Enteroviral Pathogenesis of Type 1 Diabetes: The Role of Natural Killer Cells


Abstract
1. Introduction
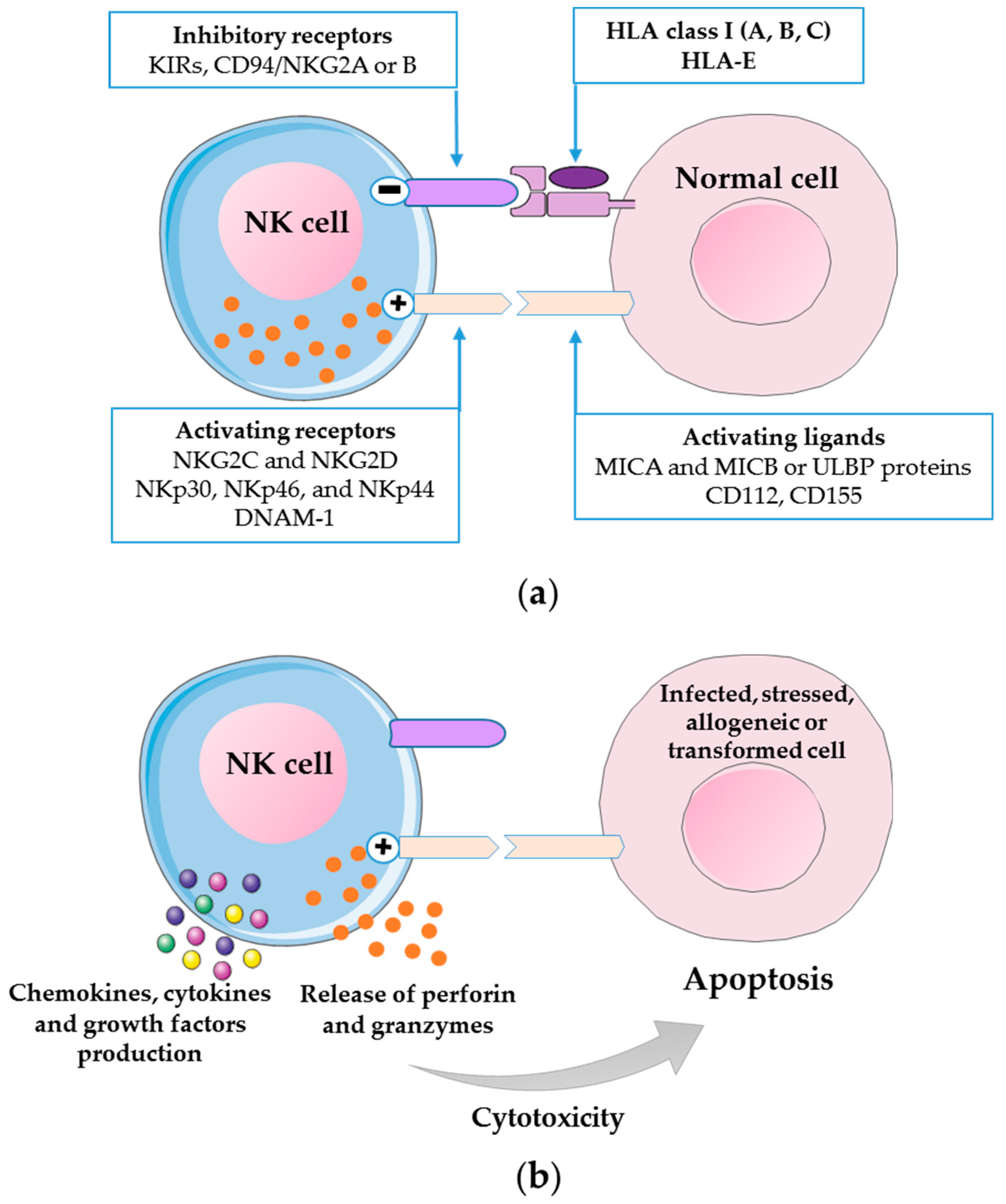
2. Biology of Human NK Cells
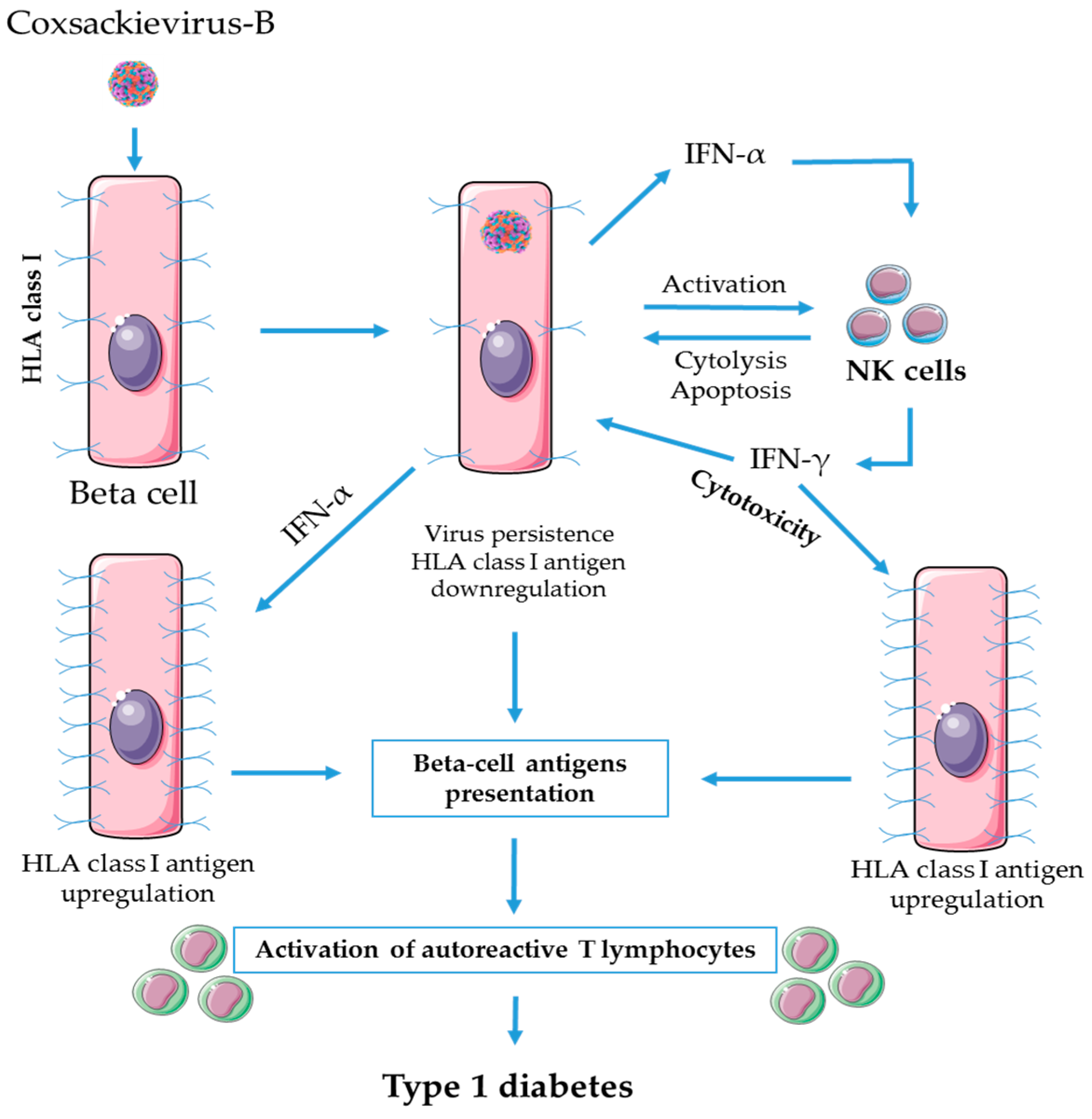
3. Role of NK Cells in Enteroviral Pathogenesis of T1D
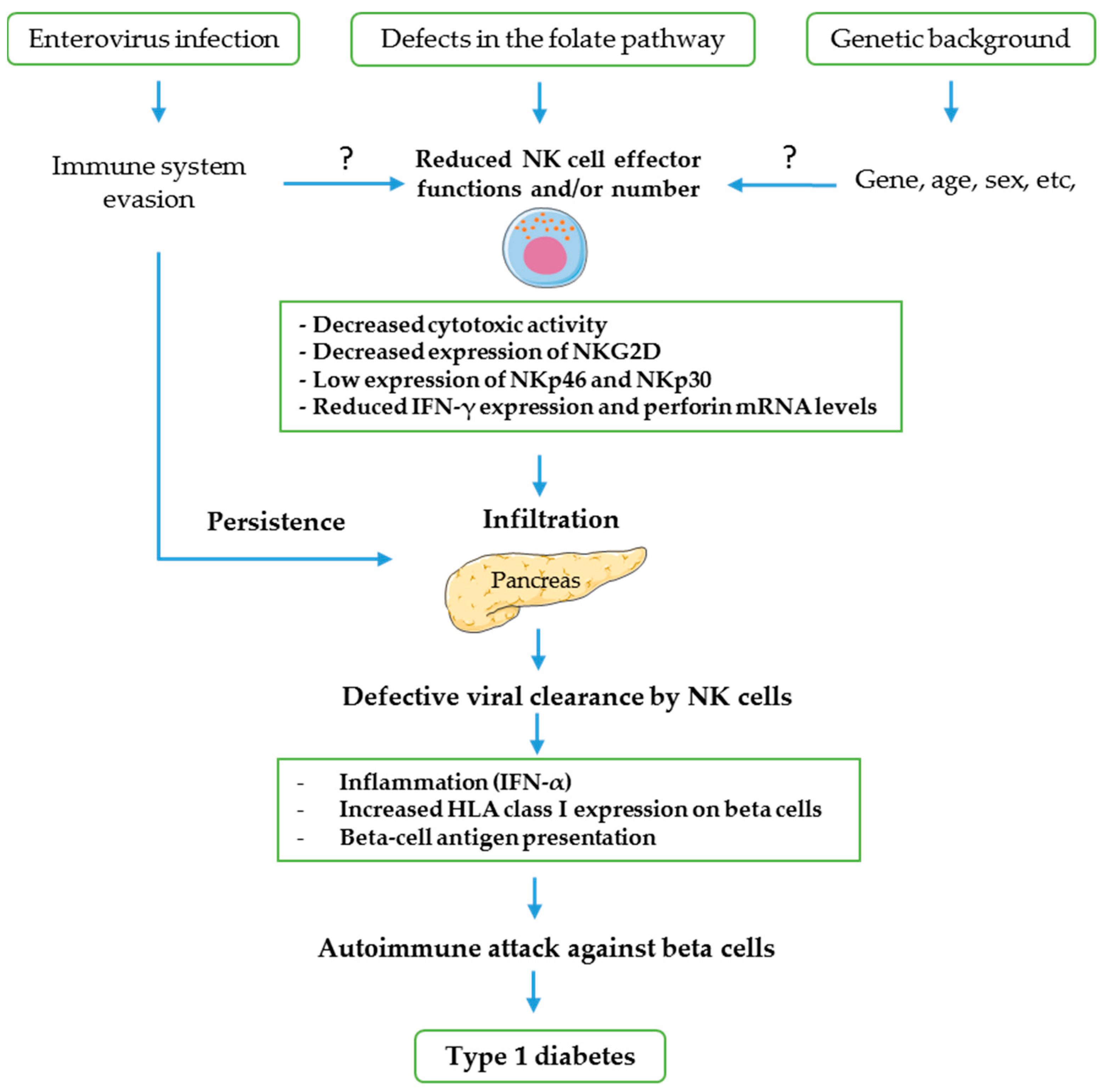
4. Possible Mechanisms of NK Cells Involvement in CV-B-Induced T1D
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Atkinson, M.A.; Eisenbarth, G.S. Type 1 diabetes: New perspectives on disease pathogenesis and treatment. Lancet 2001, 358, 221–229. [Google Scholar] [CrossRef]
- Von Herrath, M.G. Regulation of virally induced autoimmunity and immunopathology: Contribution of LCMV transgenic models to understanding autoimmune insulin-dependent diabetes mellitus. Curr. Top. Microbiol. Immunol. 2002, 263, 145–175. [Google Scholar] [PubMed]
- Gale, E.A.M. The rise of childhood type 1 diabetes in the 20th century. Diabetes 2002, 51, 3353–3361. [Google Scholar] [CrossRef] [PubMed]
- Hober, D.; Sauter, P. Pathogenesis of type 1 diabetes mellitus: Interplay between enterovirus and host. Nat. Rev. Endocrinol. 2010, 6, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Yeung, W.-C.G.; Rawlinson, W.D.; Craig, M.E. Enterovirus infection and type 1 diabetes mellitus: Systematic review and meta-analysis of observational molecular studies. BMJ 2011, 342, d35. [Google Scholar] [CrossRef]
- Hober, D.; Alidjinou, E.K. Enteroviral pathogenesis of type 1 diabetes: Queries and answers. Curr. Opin. Infect. Dis. 2013, 26, 263–269. [Google Scholar] [CrossRef]
- Allen, D.W.; Kim, K.W.; Rawlinson, W.D.; Craig, M.E. Maternal virus infections in pregnancy and type 1 diabetes in their offspring: Systematic review and meta-analysis of observational studies. Rev. Med. Virol. 2018, 28, e1974. [Google Scholar] [CrossRef]
- Ilonen, J.; Lempainen, J.; Veijola, R. The heterogeneous pathogenesis of type 1 diabetes mellitus. Nat. Rev. Endocrinol. 2019, 15, 635–650. [Google Scholar] [CrossRef]
- Zell, R.; Delwart, E.; Gorbalenya, A.E.; Hovi, T.; King, A.M.Q.; Knowles, N.J.; Lindberg, A.M.; Pallansch, M.A.; Palmenberg, A.C.; Reuter, G.; et al. ICTV Virus Taxonomy Profile: Picornaviridae. J. Gen. Virol. 2017, 98, 2421–2422. [Google Scholar] [CrossRef]
- Tapparel, C.; Siegrist, F.; Petty, T.J.; Kaiser, L. Picornavirus and enterovirus diversity with associated human diseases. Infect. Genet. Evol. 2013, 14, 282–293. [Google Scholar] [CrossRef]
- Harvala, H.; Kalimo, H.; Dahllund, L.; Santti, J.; Hughes, P.; Hyypiä, T.; Stanway, G. Mapping of tissue tropism determinants in coxsackievirus genomes. J. Gen. Virol. 2002, 83, 1697–1706. [Google Scholar] [CrossRef] [PubMed]
- Pallansch, M.; Obserte, M.; Whitton, J. Enteroviruses: Polioviruses, coxsackieviruses, echoviruses, and newer enteroviruses. In Fields Virology; DM, K., Howley, P., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013. [Google Scholar]
- Romero, J.R. Pediatric Group B Coxsackievirus Infections. In Group B Coxsackieviruses; Springer: Berlin/Heidelberg, Germany, 2008; Volume 323, pp. 223–239. [Google Scholar]
- Casas-Alba, D.; De Sevilla, M.F.; Valero-Rello, A.; Fortuny, C.; García-García, J.J.; Ortez, C.; Muchart, J.; Armangué, T.; Jordan, I.; Luaces, C.; et al. Outbreak of brainstem encephalitis associated with enterovirus-A71 in Catalonia, Spain (2016): A clinical observational study in a children’s reference centre in Catalonia. Clin. Microbiol. Infect. 2017, 23, 874–881. [Google Scholar] [CrossRef] [PubMed]
- Solomon, T.; Lewthwaite, P.; Perera, D.; Cardosa, M.J.; McMinn, P.; Ooi, M.H. Virology, epidemiology, pathogenesis, and control of enterovirus 71. Lancet Infect. Dis. 2010, 10, 778–790. [Google Scholar] [CrossRef]
- Jaïdane, H.; Sauter, P.; Sane, F.; Goffard, A.; Gharbi, J.; Hober, D. Enteroviruses and type 1 diabetes: Towards a better understanding of the relationship. Rev. Med. Virol. 2010, 20, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Racaniello, V.R. Picornaviridae: The Viruses and Their Replication. In Fields Virology; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 453–490. [Google Scholar]
- Yin, H.; Berg, A.-K.; Westman, J.; Hellerström, C.; Frisk, G. Complete nucleotide sequence of a Coxsackievirus B-4 strain capable of establishing persistent infection in human pancreatic islet cells: Effects on insulin release, proinsulin synthesis, and cell morphology. J. Med. Virol. 2002, 68, 544–557. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Berg, A.K.; Tuvemo, T.; Frisk, G. Enterovirus RNA is found in peripheral blood mononuclear cells in a majority of type 1 diabetic children at onset. Diabetes 2002, 51, 1964–1971. [Google Scholar] [CrossRef]
- Oikarinen, M.; Tauriainen, S.; Oikarinen, S.; Honkanen, T.; Collin, P.; Rantala, I.; Mäki, M.; Kaukinen, K.; Hyöty, H. Type 1 diabetes is associated with enterovirus infection in gut mucosa. Diabetes 2012, 61, 687–691. [Google Scholar] [CrossRef] [PubMed]
- Oikarinen, S.; Martiskainen, M.; Tauriainen, S.; Huhtala, H.; Ilonen, J.; Veijola, R.; Simell, O.; Knip, M.; Hyöty, H. Enterovirus RNA in blood is linked to the development of type 1 diabetes. Diabetes 2011, 60, 276–279. [Google Scholar] [CrossRef]
