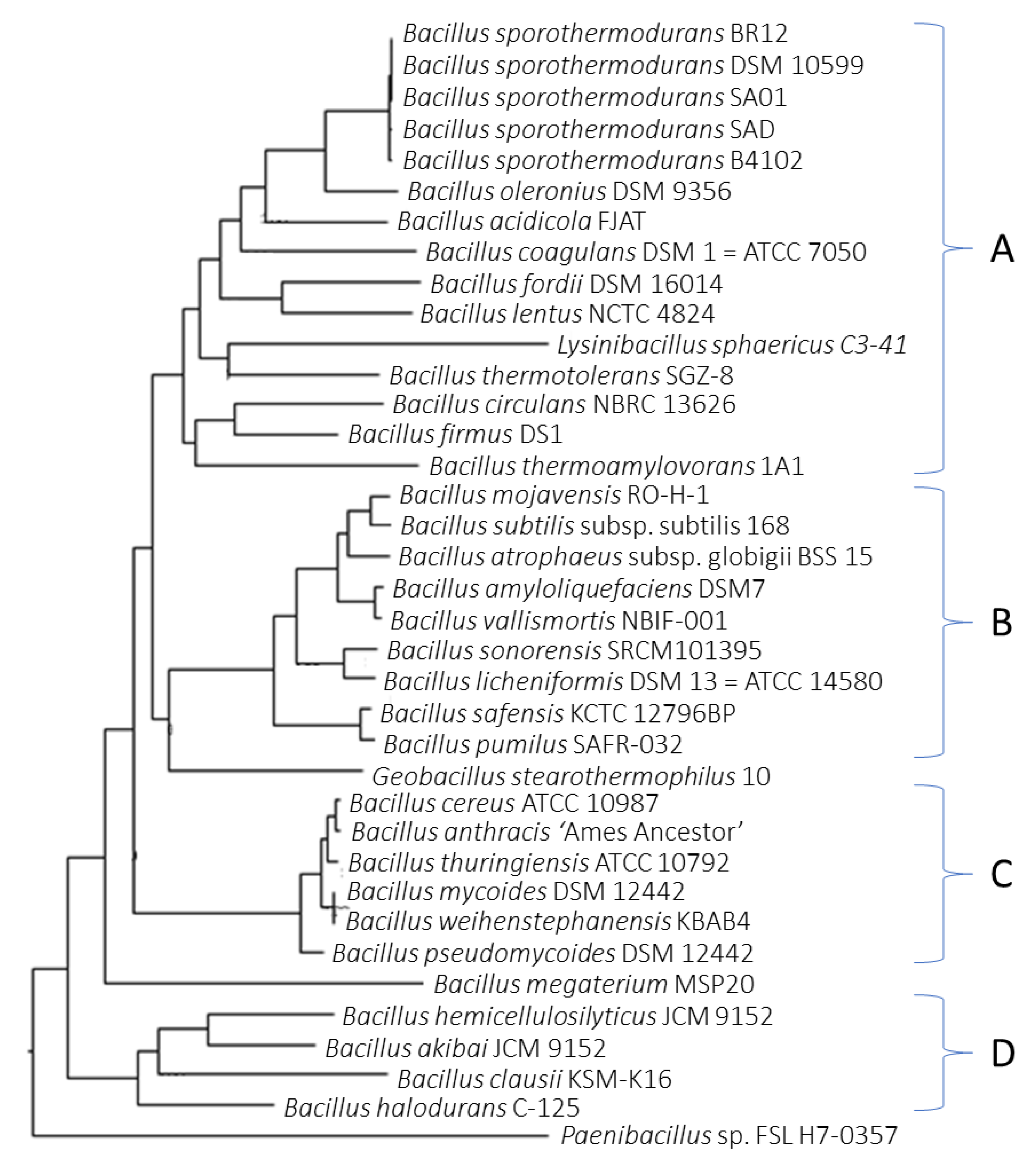
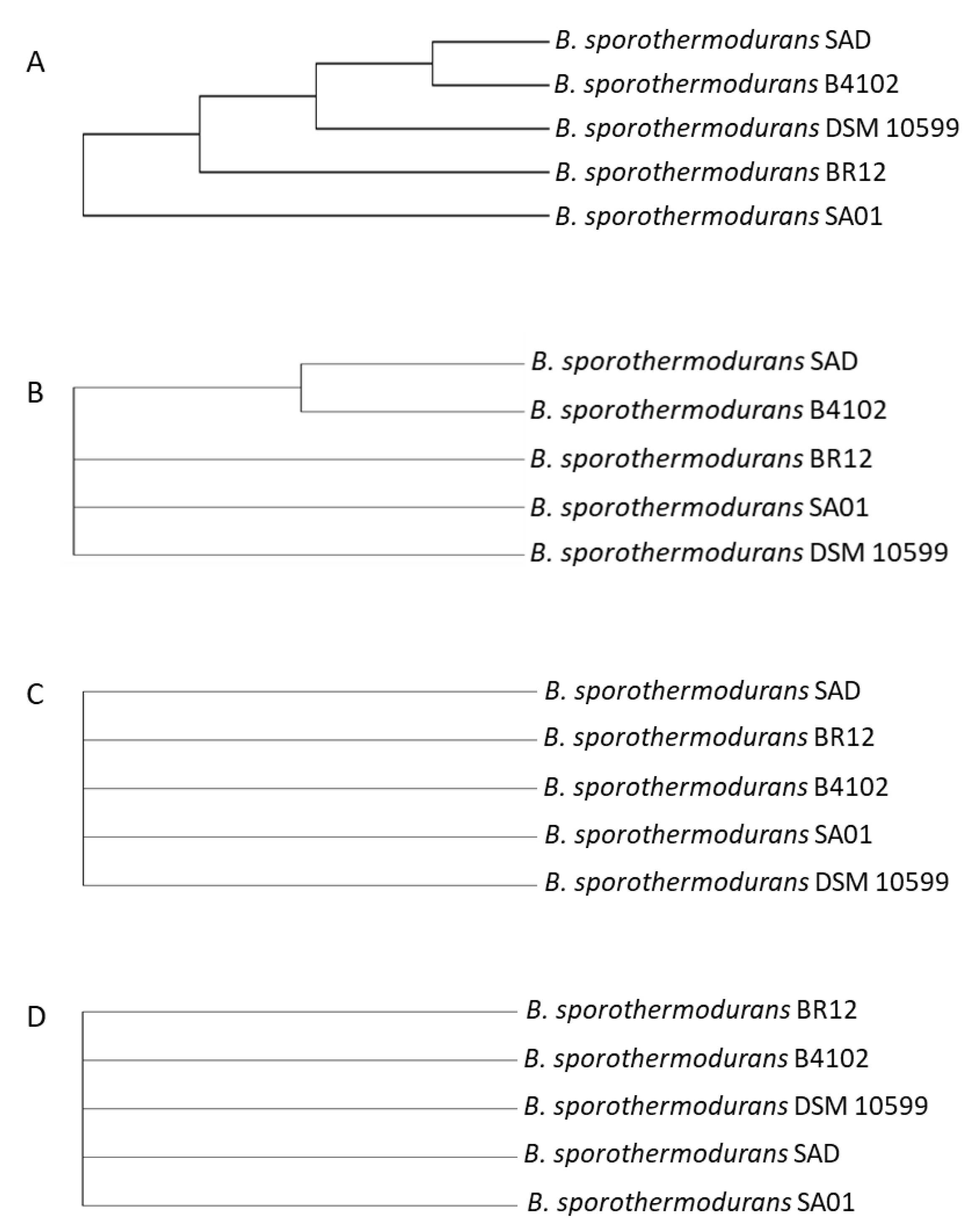
3.2. Phylogenetic Analysis Depicts Distinct Clusters of B. sporothermodurans Separate from the B. subtilis Group
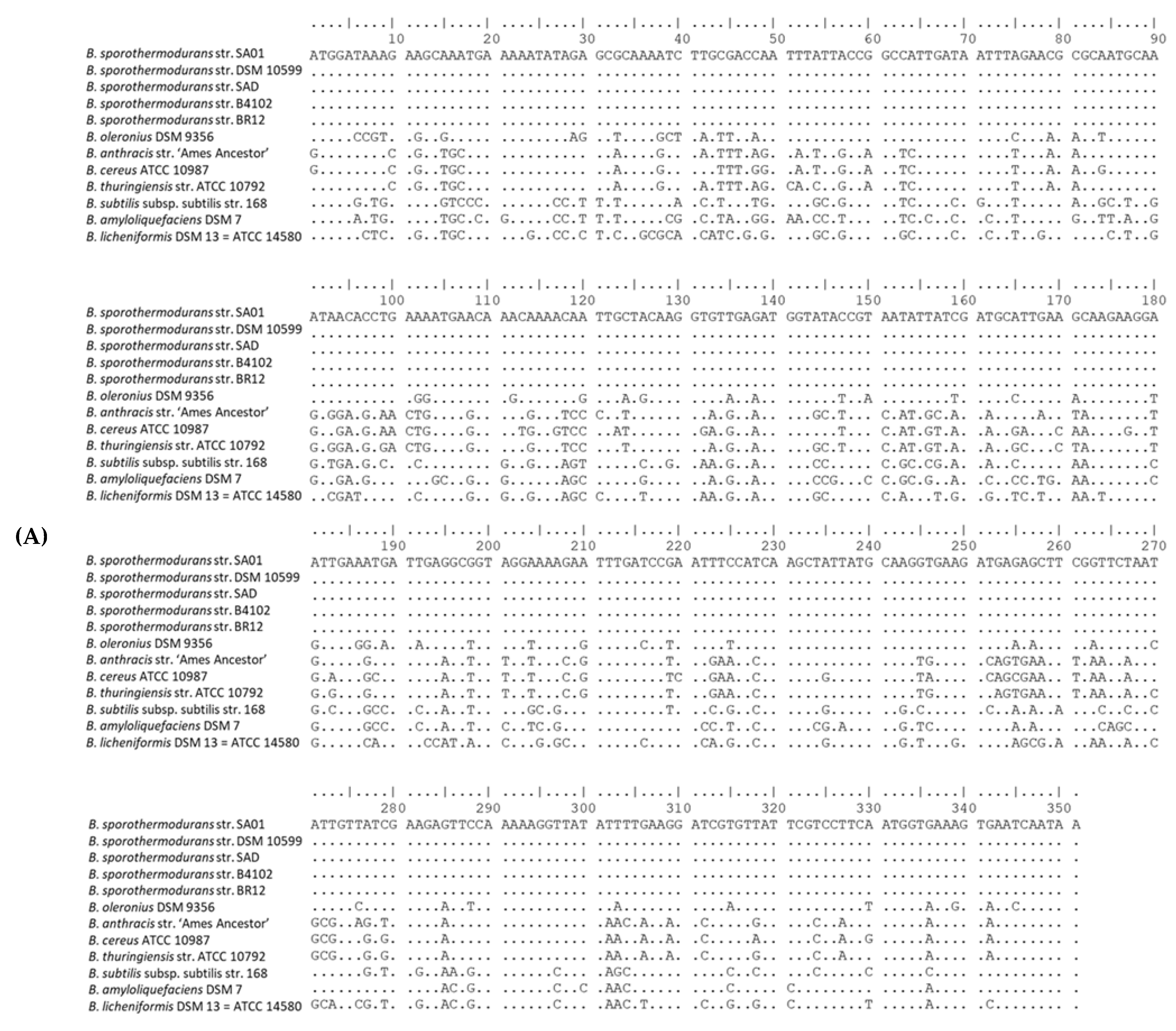
3.3. SNP Analysis
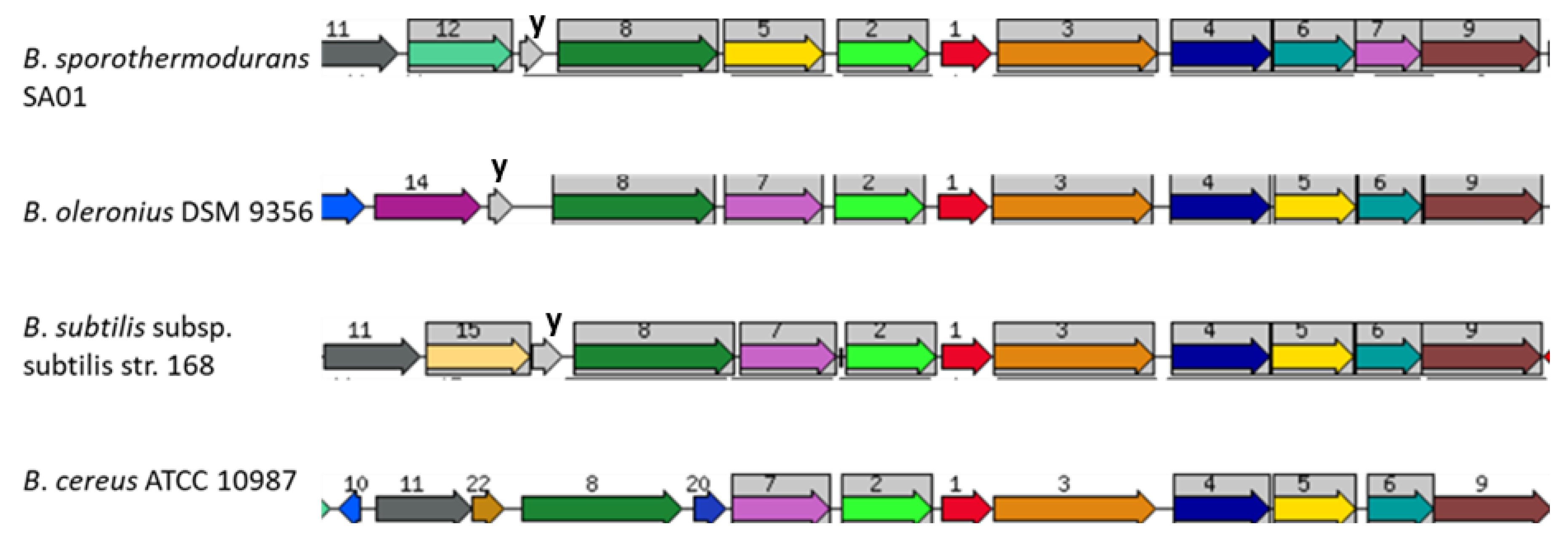
3.4. Protein Clusters Involved in Heat Resistance
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Patel, S. Drivers of bacterial genomes plasticity and roles they play in pathogen virulence, persistence and drug resistance. Infect. Genet. Evol. 2016, 45, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Magno-Perez-Bryan, M.C.; Martınez-Garcia, P.M.; Hierrezuelo, J.; Rodriguez-Palenzuela, P.; Arrebola, E.; Ramos, C.; de Vicente, A.; Perez-Garcia, A.; Romero, D. Comparative genomics within the Bacillus Genus reveal the singularities of two robust Bacillus amyloliquefaciens biocontrol strains. MPMI 2015, 28, 1102–1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tettelin, H.; Riley, D.; Cattuto, C.; Medini, D. Comparative genomics: The bacterial pan-genome. Curr. Opin. Microbiol. 2008, 11, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Loman, N.J.; Pallen, M.J. Twenty years of bacterial genome sequencing. Nat. Rev. Microbiol. 2015, 13, 787–794. [Google Scholar] [CrossRef]
- Land, M.; Hauser, L.; Jun, S.R.; Nookaew, I.; Leuze, M.R.; Ahn, T.H.; Karpinets, T.; Lund, O.; Kora, G.; Wassenaar, T.; et al. Insights from 20 years of bacterial genome sequencing. Funct. Integr. Genom. 2015, 15, 141–161. [Google Scholar] [CrossRef] [Green Version]
- Zwick, M.E.; Joseph, S.J.; Didelot, X.; Chen, P.E.; Bishop-Lilly, K.A.; Stewart, A.C.; Willner, K.; Nolan, N.; Lentz, S.; Thomason, M.K.; et al. Genomic characterization of the Bacillus cereus sensu lato species: Backdrop to the evolution of Bacillus anthracis. Genome Res. 2012, 22, 1512–1524. [Google Scholar] [CrossRef] [Green Version]
