1. Introduction
In recent years, new discoveries regarding the composition and functionality of the human skin microbiota have been made, that have enabled a more comprehensive description of this ecosystem [
1,
2,
3]. Studies highlighted the diversity and uniqueness of the collection of skin microorganisms with essential roles in protection against harmful pathogens, maintaining skin homeostasis, and priming our immune system [
3,
4,
5,
6].
Coagulase-negative staphylococci (CoNS) constitute an important part of the human skin microbiota. Culture-dependent and -independent studies have highlighted the ubiquity of CoNS, which colonize mostly moist and sebaceous areas of the skin. In this regard, the CoNS species
Staphylococcus epidermidis, Staphylococcus capitis, and
Staphylococcus hominis occupy many human skin sites [
1,
2,
6,
7,
8,
9]. Other CoNS species, such as
Staphylococcus haemolyticus, Staphylococcus lugdunensis, and
Staphylococcus warneri, can be found in lower amounts, varying from person to person and from skin site to skin site [
1,
2,
9,
10]. Some other CoNS species, such as
Staphylococcus equorum, are primarily found in food products [
11], but are also transient colonizers of human skin.
Culture-dependent approaches have been often applied in the past to isolate major skin residents. Such approaches are often biased, as cultivation results do not reflect the true distribution of the individual members of the microbiota [
12,
13]. The bias is introduced due to the chosen growth media, as well as the conditions of growth, such as O
2 and CO
2 concentrations, growth temperature, and cultivation time. Fast-growing microorganisms have a growth advantage, and directly or indirectly inhibit the growth of slow-growing microorganisms [
6]. Therefore, culture-independent studies employing next generation sequencing (NGS)-based approaches are more frequently used in recent years. Using 16S rRNA gene amplicon-NGS, it was shown that the genus
Staphylococcus is the third most abundant genus on the skin [
14]. In addition, shotgun NGS studies further unraveled the diversity and individuality of staphylococcal species on the skin, and also provided insights into the strain level distribution of these CoNS species [
1,
2]. Such studies also highlighted the existence of microbial dark matter in the form of unidentified bacterial skin residents. For instance, the study of Oh et al. [
1] has identified several uncharacterized genomes (assembled from shotgun NGS data) of unknown species, possibly belonging to the genera
Corynebacterium, Cutibacterium, and
Staphylococcus. Thus, it can be expected that species exist on the skin that cannot be easily cultivated by standard conditions.
In this context, we have recently described the genomes of seven strains of
Staphylococcus saccharolyticus, an unusual CoNS species, regarding its growth properties [
15]. Unlike almost all other CoNS species known to date,
S. saccharolyticus largely depends on anaerobic conditions for growth, and requires fastidious growth media and prolonged cultivation time (>3 days). To date, this species has been relatively uncharacterized, with limited reports on its association with implant-associated infections [
15] and bacteremia [
16]. Interestingly, however, culture-dependent studies have suggested that this species may also be a resident of the skin microbiota [
17,
18].