- Alidjinou, E.K.; Sané, F.; Engelmann, I.; Geenen, V.; Hober, D. Enterovirus persistence as a mechanism in the pathogenesis of type 1 diabetes. Discov. Med. 2014, 18, 273–282. [Google Scholar]
- Krogvold, L.; Edwin, B.; Buanes, T.; Frisk, G.; Skog, O.; Anagandula, M.; Korsgren, O.; Undlien, D.; Eike, M.C.; Richardson, S.J.; et al. Detection of a low-grade enteroviral infection in the islets of langerhans of living patients newly diagnosed with type 1 diabetes. Diabetes 2015, 64, 1682–1687. [Google Scholar] [CrossRef]
- Alidjinou, E.K.; Sané, F.; Trauet, J.; Copin, M.C.; Hober, D. Coxsackievirus B4 can infect human peripheral blood-derived macrophages. Viruses 2015, 7, 6067–6079. [Google Scholar] [CrossRef] [PubMed]
- Badia-Boungou, F.; Sane, F.; Alidjinou, E.K.; Ternois, M.; Opoko, P.A.; Haddad, J.; Stukens, C.; Lefevre, C.; Gueorguieva, I.; Hamze, M.; et al. Marker of coxsackievirus-B4 infection in saliva of patients with type 1 diabetes. Diabetes Metab. Res. Rev. 2017, 33, e2916. [Google Scholar] [CrossRef] [PubMed]
- Nekoua, M.P.; Yessoufou, A.; Alidjinou, E.K.; Badia-Boungou, F.; Moutairou, K.; Sane, F.; Hober, D. Salivary anti-coxsackievirus-B4 neutralizing activity and pattern of immune parameters in patients with type 1 diabetes: A pilot study. Acta Diabetol. 2018, 55, 827–834. [Google Scholar] [CrossRef]
- Dotta, F.; Censini, S.; Van Halteren, A.G.S.; Marselli, L.; Masini, M.; Dionisi, S.; Mosca, F.; Boggi, U.; Muda, A.O.; Del Prato, S.; et al. Coxsackie B4 virus infection of β cells and natural killer cell insulitis in recent-onset type 1 diabetic patients. Proc. Natl. Acad. Sci. USA 2007, 104, 5115–5120. [Google Scholar] [CrossRef] [PubMed]
- Richardson, S.J.; Willcox, A.; Bone, A.J.; Foulis, A.K.; Morgan, N.G. The prevalence of enteroviral capsid protein vp1 immunostaining in pancreatic islets in human type 1 diabetes. Diabetologia 2009, 52, 1143–1151. [Google Scholar] [PubMed]
- Richardson, S.J.; Leete, P.; Bone, A.J.; Foulis, A.K.; Morgan, N.G. Expression of the enteroviral capsid protein VP1 in the islet cells of patients with type 1 diabetes is associated with induction of protein kinase R and downregulation of Mcl-1. Diabetologia 2013, 56, 185–193. [Google Scholar] [CrossRef]
- Chehadeh, W.; Kerr-Conte, J.; Pattou, F.; Alm, G.; Lefebvre, J.; Wattré, P.; Hober, D. Persistent Infection of Human Pancreatic Islets by Coxsackievirus B Is Associated with Alpha Interferon Synthesis in β Cells. J. Virol. 2000, 74, 10153–10164. [Google Scholar] [CrossRef]
- Nekoua, M.P.; Bertin, A.; Sane, F.; Alidjinou, E.K.; Lobert, D.; Trauet, J.; Hober, C.; Engelmann, I.; Moutairou, K.; Yessoufou, A.; et al. Pancreatic beta cells persistently infected with coxsackievirus B4 are targets of NK cell-mediated cytolytic activity. Cell. Mol. Life Sci. 2020, 77, 179–194. [Google Scholar] [CrossRef]
- De Beeck, A.O.; Eizirik, D.L. Viral infections in type 1 diabetes mellitus-why the β cells? Nat. Rev. Endocrinol. 2016, 12, 263–273. [Google Scholar] [CrossRef]
- Silfka, M.K.; Rodriguez, F.; Whitton, J.L. Rapid on/off cycling of cytokine production by virus-specific CD8+ T cells. Nature 1999, 401, 76–79. [Google Scholar] [CrossRef]
- Richardson, S.J.; Morgan, N.G.; Foulis, A.K. Pancreatic pathology in type 1 diabetes mellitus. Endocr. Pathol. 2014, 25, 80–92. [Google Scholar] [CrossRef]
- Seliger, B.; Ritz, U.; Ferrone, S. Molecular mechanisms of HLA class I antigen abnormalities following viral infection and transformation. Int. J. Cancer 2006, 118, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Biron, C.A.; Nguyen, K.B.; Pien, G.C.; Cousens, L.P.; Salazar-Mather, T.P. NATURAL KILLER CELLS IN ANTIVIRAL DEFENSE: Function and Regulation by Innate Cytokines. Annu. Rev. Immunol. 1999, 17, 189–220. [Google Scholar] [CrossRef] [PubMed]
- Smyth, M.J.; Cretney, E.; Kelly, J.M.; Westwood, J.A.; Street, S.E.A.; Yagita, H.; Takeda, K.; Dommelen, S.L.H.V.; Degli-Esposti, M.A.; Hayakawa, Y. Activation of NK cell cytotoxicity. Mol. Immunol. 2005, 42, 501–510. [Google Scholar] [CrossRef]
- Vitale, C.; Chiossone, L.; Morreale, G.; Lanino, E.; Cottalasso, F.; Moretti, S.; Dini, G.; Moretta, L.; Mingari, M.C. Human natural killer cells undergoing in vivo differentiation after allogeneic bone marrow transplantation: Analysis of the surface expression and function of activating NK receptors. Mol. Immunol. 2005, 42, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Warren, H.S.; Smyth, M.J. NK cells and apoptosis. Immunol. Cell Biol. 1999, 77, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.G.; Anderson, J.; Shenton, B.K.; White, M.D.; Taylor, R.M.; Proud, G. Natural killer cells in insulin dependent diabetes mellitus. Br. Med. J. (Clin. Res. Ed.). 1986, 293, 244. [Google Scholar] [CrossRef][Green Version]
- Hussain, M.J.; Alviggi, L.; Millward, B.A.; Leslie, R.D.; Pyke, D.A.; Vergani, D. Evidence that the reduced number of natural killer cells in type 1 (insulin-dependent) diabetes may be genetically determined. Diabetologia 1987, 30, 907–911. [Google Scholar] [CrossRef]