- McNally, A.; Oren, Y.; Kelly, D.; Pascoe, B.; Dunn, S.; Sreecharan, T.; Vehkala, M.; Välimäki, N.; Prentice, M.B.; Ashour, A.; et al. Combined Analysis of Variation in Core, Accessory and Regulatory Genome Regions Provides a Super-Resolution View into the Evolution of Bacterial Populations. PLoS Genet. 2016, 12, 1–16. [Google Scholar] [CrossRef]
- Alcaraz, L.; Moreno-Hagelsieb, G.; Eguiarte, L.E.; Souza, V.; Herrera-Estrella, L.; Olmedo, G. Understanding the evolutionary relationships and major traits of Bacillus through comparative genomics. BMC Genom. 2010, 11, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Satyanarayana, T. Comparative Genomics of Bacillus species and its Relevance in Industrial Microbiology. Genom. Insights 2013, 6, 25–36. [Google Scholar] [CrossRef]
- Nicholson, W.L.; Munakata, N.; Horneck, G.; Melosh, H.J.; Setlow, P. Resistance of Bacillus endospores to extreme terrestrial and extraterrestrial environments. Microbiol. Mol. Biol. Rev. 2000, 64, 548–572. [Google Scholar] [CrossRef] [Green Version]
- Schallmey, M.; Singh, A.; Ward, O.P. Developments in the use of Bacillus species for industrial production. Can. J. Microbiol. 2004, 50, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Scheldeman, P.; Herman, L.; Goris, J.; De Vos, P.; Heyndrickx, M.; De Vos, P.; Heyndrickx, M. Polymerase chain reaction identification of Bacillus sporothermodurans from dairy sources. J. Appl. Microbiol. 2002, 92, 983–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herman, L.M.F.; Vaerewijck, M.J.M.; Moermans, R.J.B.; Waes, G.M.A.V.J.; Waes, G.M.A.V.J. Identification and detection of Bacillus sporothermodurans spores in 1, 10, and 100 milliliters of raw milk by PCR. Appl. Environ. Microbiol. 