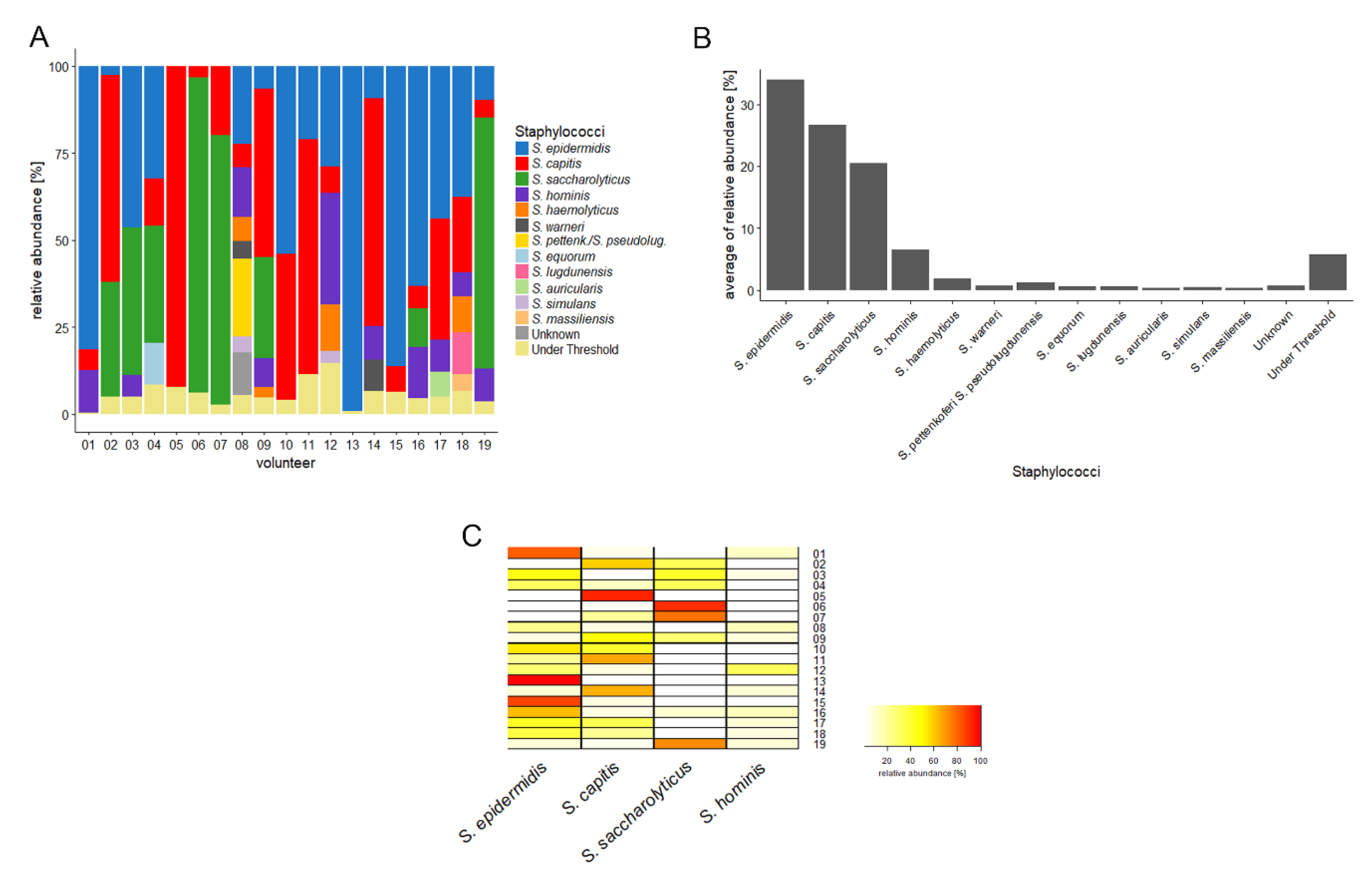
Here, we further investigated the composition and relative abundance of staphylococcal species on human skin. For this, we applied an amplicon-NGS approach based on a Staphylococcus-specific gene fragment, to survey its presence on human back skin samples from healthy volunteers. We detected an unprecedented high relative abundance of S. saccharolyticus in these samples. Supported by investigation of existing skin-derived metagenomic datasets, we posit that S. saccharolyticus constitutes a common member of the human skin microbiota.
2. Materials and Methods
2.1. Study Design and Sampling
Skin swab samples from 19 volunteers (female, n = 11; male, n = 8) with an age range of 22–43 years were taken from the upper back. None of the volunteers had a history of skin disease; none had undergone treatment with topical medicine or antibiotics during the last six months. Written informed consent was obtained from all volunteers, and the study was approved by International Medical & Dental Ethics Commission GmbH (IMDEC).
An area of 25 cm2 on the upper back was sampled with a cotton swab pre-moistened in aqueous sampling buffer containing disodium phosphate (12.49 g/L, Merck, Darmstadt, Germany), potassium dihydrogen phosphate (0.63 g/L, Merck, Darmstadt, Germany), and Triton X-100 (1 g/L, Merck, Darmstadt, Germany). After sampling, the swab was transferred into a sterile tube containing 2 mL of sampling buffer; the swab was vigorously shaken in the sampling buffer and then removed. Skin swab material was stored at -20 °C until further processing.