- Negishi, K.; Waldeck, N.; Chandy, G.; Buckingham, B.; Kershnar, A.; Fisher, L.; Gupta, S.; Charles, M.A. Natural killer cell and islet killer cell activities in type 1 (insulin-dependent) diabetes. Diabetologia 1986, 29, 352–357. [Google Scholar] [CrossRef]
- Lorini, R.; Moretta, A.; Valtorta, A.; D’Annunzio, G.; Cortona, L.; Vitali, L.; Bozzola, M.; Severi, F. Cytotoxic activity in children with insulin-dependent diabetes mellitus. Diabetes Res. Clin. Pract. 1994, 23, 37–42. [Google Scholar] [CrossRef]
- Qin, H.; Lee, I.-F.; Panagiotopoulos, C.; Wang, X.; Chu, A.D.; Utz, P.J.; Priatel, J.J.; Tan, R. Natural Killer Cells From Children With Type 1 Diabetes Have Defects in NKG2D-Dependent Function and Signaling. Diabetes 2011, 60, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Flodström, M.; Shi, F.D.; Sarvetnick, N.; Ljunggren, H.G. The natural killer cell—Friend or foe in autoimmune disease? Scand. J. Immunol. 2002, 55, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Rodacki, M.; Svoren, B.; Butty, V.; Besse, W.; Laffel, L.; Benoist, C.; Mathis, D. Altered natural killer cells in type 1 diabetic patients. Diabetes 2007, 56, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Marca, V.; Gianchecchi, E.; Fierabracci, A. Type 1 diabetes and its multi-factorial pathogenesis: The putative role of NK cells. Int. J. Mol. Sci. 2018, 19, 794. [Google Scholar] [CrossRef]
- Godeny, E.K.; Gauntt, C.J. Involvement of natural killer cells in coxsackievirus B3-induced murine myocarditis. J. Immunol. 1986, 137, 1695–1702. [Google Scholar]
- Godeny, E.K.; Gauntt, C.J. Murine natural killer cells limit coxsackievirus B3 replication. J. Immunol. 1987, 139, 913–918. [Google Scholar]
- Hühn, M.H.; Hultcrantz, M.; Lind, K.; Ljunggren, H.G.; Malmberg, K.J.; Flodström-Tullberg, M. IFN-γ production dominates the early human natural killer cell response to Coxsackievirus infection. Cell. Microbiol. 2008, 10, 426–436. [Google Scholar] [CrossRef][Green Version]
- Cooper, M.A.; Elliott, J.M.; Keyel, P.A.; Yang, L.; Carrero, J.A.; Yokoyama, W.M. Cytokine-induced memory-like natural killer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 1915–1919. [Google Scholar] [CrossRef]
- Sun, J.C.; Beilke, J.N.; Lanier, L.L. Adaptive immune features of natural killer cells. Nature 2009, 457, 557–561. [Google Scholar] [CrossRef]
- Gianchecchi, E.; Delfino, D.V.; Fierabracci, A. NK cells in autoimmune diseases: Linking innate and adaptive immune responses. Autoimmun. Rev. 2018, 17, 142–154. [Google Scholar] [CrossRef]
- Johansson, S.; Berg, L.; Hall, H.; Höglund, P. NK cells: Elusive players in autoimmunity. Trends Immunol. 2005, 26, 613–618. [Google Scholar] [CrossRef]
- Johansson, S.; Hall, H.; Berg, L.; Höglund, P. NK cells in autoimmune disease. Curr. Top. Microbiol. Immunol. 2005, 298, 259–277. [Google Scholar]
- Hamerman, J.A.; Ogasawara, K.; Lanier, L.L. NK cells in innate immunity. Curr. Opin. Immunol. 2005, 17, 29–35. [Google Scholar] [CrossRef]
- Baxter, A.G.; Smyth, M.J. The role of NK cells in autoimmune disease. Autoimmunity 2002, 35, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.A.; Fehniger, T.A.; Caligiuri, M.A. The biology of human natural killer-cell subsets. Trends Immunol. 2001, 22, 633–640. [Google Scholar] [CrossRef]
- Perricone, R.; Perricone, C.; De Carolis, C.; Shoenfeld, Y. NK cells in autoimmunity: A two-edged weapon of the immune system. Autoimmun. Rev. 2008, 7, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Mah, A.Y.; Cooper, M.A. Metabolic regulation of natural killer cell IFN-γ production. Crit. Rev. Immunol. 2016, 36, 131–147. [Google Scholar] [CrossRef] [PubMed]
- Degli-Esposti, M. To die or not to die—The quest of the TRAIL receptors. J. Leukoc. Biol. 1999, 65, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef]
- Vivier, E.; Raulet, D.H.; Moretta, A.; Caligiuri, M.A.; Zitvogel, L.; Lanier, L.L.; Yokoyama, W.M.; Ugolini, S. Innate or adaptive immunity? The example of natural killer cells. Science 2011, 331, 44–49. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, J.; Tian, Z. The regulatory effect of natural killer cells: Do “NK-reg cells” exist? Cell. Mol. Immunol. 2006, 3, 241–254. [Google Scholar] [PubMed]
- Moretta, A.; Bottino, C.; Vitale, M.; Pende, D.; Cantoni, C.; Mingari, M.C.; Biassoni, R.; Moretta, L. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu. Rev. Immunol. 2001, 19, 197–223. [Google Scholar] [CrossRef] [PubMed]
- Lanier, L.L. Up on the tightrope: Natural killer cell activation and inhibition. Nat. Immunol. 2008, 9, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Di Vito, C.; Mikulak, J.; Mavilio, D. On the way to become a natural killer cell. Front. Immunol. 2019, 10, 1812. [Google Scholar] [CrossRef] [PubMed]
- Fogler, W.E.; Volker, K.; McCormick, K.L.; Watanabe, M.; Ortaldo, J.R.; Wiltrout, R.H. NK cell infiltration into lung, liver, and subcutaneous B16 melanoma is mediated by VCAM-1/VLA-4 interaction. J. Immunol. 1996, 156, 4707–4714. [Google Scholar]