1997, 63, 3139–3143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heyndrickx, M.; Coorevits, A.; Scheldeman, P.; Lebbe, L.; Schumann, P.; Rodŕiguez-Diaz, M.; Forsyth, G.; Dinsdale, A.; Heyrman, J.; Logan, N.A.; et al. Emended descriptions of Bacillus sporothermodurans and Bacillus oleronius with the inclusion of dairy farm isolates of both species. Int. J. Syst. Evol. Microbiol. 2012, 62, 307–314. [Google Scholar] [CrossRef]
- Owusu-Darko, R.; Allam, M.; de Oliveira, S.D.; Ferreira, C.A.S.; Grover, S.; Mtshali, S.; Ismail, A.; Mallappa, R.H.; Tabit, F.; Buys, E.M. Genome Sequences of Bacillus sporothermodurans Strains Isolated from Ultra-High-Temperature Milk. Microbiol. Resour. Announc. 2019, 8, e00145-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owusu-Darko, R.; Allam, M.; Mtshali, S.; Ismail, A.; Buys, E.M. Draft genome sequence of Bacillus oleronius DSM 9356 isolated from the termite Reticulitermes santonensis. Genom. Data 2017, 12, 76–78. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid Annotations using Subsystems Technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [Green Version]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, D206–D214. [Google Scholar] [CrossRef]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Wattam, A.R.; Abraham, D.; Dalay, O.; Disz, T.L.; Driscoll, T.; Gabbard, J.L.; Gillespie, J.J.; Gough, R.; Hix, D.; Kenyon, R.; et al. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 2014, 42, 581–591. [Google Scholar] [CrossRef] [Green Version]