2.2. DNA Extraction
DNA from the 2 mL sample was extracted using the DNeasy PowerSoil Kit (QIAGEN, Hilden, Germany) following the manufacturer’s protocol, with an additional cell lysis step using lysostaphin (0.05 mg/mL, Merck, Darmstadt, Germany) and lysozyme (9.5 mg/mL, Merck, Darmstadt, Germany) prior to extraction. DNA concentrations were measured using the Qubit dsDNA HS Assay (ThermoFisher Scientific, Waltham, MA, USA) with a Qubit fluorometer following the manufacturer’s instructions.
2.3. Amplicon PCR
A fragment of the
tuf gene present in the genomes of all staphylococcal species available in GenBank (as of December 2019) was used for species identification, analogous to a previous study using a different
tuf gene fragment [
19]. The target sequence was amplified using
tuf-specific primers that contained MiSeq adapter sequences: tuf2_F, 5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGACAGGCCGTGTTGAACGTG-3′; tuf2_R, 5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGACAGTACGTCCACCTTCACG-3′.
PCR reaction mixtures were made in a total volume of 25 µl and comprised 5 µl of DNA sample, 2.5 µl AccuPrime PCR Buffer II (Invitrogen, Waltham, MA, USA), 1.5 µl of each primer (10 µM) (DNA Technology, Risskov, Denmark), 0.15 µl AccuPrime Taq DNA Polymerase High Fidelity (Invitrogen, Waltham, MA, USA), and 14.35 µl of PCR grade water. The PCR reaction was performed using the following cycle conditions: an initial denaturation at 94 °C for 2 min, followed by 35 cycles of denaturation at 94 °C for 20 sec, annealing at 55 °C for 30 sec, elongation at 68 °C for 1 min, and a final elongation step at 72 °C for 5 min. PCR products were verified on an agarose gel and purified using the Qiagen GenereadTM Size Selection kit (Qiagen, Hilden, Germany). The concentration of the purified PCR products was measured with a NanoDrop 2000 spectrophotometer (ThermoFisher Scientific, Waltham, MA, USA).
2.4. Amplicon Next Generation Sequencing
PCR products were used to attach indices and Illumina sequencing adapters using the Nextera XT Index kit (Illumina, San Diego, CA, USA). Index PCR was performed using 5 µl of template PCR product, 2.5 µl of each index primer, 12.5 µl of 2x KAPA HiFi HotStart ReadyMix, and 2.5 µl PCR grade water. The thermal cycling scheme was as follows: 95 °C for 3 min, 8 cycles of 30 sec at 95 °C, 30 sec at 55 °C, and 30 sec at 72 °C, and a final extension at 72 °C for 5 min. Quantification of the products was performed using the Quant-iT dsDNA HS assay kit (ThermoFisher Scientific, Waltham, MA, USA) and a Qubit fluorometer, following the manufacturer’s instructions. MagSi-NGSPREP Plus Magnetic beads (Steinbrenner Laborsysteme GmbH, Wiesenbach, Germany) were used for purification of the indexed products as recommended by the manufacturer, and normalization was performed using the Janus Automated Workstation from Perkin Elmer (Perkin Elmer, Waltham, MA, USA). Sequencing was conducted using an Illumina MiSeq platform with dual indexing and the MiSeq reagent kit v3 (600 cycles), as recommended by the manufacturer.
2.5. Bioinformatics
FASTQ sequences obtained after demultiplexing the reads and trimming the primers were imported into QIIME2 (v. 2019.7) [
20]. Sequences with an average quality score lower than 20 or containing unresolved nucleotides were removed from the dataset with the split_libraries_fastq.py script from QIIME. The paired-end reads were denoised and chimeras removed with DADA2 via QIIME2, and a feature table was generated [
21]. These features were then clustered with VSEARCH at a threshold of 99% identity against an in-house generated
tuf allele database that contained all
tuf alleles from all staphylococcal genomes available in GenBank (as of December 2019). Data were normalized, and figures were prepared in R with the packages ggplot2 and gplots.