- Glas, R.; Franksson, L.; Une, C.; Eloranta, M.L.; Öhlén, C.; Örn, A.; Kärre, K. Recruitment and activation of natural killer (NK) cells in vivo determined by the target cell phenotype: An adaptive component of NK cell- mediated responses. J. Exp. Med. 2000, 191, 129–138. [Google Scholar] [CrossRef]
- Moretta, A.; Bottino, C.; Vitale, M.; Pende, D.; Biassoni, R.; Mingari, M.C.; Moretta, L. Receptors for HLA class-I molecules in human natural killer cells. Annu. Rev. Immunol. 1996, 14, 619–648. [Google Scholar] [CrossRef]
- Sivori, S.; Vitale, M.; Bottino, C.; Marcenaro, E.; Sanseverino, L.; Parolini, S.; Moretta, L.; Moretta, A. CD94 functions as a natural killer cell inhibitory receptor for different HLA class I alleles: Identification of the inhibitory form of CD94 by the use of novel monoclonal antibodies. Eur. J. Immunol. 1996, 26, 2487–2492. [Google Scholar] [CrossRef]
- Moretta, L.; Moretta, A. Unravelling natural killer cell function: Triggering and inhibitory human NK receptors. EMBO J. 2004, 23, 255–259. [Google Scholar] [CrossRef]
- Moretta, L.; Ciccone, E.; Moretta, A.; Höglund, P.; Öhlén, C.; Kärre, K. Allorecognition by NK cells: Nonself or no self? Immunol. Today 1992, 13, 300–306. [Google Scholar] [CrossRef]
- Sutherland, C.L.; Jan Chalupny, N.; Cosman, D. The UL16-binding proteins, a novel family of MHC class I-related ligands for NKG2D, activate natural killer cell functions. Immunol. Rev. 2001, 181, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Kärre, K. Natural killer cell recognition of missing self. Nat. Immunol. 2008, 9, 477–480. [Google Scholar] [CrossRef]
- Del Zotto, G.; Marcenaro, E.; Vacca, P.; Sivori, S.; Pende, D.; Della Chiesa, M.; Moretta, F.; Ingegnere, T.; Mingari, M.C.; Moretta, A.; et al. Markers and function of human NK cells in normal and pathological conditions. Cytom. Part B Clin. Cytom. 2017, 92, 100–114. [Google Scholar] [CrossRef] [PubMed]
- Vitale, M.; Della Chiesa, M.; Carlomagno, S.; Romagnani, C.; Thiel, A.; Moretta, L.; Moretta, A. The small subset of CD56brightCD16- natural killer cells is selectively responsible for both cell proliferation and interferon-gamma production upon interaction with dendritic cells. Eur. J. Immunol. 2004, 34, 1715–1722. [Google Scholar] [CrossRef] [PubMed]
- Raulet, D.H. Interplay of natural killer cells and their receptors with the adaptive immune response. Nat. Immunol. 2004, 5, 996–1002. [Google Scholar] [CrossRef] [PubMed]
- Backström, E.; Kristensson, K.; Ljunggren, H.-G. Activation of natural killer cells: Underlying molecular mechanisms revealed. Scand. J. Immunol. 2004, 60, 14–22. [Google Scholar] [CrossRef]
- Walzer, T.; Dalod, M.; Robbins, S.H.; Zitvogel, L.; Vivier, E. Natural-killer cells and dendritic cells: “L’union fait la force”. Blood 2005, 106, 2252–2258. [Google Scholar] [CrossRef]
- Strowig, T.; Brilot, F.; Münz, C. Noncytotoxic Functions of NK Cells: Direct Pathogen Restriction and Assistance to Adaptive Immunity. J. Immunol. 2008, 180, 7785–7791. [Google Scholar] [CrossRef]
- Yokoyama, W.M. Mistaken notions about natural killer cells. Nat. Immunol. 2008, 9, 481–485. [Google Scholar] [CrossRef]
- Moretta, A.; Marcenaro, E.; Parolini, S.; Ferlazzo, G.; Moretta, L. NK cells at the interface between innate and adaptive immunity. Cell Death Differ. 2008, 15, 226–233. [Google Scholar] [CrossRef]
- Moretta, L.; Locatelli, F. Innate lymphoid cells in normal and disease: An introductory overview. Immunol. Lett. 2016, 179, 1. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.A.; Caligiuri, M.A. Isolation and Characterization of Human Natural Killer Cell Subsets. Curr. Protoc. Immunol. 2004, 60, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.A.; Fehniger, T.A.; Turner, S.C.; Chen, K.S.; Ghaheri, B.A.; Ghayur, T.; Carson, W.E.; Caligiuri, M.A. Human natural killer cells: A unique innate immunoregulatory role for the CD56bright subset. Blood 2001, 97, 3146–3151. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.; Ley, K. Trafficking of Natural Killer Cells. Curr. Mol. Med. 2005, 4, 431–438. [Google Scholar] [CrossRef]
- Santoni, A.; Carlino, C.; Gismondi, A. Uterine NK cell development, migration and function. Reprod. Biomed. Online 2008, 16, 202–210. [Google Scholar] [CrossRef]
- Carrega, P.; Ferlazzo, G. Natural killer cell distribution and trafficking in human tissues. Front. Immunol. 2012, 3, 347. [Google Scholar] [CrossRef]
- Bernardini, G.; Santoni, A. The pathophysiological role of Chemokines in the regulation of NK cell tissue homing. Crit. Rev. Oncog. 2014, 19, 77–90. [Google Scholar] [CrossRef]
- Moffett, A.; Colucci, F. Uterine NK cells: Active regulators at the maternal-fetal interface. J. Clin. Investig. 2014, 124, 1872–1879. [Google Scholar] [CrossRef]
- Caligiuri, M.A. Human natural killer cells. Blood 2008, 112, 461–469. [Google Scholar] [CrossRef]
- Ferlazzo, G.; Thomas, D.; Lin, S.-L.; Goodman, K.; Morandi, B.; Muller, W.A.; Moretta, A.; Münz, C. The Abundant NK Cells in Human Secondary Lymphoid Tissues Require Activation to Express Killer Cell Ig-Like Receptors and Become Cytolytic. J. Immunol. 2004, 172, 1455–1462. [Google Scholar] [CrossRef]