- Wattam, A.R.; Davis, J.J.; Assaf, R.; Boisvert, S.; Brettin, T.; Bun, C.; Conrad, N.; Dietrich, E.M.; Disz, T.; Gabbard, J.L.; et al. Improvements to PATRIC, the all-bacterial bioinformatics database and analysis resource center. Nucleic Acids Res. 2017, 45, D535–D542. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A. RAxML Version 8: A tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30, 33–34. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A. FigTree v1.4.3. Available online: https://tree.bio.ed.ac.uk/software/figtree/ (accessed on 6 February 2020).
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, T. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Capra, J.A.; Singh, M. Predicting functionally important residues from sequence conservation. Bioinformatics 2007, 23, 1875–1882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaas, R.S.; Leekitcharoenphon, P.; Aarestrup, F.M.; Lund, O. Solving the problem of comparing whole bacterial genomes across different sequencing platforms. PLoS ONE 2014, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices. Comput. Appl. Biosci. 1992, 8, 275–282. [Google Scholar]
- Felsenstein, J. Confidence Limits on Phylogenies: An Approach Using the Bootstrap. Evolution 1985, 39, 783. [Google Scholar] [CrossRef]
- Kuhnigk, T.; Borst, E.-M.; Breunig, A.; König, H.; Collins, M.D.; Hutson, R.A.; Kämpfer, P. Bacillus oleronius sp.nov., a member of the hindgut flora of the termite Reticulitermes santonensis (Feytaud). Can. J. Microbiol. 1995, 41, 699–706. [Google Scholar] [CrossRef]
- Flint, S.; Gonzaga, Z.J.; Good, J.; Palmer, J. Bacillus thermoamylovorans—A new threat to the dairy industry—A review. Int. Dairy J. 2016, 65, 38–43. [Google Scholar] [CrossRef]