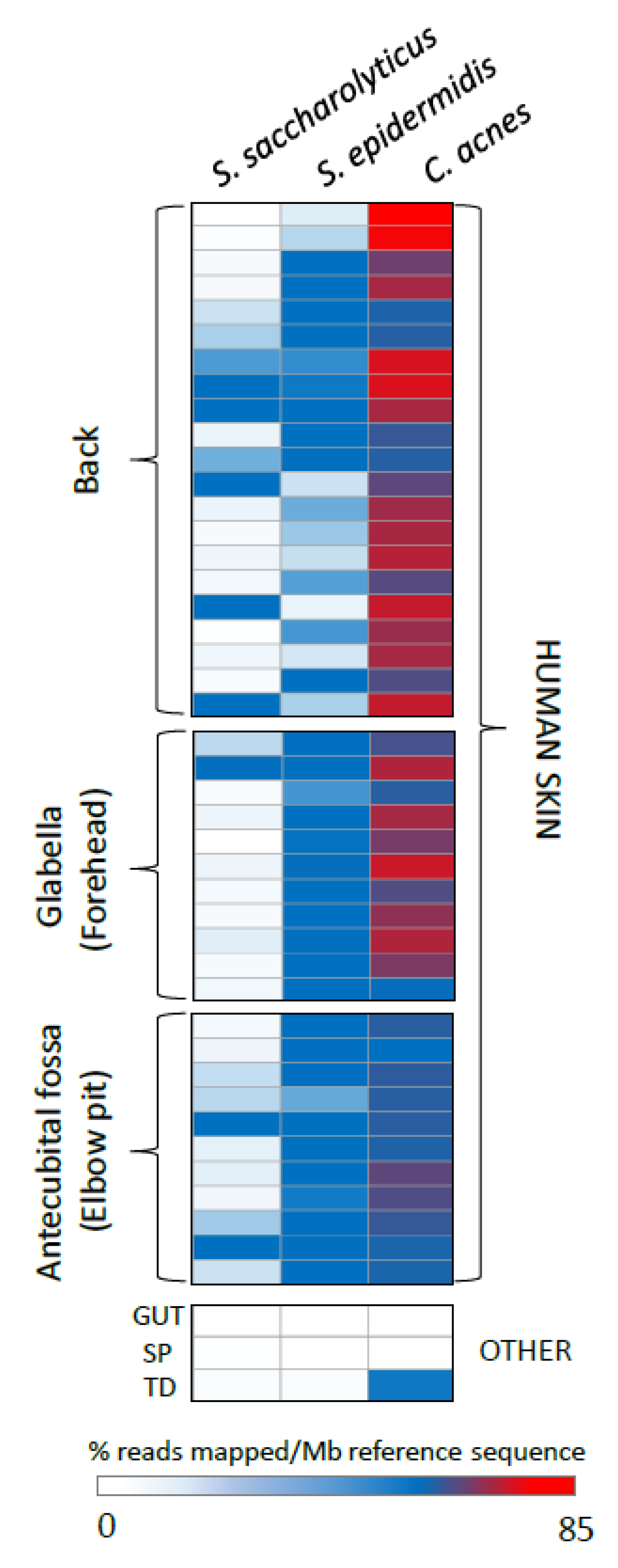
2.6. Metagenome Database Search Strategy
The presence of sequences similar to
S. saccharolyticus within available metagenomes deposited in the Sequence Read Archive (SRA) was initially assessed using the online tool at
www.searchsra.org to provide a broad overview of datasets with matches. A more detailed investigation of the level of representation of
S. saccharolyticus in existing human skin and other human-associated metagenomic datasets (identified within the initial search of the SRA) was then conducted by mapping pooled sequencing reads from metagenomic datasets against the
S. saccharolyticus genome sequence (strain 05B0362; GenBank accession number: QHKH00000000). Sequencing reads were obtained from the SRA and processed using Geneious Prime 2020 to remove low quality reads (Trim using BBDuk; min. 50 bp) and duplicates (Dedupe from BBTools), with default parameters. The resulting collections of high-quality reads were mapped against the genome sequences of
S. saccharolyticus 05B0362,
Cutibacterium acnes strain ATCC 6919 (NZ_CP023676.1) and
Staphylococcus epidermidis strain ATCC 14990 (NZ_CP035288.1) using the Geneious Prime 2020 map to reference tool with the following criteria: 100% identity; no gaps or mismatches; maximum ambiguity = 1. For each metagenomic dataset mapped, the total number of reads mapped to each reference genome was normalized by the total size of the dataset, to provide reads mapped per megabase DNA.