- Loza, M.J.; Perussia, B. The IL-12 Signature: NK Cell Terminal CD56 +high Stage and Effector Functions. J. Immunol. 2004, 172, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Romagnani, C.; Juelke, K.; Falco, M.; Morandi, B.; D’Agostino, A.; Costa, R.; Ratto, G.; Forte, G.; Carrega, P.; Lui, G.; et al. CD56 bright CD16—Killer Ig-Like Receptor—NK Cells Display Longer Telomeres and Acquire Features of CD56 dim NK Cells upon Activation. J. Immunol. 2007, 178, 4947–4955. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.; Hong, D.-L.; Atzberger, A.; Kollnberger, S.; Filer, A.D.; Buckley, C.D.; McMichael, A.; Enver, T.; Bowness, P. CD56 bright Human NK Cells Differentiate into CD56 dim Cells: Role of Contact with Peripheral Fibroblasts. J. Immunol. 2007, 179, 89–94. [Google Scholar] [CrossRef]
- Yu, J.; Mao, H.C.; Wei, M.; Hughes, T.; Zhang, J.; Park, I.K.; Liu, S.; McClory, S.; Marcucci, G.; Trotta, R.; et al. CD94 surface density identifies a functional intermediary between the CD56bright and CD56dim human NK-cell subsets. Blood 2010, 115, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Correa, B.; Bergua, J.M.; Pera, A.; Campos, C.; Arcos, M.J.; Bañas, H.; Duran, E.; Solana, R.; Tarazona, R. In vitro culture with interleukin-15 leads to expression of activating receptors and recovery of natural killer cell function in acute myeloid leukemia patients. Front. Immunol. 2017, 8, 931. [Google Scholar] [CrossRef]
- Andre, P. Modification of P-selectin glycoprotein ligand-1 with a natural killer cell-restricted sulfated lactosamine creates an alternate ligand for L-selectin. Proc. Natl. Acad. Sci. USA 2000, 97, 3400–3405. [Google Scholar] [CrossRef]
- Caligiuri, M.A.; Zmuidzinas, A.; Manley, T.J.; Levine, H.; Smith, K.A.; Ritz, J. Functional consequences of interleukin 2 receptor expression on resting human lymphocytes: Identification of a novel natural killer cell subset with high affinity receptors. J. Exp. Med. 1990, 171, 1509–1526. [Google Scholar] [CrossRef] [PubMed]
- Nagler, A.; Lanier, L.L.; Phillips, J.H. Constitutive expression of high affinity interleukin 2 receptors on human CD16− natural killer cells in vivo. J. Exp. Med. 1990, 171, 1527–1533. [Google Scholar] [CrossRef]
- Baume, D.M.; Robertson, M.J.; Levine, H.; Manley, T.J.; Schow, P.W.; Ritz, J. Differential responses to interleukin 2 define functionally distinct subsets of human natural killer cells. Eur. J. Immunol. 1992, 22, 1–6. [Google Scholar] [CrossRef]
- Caligiuri, M.A.; Murray, C.; Robertson, M.J.; Wang, E.; Cochran, K.; Cameron, C.; Schow, P.; Ross, M.E.; Klumpp, T.R.; Soiffer, R.J.; et al. Selective modulation of human natural killer cells in vivo after prolonged infusion of low dose recombinant interleukin 2. J. Clin. Investig. 1993, 91, 123–132. [Google Scholar] [CrossRef]
- Foulis, A.K.; Liddle, C.N.; Farquharson, M.A.; Richmond, J.A.; Weir, R.S. The histopathology of the pancreas in Type I (insulin-dependent) diabetes mellitus: A 25-year review of deaths in patients under 20 years of age in the United Kingdom. Diabetologia 1986, 29, 267–274. [Google Scholar] [CrossRef]
- André, I.; Gonzalez, A.; Wang, B.; Katz, J.; Benoist, C.; Mathis, D. Checkpoints in the progression of autoimmune disease: Lessons from diabetes models. Proc. Natl. Acad. Sci. USA 1996, 93, 2260–2263. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Korganow, A.S.; Mangialaio, S.; Höglund, P.; André, I.; Lühder, F.; Gonzales, A.; Poirot, L.; Benoist, C.; Mathis, D. Different modes of pathogenesis in T-cell-dependent autoimmunity: Clues from two TCR transgenic systems. Immunol. Rev. 1999, 169, 139–146. [Google Scholar] [CrossRef]
- Flodström-Tullberg, M.; Bryceson, Y.T.; Shi, F.D.; Höglund, P.; Ljunggren, H.G. Natural killer cells in human autoimmunity. Curr. Opin. Immunol. 2009, 21, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.D.; Ljunggren, H.G.; Sarvetnick, N. Innate immunity and autoimmunity: From self-protection to self-destruction. Trends Immunol. 2001, 22, 97–101. [Google Scholar] [CrossRef]
- Pazmany, L. Do NK cells regulate human autoimmunity? Cytokine 2005, 32, 76–80. [Google Scholar] [CrossRef]
- Shi, F.D.; Van Kaer, L. Reciprocal regulation between natural killer cells and autoreactive T cells. Nat. Rev. Immunol. 2006, 6, 751–760. [Google Scholar] [CrossRef]
- Todd, D.J.; Forsberg, E.M.; Greiner, D.L.; Mordes, J.P.; Rossini, A.A.; Bortell, R. Deficiencies in Gut NK Cell Number and Function Precede Diabetes Onset in BB Rats. J. Immunol. 2004, 172, 5356–5362. [Google Scholar] [CrossRef]
- Poirot, L.; Benoist, C.; Mathis, D. Natural killer cells distinguish innocuous and destructive forms of pancreatic islet autoimmunity. Proc. Natl. Acad. Sci. USA 2004, 101, 8102–8107. [Google Scholar] [CrossRef]
- Brauner, H.; Elemans, M.; Lemos, S.; Broberger, C.; Holmberg, D.; Flodström-Tullberg, M.; Kärre, K.; Höglund, P. Distinct Phenotype and Function of NK Cells in the Pancreas of Nonobese Diabetic Mice. J. Immunol. 2010, 184, 2272–2280. [Google Scholar] [CrossRef]