- Yang, G.; Zhou, X.; Zhou, S.; Yang, D.; Wang, Y.; Wang, D. Bacillus thermotolerans sp. nov., a thermophilic bacterium capable of reducing humus. Int. J. Syst. Evol. Microbiol. 2013, 63, 3672–3678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, A.; Pal, Y.; Khatri, I.; Ojha, A.K.; Gruber-Vodicka, H.; Schumann, P.; Dastager, S.; Subramanian, S.; Mayilraj, S.; Krishnamurthi, S. Examination into the taxonomic position of Bacillus thermotolerans Yang et al., 2013, proposal for its reclassification into a new genus and species Quasibacillus thermotolerans gen. nov., comb. nov. and reclassification of B. encimensis Dastager et al. Syst. Appl. Microbiol. 2017, 40, 411–422. [Google Scholar] [CrossRef]
- Bobay, L.M.; Ochman, H. The evolution of bacterial genome architecture. Front. Genet. 2017, 8, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheldeman, P.; Herman, L.; Foster, S.; Heyndrickx, M. Bacillus sporothermodurans and other highly heat-resistant spore formers in milk. J. Appl. Microbiol. 2006, 101, 542–555. [Google Scholar] [CrossRef] [PubMed]
- Mcinerney, J.O.; Mcnally, A.; Connell, M.J.O. Why prokaryotes have pangenomes. Nat. Publ. Gr. 2017, 2, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Mcinerney, J.O.; Mcnally, A.; Connell, M.J.O. Reply to ‘The population genetics of pangenomes’. Nat. Microbiol. 2017, 2, 41564. [Google Scholar] [CrossRef]
- Raskin, D.M.; Seshadri, R.; Pukatzki, S.U.; Mekalanos, J.J. Bacterial genomics and pathogen evolution. Cell 2006, 124, 703–714. [Google Scholar] [CrossRef] [Green Version]
- LaBreck, C.J.; May, S.; Viola, M.G.; Conti, J.; Camberg, J.L. The Protein Chaperone ClpX Targets Native and Non-native Aggregated Substrates for Remodeling, Disassembly, and Degradation with ClpP. Front. Mol. Biosci. 2017, 4, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Bukau, B.; Horwich, A.L. The Hsp70 and Hsp60 chaperone machines. Cell 1998, 92, 351–366. [Google Scholar] [CrossRef] [Green Version]