A more targeted search strategy was also applied using an
S. saccharolyticus-specific gene as a query to search metagenomic datasets derived from back skin [
1]. The
hya gene encoding a hyaluronate lysase was chosen (locus tag DMB78_01130 in the genome of strain 05B0362), due to its low average nucleotide identify with
hya genes from other staphylococcal species. The search was performed as an SRA nucleotide BLAST. The gene was considered as being present when the coverage exceeded 40%.
2.7. S. Saccharolyticus Growth
S. saccharolyticus (strain DVP5-16-4677) was grown on Fastidious anaerobic agar (FAA) plates (LAB M, Bury, UK) and incubated anaerobically at 37 °C for 4 days. For liquid growth, brain-heart infusion-yeast broth supplemented with 0.05% (w/vol) cysteine (BHCY broth) was used. The following growth conditions were evaluated and performed at 37 °C: anoxic conditions (Oxoid AnaeroGen System; ThermoFisher Scientific, Waltham, MA, USA), and oxic conditions with and without CO2-supplementation (Oxoid CO2 Gen system; ThermoFisher Scientific, Waltham, MA, USA). Optical density (OD) data at 600 nm was determined until the stationary growth phase.
4. Discussion
The skin microbiome affects the health-state of our skin. Understanding the bacterial composition on non-diseased skin is therefore of importance. Here, we focused on the staphylococcal composition of the human back skin. Similar to most previous studies [
1,
2],
S. epidermidis was found to be the most abundant staphylococcal species on human back skin, followed by
S. capitis. Surprisingly,
S. saccharolyticus ranked third. This species has previously not been reported to be abundant on human skin. However, two studies from 1978 reported the presence of
S. saccharolyticus, formerly named
Peptococcus saccharolyticus, in forehead and armpit skin samples [
17,
18]. In these studies, around 20% of samples were found to be positive for
S. saccharolyticus. The organism grew on TSY (supplemented with 0.5% Tween-80) agar plates after 4–7 days of incubation, with preference for anaerobic conditions. Identification (and differentiation from other CoNS) was based on cell and colony morphology, anaerobic growth preference, and a weak catalase activity. Biochemically,
S. saccharolyticus cannot produce lactic acid from glucose (in contrast to other staphylococci); it can ferment glucose, fructose, and glycerol, but not maltose. Interestingly, Evans et al. [
17] stated that it was puzzling that the organism was not recognized in past studies “in view of its prevalence”. The authors also suggested that the reason that previous skin studies have overlooked this organism was due to (i) the choice of the culture media, (ii) the need for prolonged incubation time, (iii) the preference for anaerobic culture conditions, and (iv) misidentification. Indeed, 40 years later, not much has changed in this regard. Most culture-dependent skin microbiota studies do overlook this microorganism, possibly due to inappropriate growth and cultivation conditions, as outlined by Evans et al. [
17,
18]. In addition, fast-growing species such as
S. epidermidis might outcompete
S. saccharolyticus on (standard) agar plates. This could explain why
S. saccharolyticus was overlooked in culture-dependent studies.
However, this does not explain why
S. saccharolyticus was previously not detected in culture-independent studies, which are nowadays more frequently conducted. Many culture-independent studies are carried out using 16S rRNA gene amplicon sequencing, which relies on sufficient differences in the 16S rRNA gene to distinguish species. However, the 16S rRNA gene of
S. saccharolyticus does not carry many single nucleotide polymorphisms (SNPs) that can easily distinguish it from other CoNS, namely
S. capitis [
15]. Thus, depending on the 16S rRNA gene amplification strategy (amplifying the V1, V2, V3, V4, V5, and the V6 region, respectively, or a combination thereof),