- Ogasawara, K.; Hamerman, J.A.; Hsin, H.; Chikuma, S.; Bour-Jordan, H.; Chen, T.; Pertel, T.; Carnaud, C.; Bluestone, J.A.; Lanier, L.L. Impairment of NK cell function by NKG2D modulation in NOD mice. Immunity 2003, 18, 41–51. [Google Scholar] [CrossRef]
- Flodström, M.; Maday, A.; Balakrishna, D.; Cleary, M.M.; Yoshimura, A.; Sarvetnick, N. Target cell defense prevents the development of diabetes after viral infection. Nat. Immunol. 2002, 3, 373–382. [Google Scholar] [CrossRef]
- Sadelain, M.W.J.; Qin, H.Y.; Lauzon, J.; Singh, B. Prevention of type I diabetes in NOD mice by adjuvant immunotherapy. Diabetes 1990, 39, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.-F.; Qin, H.; Trudeau, J.; Dutz, J.; Tan, R. Regulation of Autoimmune Diabetes by Complete Freund’s Adjuvant Is Mediated by NK Cells. J. Immunol. 2004, 172, 937–942. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.F.; Qin, H.; Priatel, J.J.; Tan, R. Critical role for IFN-γ in natural killer cell-mediated protection from diabetes. Eur. J. Immunol. 2008, 38, 82–89. [Google Scholar] [CrossRef]
- Zhou, R.; Wei, H.; Tian, Z. NK3-Like NK Cells Are Involved in Protective Effect of Polyinosinic-Polycytidylic Acid on Type 1 Diabetes in Nonobese Diabetic Mice. J. Immunol. 2007, 178, 2141–2147. [Google Scholar] [CrossRef] [PubMed]
- Åkesson, C.; Uvebrant, K.; Oderup, C.; Lynch, K.; Harris, R.A.; Lernmark, Å.; Agardh, C.D.; Cilio, C.M. Altered natural killer (NK) cell frequency and phenotype in latent autoimmune diabetes in adults (LADA) prior to insulin deficiency. Clin. Exp. Immunol. 2010, 161, 48–56. [Google Scholar] [CrossRef]
- Hunt, P.; Eardley, D. Suppressive Effects of Insulin and Insulin-Like Growth factor-1 (IGF1) on Immune Responses. J Immunol. 1986, 136, 3994–3999. [Google Scholar]
- Dandona, P.; Aljada, A.; Mohanty, P.; Ghanim, H.; Hamouda, W.; Assian, E.; Ahmad, S. Insulin inhibits intranuclear nuclear factor κB and stimulates IκB in mononuclear cells in obese subjects: Evidence for an anti-inflammatory effect? J. Clin. Endocrinol. Metab. 2001, 86, 3257–3265. [Google Scholar] [CrossRef]
- Jeschke, M.G.; Klein, D.; Herndon, D.N. Insulin Treatment Improves the Systemic Inflammatory Reaction to Severe Trauma. Ann. Surg. 2004, 239, 553–560. [Google Scholar] [CrossRef]
- Deng, H.; Chai, J. The effects and mechanisms of insulin on systemic inflammatory response and immune cells in severe trauma, burn injury, and sepsis. Int. Immunopharmacol. 2009, 9, 1251–1259. [Google Scholar] [CrossRef] [PubMed]
- Xiu, F.; Stanojcic, M.; Diao, L.; Jeschke, M.G. Stress hyperglycemia, insulin treatment, and innate immune cells. Int. J. Endocrinol. 2014, 2014, 486403. [Google Scholar] [CrossRef] [PubMed]
- Nekoua, M.P.; Fachinan, R.; Fagninou, A.; Alidjinou, E.K.; Moutairou, K.; Hober, D.; Yessoufou, A. Does control of glycemia regulate immunological parameters in insulin-treated persons with type 1 diabetes? Diabetes Res. Clin. Pract. 2019, 157, 107868. [Google Scholar] [CrossRef]
- Lima, J.F.; Oliveira, L.M.S.; Pereira, N.Z.; Duarte, A.J.S.; Sato, M.N. Polyfunctional natural killer cells with a low activation profile in response to Toll-like receptor 3 activation in HIV-1-exposed seronegative subjects. Sci. Rep. 2017, 7, 524. [Google Scholar] [CrossRef] [PubMed]
- Coppieters, K.T.; Von Herrath, M.G. Histopathology of type 1 diabetes: Old paradigms and new insights. Rev. Diabet. Stud. 2009, 6, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Perona-Wright, G.; Mohrs, K.; Szaba, F.M.; Kummer, L.W.; Madan, R.; Karp, C.L.; Johnson, L.L.; Smiley, S.T.; Mohrs, M. Systemic but Not Local Infections Elicit Immunosuppressive IL-10 Production by Natural Killer Cells. Cell Host Microbe 2009, 6, 503–512. [Google Scholar] [CrossRef]
- Vivier, E.; Ugolini, S. Regulatory Natural Killer Cells: New Players in the IL-10 Anti-Inflammatory Response. Cell Host Microbe 2009, 6, 493–495. [Google Scholar] [CrossRef]
- Hofmann, P.; Schmidtke, M.; Stelzner, A.; Gemsa, D. Suppression of proinflammatory cytokines and induction of IL-10 in human monocytes after coxsackievirus B3 infection. J. Med. Virol. 2001, 64, 487–498. [Google Scholar] [CrossRef]
- Egger, D.; Gosert, R.; Bienz, K. Role of cellular structures in viral RNA replication. In Molecular Biology of Picornaviruses; Semler, B., Wimmer, E., Eds.; American Society for Microbiology: Washington, DC, USA, 2002; pp. 247–253. [Google Scholar]
- Deitz, S.B.; Dodd, D.A.; Cooper, S.; Parham, P.; Kirkegaard, K. MHC I-dependent antigen presentation is inhibited by poliovirus protein 3A. Proc. Natl. Acad. Sci. USA 2000, 97, 13790–13795. [Google Scholar] [CrossRef]
- Moffat, K.; Howell, G.; Knox, C.; Belsham, G.J.; Monaghan, P.; Ryan, M.D.; Wileman, T. Effects of foot-and-mouth disease virus nonstructural proteins on the structure and function of the early secretory pathway: 2BC but not 3A blocks endoplasmic reticulum-to-Golgi transport. J. Virol. 2005, 79, 4382–4395. [Google Scholar] [CrossRef]
- Kirkegaard, K.; Taylor, M.P.; Jackson, W.T. Cellular autophagy: Surrender, avoidance and subversion by microorganisms. Nat. Rev. Microbiol. 2004, 2, 301–314. [Google Scholar] [CrossRef] [PubMed]