- Wickner, S.; Maurizi, M.R.; Gottesman, S. Posttranslational Quality Control: Folding, Refolding, and Degrading Proteins. Science 1999, 286, 1888–1893. [Google Scholar] [CrossRef] [PubMed]
- Stoecklin, G.; Bukau, B. Telling right from wrong in life—Cellular quality control. Nat. Rev. Mol. Cell Biol. 2013, 14, 613. [Google Scholar] [CrossRef] [PubMed]
- Beeby, M.; Connor, B.D.O.; Ryttersgaard, C.; Boutz, D.R.; Perry, L.J.; Yeates, T.O. The Genomics of Disulfide Bonding and Protein Stabilization in Thermophiles. PLoS Biol. 2005, 3, 1549–1558. [Google Scholar] [CrossRef] [Green Version]
- Ladenstein, R.; Ren, B. Protein disulfides and protein disulfide oxidoreductases in hyperthermophiles. FEBS J. 2006, 273, 4170–4185. [Google Scholar] [CrossRef] [PubMed]
- Hecker, M.; Schumann, W.; Völker, U. Heat-shock and general stress response in Bacillus subtilis. Mol. Microbiol. 1996, 19, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Liberek, K.; Georgopoulos, C. Autoregulation of the Escherichia coli heat shock response by the DnaK and DnaJ heat shock proteins. Proc. Natl. Acad. Sci. USA 1993, 90, 11019–11023. [Google Scholar] [CrossRef] [Green Version]
- Schroder, H.; Langer, T.; Hartl, F.; Bukaul, B. DnaK, DnaJ and GrpE form a cellular chaperone machinery capable of repairing heat-induced protein damage. EMBO J. 1993, 12, 4137–4144. [Google Scholar] [CrossRef]
- Gamer, J.; Multhaup, G.; Tomoyasu, T.; Mccarty, J.S.; Rudiger, S.; Schonfeld, H.-J.; Schirra, C.; Bujard, H.; Bukau, B. A cycle of binding and release of the DnaK, DnaJ and GrpE chaperones regulates activity of the Escherichia coli heat shock transcription factor sigma32. EMBO J. 1996, 15, 607–617. [Google Scholar] [CrossRef]