S. saccharolyticus can be indistinguishable from
S. capitis.
In recent years, shotgun sequencing was employed to identify the skin metagenome [
1,
2]. However, meaningful analyses of shotgun sequencing data rely on reference genomes of all skin microorganisms. Regarding
S. saccharolyticus, no such reference genome was available before March 2019. Adding to the confusion, three genomes assigned to
S. saccharolyticus were publicly available in GenBank before 2019, but these were wrongly classified as
S. saccharolyticus, and actually belong to
S. capitis, as previously noted [
15]. They were recently correctly reassigned to
S. capitis. Besides the genomes of seven
S. saccharolyticus strains that have been sequenced during our previous study [
15], the type strain of
S. saccharolyticus (ATCC 14953/NCTC 11807) has been sequenced by two independent teams (WGS projects UHDZ01 and RXWW01), resulting in nearly identical genome sequences. Taken together, before March 2019 there was no correct reference genome sequence of
S. saccharolyticus available; this has been now resolved. Thus, current and future shotgun sequencing studies should be able to identify
S. saccharolyticus correctly.
Here, we employed an amplicon sequencing method that is a modification from an existing method by Strube et al. [
19]. The method is based on the amplification of a
tuf gene fragment, with primers that were designed by Martineau et al. [
22]. The
tuf gene, encoding the elongation factor Tu, is a highly conserved gene in all staphylococcal species. We modified this method by choosing different amplification primers, since we noticed that the reverse primer designed by Martineau et al. [
22] has two mismatches with the corresponding
tuf gene sequence in the genome of
S. saccharolyticus. It is thus likely that the primers of Martineau et al. [
22] do not amplify the
tuf gene of
S. saccharolyticus, or only with reduced efficiency.
Previous studies reported that
S. saccharolyticus has a preference for anaerobic growth conditions [
17,
18]. Here, we showed that the bacterium can also grow under atmospheric conditions in broth, but with a reduced growth yield compared to anaerobic conditions. However, the growth in a CO
2-rich atmosphere is comparable to the growth under anaerobic conditions. A mechanistic explanation of the effect of CO
2 on bacterial growth was recently published by Fan et al. [
23]. The authors investigated the growth-promoting effect of CO
2 and the CO
2-dependency of small colony variants of
S. aureus, whose growth defect can be compensated by increased CO
2/bicarbonate supplementation. They found that staphylococci employ a CO
2-concentrating mechanism that enables them to grow at atmospheric CO
2 levels. More specifically, they found that the system MpsAB is crucial for
S. aureus growth at atmospheric CO
2 levels. From a set of carefully designed experiments, they concluded that the MpsAB system represents a dissolved inorganic carbon transporter, or bicarbonate concentrating system, which creates an elevated concentration of intracellular bicarbonate. Consequently, a staphylococcal species without a functional MpsAB system would only grow poorly at atmospheric CO
2 levels, and would rely on increased CO
2 concentrations for accelerated growth. As we previously noted that the genome of
S. saccharolyticus contains over 300 frameshift mutations [
15], we checked the
mpsAB genes. Indeed, the
mpsAB genes in the genome of
S. saccharolyticus are frameshifted, and thus the MpsAB system is most likely non-functional in
S. saccharolyticus. In conclusion, the lack of a functional CO
2-concentrating system in
S. saccharolyticus is a likely explanation for its insufficient growth under atmospheric conditions. This can be compensated by providing increased CO
2 concentrations.