- De Jong, A.S.; Visch, H.-J.; De Mattia, F.; Van Dommelen, M.M.; Swarts, H.G.; Luyten, T.; Callewaert, G.; Melchers, W.J.; Willems, P.H.; Van Kuppeveld, F.J. The coxsackievirus 2B protein increases efflux of ions from the endoplasmic reticulum and Golgi, thereby inhibiting protein trafficking through the Golgi. J. Biol. Chem. 2006, 281, 14144–14150. [Google Scholar] [CrossRef] [PubMed]
- Cornell, C.T.; Kiosses, W.B.; Harkins, S.; Whitton, J.L. Inhibition of protein trafficking by coxsackievirus b3: Multiple viral proteins target a single organelle. J. Virol. 2006, 80, 6637–6647. [Google Scholar] [CrossRef][Green Version]
- Cornell, C.T.; Kiosses, W.B.; Harkins, S.; Whitton, J.L. Coxsackievirus B3 proteins directionally complement each other to downregulate surface major histocompatibility complex class I. J. Virol. 2007, 81, 6785–6797. [Google Scholar] [CrossRef] [PubMed]
- Vives-Pi, M.; Rodríguez-Fernández, S.; Pujol-Autonell, I. How apoptotic β-cells direct immune response to tolerance or to autoimmune diabetes: A review. Apoptosis 2015, 20, 263–272. [Google Scholar] [CrossRef]
- Mathis, D.; Vence, L.; Benoist, C. Beta-cell death during progression to diabetes. Nature 2001, 414, 792–798. [Google Scholar] [CrossRef]
- O’Brien, B.A.; Geng, X.; Orteu, C.H.; Huang, Y.; Ghoreishi, M.; Zhang, Y.; Bush, J.A.; Li, G.; Finegood, D.T.; Dutz, J.P. A deficiency in the in vivo clearance of apoptotic cells is a feature of the NOD mouse. J. Autoimmun. 2006, 26, 104–115. [Google Scholar] [CrossRef]
- Eizirik, D.L.; Grieco, F.A. On the Immense Variety and Complexity of Circumstances Conditioning Pancreatic-Cell Apoptosis in Type 1 Diabetes. Diabetes 2012, 61, 1661–1663. [Google Scholar] [CrossRef][Green Version]
- Hodge, D.; Berthet, C.; Coppola, V.; Kastenmüller, W.; Buschman, M.; Schaughency, P.; Shirota, H.; Scarzello, A.; Subleski, J.; Anver, M.; et al. IFN-gamma AU-rich element removal promotes chronic IFN-gamma expression and autoimmunity in mice. J. Autoimmun. 2014, 53, 33–45. [Google Scholar] [CrossRef]
- Lees, J.R. Interferon gamma in autoimmunity: A complicated player on a complex stage. Cytokine 2015, 74, 18–26. [Google Scholar] [CrossRef]
- Samuel, C.E. Antiviral actions of interferons. Clin. Microbiol. Rev. 2001, 14, 778–809. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, L.G.; Chisari, F.V. Noncytolytic control of viral infections by the innate and adaptive immune response. Annu. Rev. Immunol. 2001, 19, 65–91. [Google Scholar] [CrossRef] [PubMed]
- Chesler, D.A.; Reiss, C.S. The role of IFN-γ in immune responses to viral infections of the central nervous system. Cytokine Growth Factor Rev. 2002, 13, 441–454. [Google Scholar] [CrossRef]
- Hultcrantz, M.; Hühn, M.H.; Wolf, M.; Olsson, A.; Jacobson, S.; Williams, B.R.; Korsgren, O.; Flodström-Tullberg, M. Interferons induce an antiviral state in human pancreatic islet cells. Virology 2007, 367, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Gibbert, K.; Schlaak, J.F.; Yang, D.; Dittmer, U. IFN-α subtypes: Distinct biological activities in anti-viral therapy. Br. J. Pharmacol. 2013, 168, 1048–1058. [Google Scholar] [CrossRef]
- Nair, M.P.N.; Lewis, E.W.; Schwartz, S.A. Immunoregulatory dysfunctions in type I diabetes: Natural and antibody-dependent cellular cytotoxic activities. J. Clin. Immunol. 1986, 6, 363–372. [Google Scholar] [CrossRef]
- Chen, S.A.; Shalaby, M.R.; Crase, D.R.; Palladino, M.A.; Baughman, R.A. Pharmacokinetics of Recombinant Murine Interferon-γ and Human Interferon-αA/D(Bgl) Administered in Concert and Their Influence on Natural Killer Cell Function in Mice. J. Interferon Res. 1988, 8, 597–608. [Google Scholar] [CrossRef]
- Semenzato, G.; Pizzolo, G.; Agostini, C.; Ambrosetti, A.; Zambello, R.; Trentin, L.; Luca, M.; Masciarelli, M.; Chilosi, M.; Vinante, F. α-Interferon activates the natural killer system in patients with hairy cell leukemia. Blood 1986, 68, 293–296. [Google Scholar] [CrossRef]
- Conti, P.; Reale, M.; Angeletti, P.U.; Dinarello, C.A. Restoration of anti-interleukin-1 depressed natural killer activity by human recombinant interferon α or γ, human recombinant interleukin-2 and indomethacin. Int. J. Immunopharmacol. 1988, 10, 907–911. [Google Scholar] [CrossRef]
- Sarzotti, M.; Klimpel, G.R.; Baron, S. Long-term killing of natural killer-resistant target cells by interferon-alpha-, interferon-gamma-, and interleukin-2-activated natural killer cells. Nat. Immun. Cell Growth Regul. 1989, 8, 66–75. [Google Scholar]
- Verhagen, A.; Mackay, I.; Rowley, M.; Tymms, M. Comparison of augmentation of human natural killer cell cytotoxicity by interferon-alpha subtypes. Nat. Immun. Cell Growth Regul. 1990, 9, 325–333. [Google Scholar]
- Pujol-Borrell, R.; Todd, I.; Doshi, M.; Gray, D.; Feldmann, M.; Bottazzo, G.F. Differential expression and regulation of MHC products in the endocrine and exocrine cells of the human pancreas. Clin. Exp. Immunol. 1986, 65, 128–139. [Google Scholar]
- Stewart, T.A.; Hultgren, B.; Huang, X.; Pitts-Meek, S.; Hully, J.; MacLachlan, N.J. Induction of type I diabetes by interferon-α in transgenic mice. Science 1993, 260, 1942–1946. [Google Scholar] [CrossRef] [PubMed]