- Lee, S.; Sowa, M.E.; Watanabe, Y.H.; Sigler, P.B.; Chiu, W.; Yoshida, M.; Tsai, F.T.F. The structure of ClpB: A molecular chaperone that rescues proteins from an aggregated state. Cell 2003, 115, 229–240. [Google Scholar] [CrossRef] [Green Version]
- Doyle, S.M.; Wickner, S. Hsp104 and ClpB: Protein disaggregating machines. Trends Biochem. Sci. 2009, 34, 40–48. [Google Scholar] [CrossRef]
- Kirstein, J.; Molière, N.; Dougan, D.A.; Turgay, K. Adapting the machine: Adaptor proteins for Hsp100/Clp and AAA+ proteases. Nat. Rev. Microbiol. 2009, 7, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Gerth, U.; Kirstein, J.; Mostertz, J.; Waldminghaus, T.; Miethke, M.; Kock, H.; Hecker, M. Fine-Tuning in Regulation of Clp Protein Content in Bacillus subtilis. J. Bacteriol. 2004, 186, 179–191. [Google Scholar] [CrossRef] [Green Version]
- Glover, J.R.; Lindquist, S. Hsp104, Hsp70, and Hsp40: A novel chaperone system that rescues previously aggregated proteins. Cell 1998, 94, 73–82. [Google Scholar] [CrossRef] [Green Version]
- Tomoyasu, T.; Mogk, A.; Langen, H.; Goloubinoff, P.; Bukau, B. Genetic dissection of the roles of chaperones and proteases in protein folding and degradation in the Escherichia coli cytosol. Mol. Microbiol. 2001, 40, 397–413. [Google Scholar] [CrossRef]
- Schulz, A.; Schumann, W. hrcA, the first gene of the Bacillus subtilis dnaK operon encodes a negative regulator of class I heat shock genes. These include: hrcA, the First Gene of the Bacillus subtilis dnaK Operon Encodes a Negative Regulator of Class I Heat Shock Genes. J. Bacteriol. 1996, 178, 1088–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hitomi, M.; Nishimura, H.; Tsujimoto, Y.; Matsui, H.; Watanabe, K. Identification of a helix-turn-helix motif of Bacillus thermoglucosidasius HrcA essential for binding to the CIRCE element and thermostability of the HrcA-CIRCE complex, indicating a role as a thermosensor. J. Bacteriol. 2003, 185, 381–385. [Google Scholar] [CrossRef] [Green Version]