Many questions remain, e.g., regarding the preferred niche of S. saccharolyticus on human skin and its evolutionary history that can be regarded as an example of reductive evolution, indicative of the massive genome decay [
15]. Genomic modifications such as genome decay are often a result of a relative recent lifestyle change, e.g., the adaptation to a new host or a new niche within a host, associated with a strict(er) host dependency [
24,
25,
26]. What could a scenario for the evolutionary history of
S. saccharolyticus look like? In this regard, an interesting feature of
S. saccharolyticus is the presence of a hyaluronate lyase (Hya), which is absent in other human-associated skin-resident CoNS. Closest homologs of the
hya gene of
S. saccharolyticus are present in two animal-associated staphylococci:
Staphylococcus agnetis and
Staphylococcus hyicus. S. agnetis is associated with lameness in broiler chickens, and
S. hyicus causes skin diseases, such as exudative dermatitis in piglets [
27,
28]. It is tempting to suggest that the (horizontal) acquisition of
hya, possibly from an animal-associated staphylococcal species, contributed to a lifestyle switch of
S. saccharolyticus. Hyaluronate lyases degrade hyaluronic acid, a major polysaccharide of the extracellular matrix of tissues [
29]. In the epidermis, hyaluronic acid is found in high concentrations, in particular in deeper layers of the epidermis, such as the stratum spinosum [
30]. Thus, a hyaluronidase-producing
S. saccharolyticus is likely better equipped to penetrate and propagate in deeper layers of the epidermis. We further speculate that a strong host association in deeper layers might have been established, where
S. saccharolyticus would have access to a range of host-derived compounds including amino acids and cofactors. This in turn would render bacterial genes to synthesize such compounds dispensable. As a consequence, genome decay would be accelerated, aiming at a slimmer, less energy-consuming lifestyle that is adapted to an oxygen-depleted niche, i.e., the epidermis below the stratum corneum.
Another open question remains regarding the clinical significance of these findings. At present, only few studies, mainly case reports, have reported the involvement of
S. saccharolyticus in human disease. The microorganism has been described as the etiologic agent of infective endocarditis, empyema and bone and joint infections such as shoulder synovitis and vertebral osteomyelitis [
31,
32,
33,
34,
35,
36]. In addition, the bacterium was reported to be responsible for nosocomial bloodstream infections in a German hospital [
16]. A few reports have found
S. saccharolyticus in implant-associated infections, such as prosthetic valve endocarditis [
37] and we recently described eight cases of prosthetic joint infections where
S. saccharolyticus was identified from tissue biopsies [
15]. If
S. saccharolyticus is widespread on human skin, as our study results suggest, one would expect to see more reports regarding the potential disease association of this bacterium, as seen for example for other skin-resident CoNS, such as
S. epidermidis and
S. capitis. As outlined above in detail, we hypothesize that mainly due to the fastidious growth conditions,
S. saccharolyticus was overlooked in numerous disease cases, in particular in implant-associated infections, as such infections are often caused by skin-derived bacteria, including CoNS. In several cases,
S. saccharolyticus has been identified, but was labeled as contaminant [
38]. As also true for other CoNS, assigning an etiological role to
S. saccharolyticus in disease requires thorough investigations to exclude skin-derived contamination of the biopsy material or contamination during subsequent sample processing steps. Carefully designed and executed future studies are needed to elucidate the etiology and frequency of
S. saccharolyticus in human disease.
The study has some limitations, most importantly the small sample size (
n = 19) and the focus on back skin. Moreover, only relatively young participants were investigated in this study. Appropriate skin sampling methods have previously been discussed [
39]; due to the here applied sampling method, i.e., skin swabs, we harvested mainly the microbiome of the stratum corneum. Thus, microorganisms that potentially penetrate deeper skin layers of the epidermis might be underrepresented. In future studies, we aim at analyzing more individuals, thereby covering and comparing different age groups and diverse skin health conditions. In addition, different skin sites will be investigated, and different skin sampling methods applied, in order to determine the specific skin tissue location of
S. saccharolyticus.
In conclusion, here we found that the coagulase-negative species S. saccharolyticus is relatively often found in human skin samples, as judged from a culture-independent amplicon sequencing approach. When present, the organism can comprise a major part of the staphylococcal skin population, and is found in several different skin sites. It has yet to be shown in the future if skin that is primarily colonized with S. saccharolyticus has distinguishable features from skin colonized with S. epidermidis or S. capitis.

 ,
, 
{kind=link}
{kind=link}
{kind=link}