- Chastanet, A.; Losick, R. Engulfment during sporulation in Bacillus subtilis is governed by a multi-protein complex containing tandemly acting autolysins. Mol. Microbiol. 2007, 64, 139–152. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Owusu-Darko, R.; Allam, M.; Ismail, A.; Ferreira, C.A.S.; Oliveira, S.D.d.; Buys, E.M. Comparative Genome Analysis of Bacillus sporothermodurans with Its Closest Phylogenetic Neighbor, Bacillus oleronius, and Bacillus cereus and Bacillus subtilis Groups. Microorganisms 2020, 8, 1185. https://doi.org/10.3390/microorganisms8081185
Owusu-Darko R, Allam M, Ismail A, Ferreira CAS, Oliveira SDd, Buys EM. Comparative Genome Analysis of Bacillus sporothermodurans with Its Closest Phylogenetic Neighbor, Bacillus oleronius, and Bacillus cereus and Bacillus subtilis Groups. Microorganisms. 2020; 8(8):1185. https://doi.org/10.3390/microorganisms8081185
Chicago/Turabian StyleOwusu-Darko, Rodney, Mushal Allam, Arshad Ismail, Carlos A. S. Ferreira, Sílvia D. de Oliveira, and Elna M. Buys. 2020. "Comparative Genome Analysis of Bacillus sporothermodurans with Its Closest Phylogenetic Neighbor, Bacillus oleronius, and Bacillus cereus and Bacillus subtilis Groups" Microorganisms 8, no. 8: 1185. https://doi.org/10.3390/microorganisms8081185
APA StyleOwusu-Darko, R., Allam, M., Ismail, A., Ferreira, C. A. S., Oliveira, S. D. d., & Buys, E. M. (2020). Comparative Genome Analysis of Bacillus sporothermodurans with Its Closest Phylogenetic Neighbor, Bacillus oleronius, and Bacillus cereus and Bacillus subtilis Groups. Microorganisms, 8(8), 1185. https://doi.org/10.3390/microorganisms8081185