- MacKay, P.; Jacobson, J.; Rabinovitch, A. Spontaneous diabetes mellitus in the Bio-Breeding/Worcester rat. Evidence in vitro for natural killer cell lysis of islet cells. J. Clin. Investig. 1986, 77, 916–924. [Google Scholar] [CrossRef] [PubMed]
- Pukel, C.; Baquerizo, H.; Rabinovitch, A. Interleukin 2 activates BB/W diabetic rat lymphoid cells cytotoxic to islet cells. Diabetes 1987, 36, 1217–1222. [Google Scholar] [CrossRef] [PubMed]
- Bancroft, G.J. The role of natural killer cells in innate resistance to infection. Curr. Opin. Immunol. 1993, 5, 503–510. [Google Scholar] [CrossRef]
- Nair, S.; Leung, K.C.; Rawlinson, W.D.; Naing, Z.; Craig, M.E. Enterovirus infection induces cytokine and chemokine expression in insulin-producing cells. J. Med. Virol. 2010, 82, 1950–1957. [Google Scholar] [CrossRef]
- Foulis, A.K. The pathogenesis of beta cell destruction in type I (insulin-dependent) diabetes mellitus. J. Pathol. 1987, 152, 141–148. [Google Scholar] [CrossRef]
- Kay, T.W.H.; Campbell, I.L.; Oxbrow, L.; Harrison, L.C. Overexpression of class I major histocompatibility complex accompanies insulitis in the non-obese diabetic mouse and is prevented by anti-interferon-γ antibody. Diabetologia 1991, 34, 779–785. [Google Scholar] [CrossRef]
- Thomas, H.E.; Parker, J.L.; Schreiber, R.D.; Kay, T.W.H. IFN-γ action on pancreatic beta cells causes class I MHC upregulation but not diabetes. J. Clin. Investig. 1998, 102, 1249–1257. [Google Scholar] [CrossRef]
- Chong, M.M.W.; Thomas, H.E.; Kay, T.W.H. γ-interferon signaling in pancreatic β-cells is persistent but can be terminated by overexpression of suppressor of cytokine signaling-1. Diabetes 2001, 50, 2744–2751. [Google Scholar] [CrossRef] [PubMed]
- Chong, M.M.W.; Chen, Y.; Darwiche, R.; Dudek, N.L.; Irawaty, W.; Santamaria, P.; Allison, J.; Kay, T.W.H.; Thomas, H.E. Suppressor of Cytokine Signaling-1 Overexpression Protects Pancreatic β Cells from CD8+ T Cell-Mediated Autoimmune Destruction. J. Immunol. 2004, 172, 5714–5721. [Google Scholar] [CrossRef] [PubMed]
- Vasu, S.; McClenaghan, N.H.; McCluskey, J.T.; Flatt, P.R. Mechanisms of toxicity by proinflammatory cytokines in a novel human pancreatic beta cell line, 1.1B4. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Barral, A.M.; Thomas, H.E.; Ling, E.M.; Darwiche, R.; Rodrigo, E.; Christen, U.; Ejrnaes, M.; Wolfe, T.; Kay, T.W.; von Herrath, M.G. SOCS-1 protects from virally-induced CD8 T cell mediated type 1 diabetes. J. Autoimmun. 2006, 27, 166–173. [Google Scholar] [CrossRef]
- Potvin, D.M.; Metzger, D.W.; Lee, W.T.; Collins, D.N.; Ramsingh, A.I. Exogenous Interleukin-12 Protects against Lethal Infection with Coxsackievirus B4. J. Virol. 2003, 77, 8272–8279. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Xiao, X.; Wang, J. Innate immunity evasion by enteroviruses: Insights into virus-host interaction. Viruses 2016, 8, 22. [Google Scholar] [CrossRef] [PubMed]
- Bayer, A.L.; Fraker, C.A. The folate cycle as a cause of natural killer cell dysfunction and viral etiology in type 1 diabetes. Front. Endocrinol. 2017, 8, 315. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Reference | T1D Patients Number (Male/Female) | Age Mean (SD or Range) | NK Cells Frequency in Peripheral Blood of T1D Patients as Compared to Healthy Individuals | NK Cell Effector Functions of T1D Patients as Compared to Healthy Individuals |
|---|---|---|---|---|
| Nekoua et al. 2020 [31] | 7 (2/5) | 37.0 years (16.7) | Reduced number of NK cells in long-established T1D patients | Decreased cytotoxic activity in long-established T1D patients. Target cells: CV-B4 persistently infected human 1.1B4 pancreatic beta cells |
| Hussain et al. 1987 [41] | 34 (18/16) | 38 years (2–78) | Reduced number of NK cells in recent-onset and long-established T1D patients | Increased cytotoxic activity in recent-onset T1D patients. Target cells: human myeloid K562 cell line |
| Negishi et al. 1986 [42] | 20 (11/9) | 12 years (4–35) | Reduced number of NK cells in recent-onset T1D patients | Decreased cytotoxic activity in recent-onset T1D patients. Target cells: human myeloid K562 cell line |
| Lorini et al. 1994 [43] | 25 (14/11) | 12.2 years (4.45) | Reduced number of NK cells in recent-onset and long-established T1D patients | Decreased cytotoxic activity in recent-onset and long-established T1D patients. Target cells: human myeloid K562 cell line |
| Qin et al. 2011 [44] | 116 (67/49) | 9.3 years (4.5) | Reduced number of NK cells in long-established T1D patients | Decreased IFN-γ secretion and cytotoxic activity and NKG2D-dependent cytotoxicity in long-established T1D patients. Target cell lines: K562, Raji and Daudi |
| Rodacki et al. 2007 [46] | 133 (67/66) | 13.6 years (6.55) | Reduced number of NK cells in recent-onset T1D patients | Decreased expression of NKG2D in recent-onset and long-established T1D patients Low expression of NKp46 and NKp30, reduced IFN-γ expression and perforin mRNA levels in long-established T1D patients |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nekoua, M.P.; Dechaumes, A.; Sane, F.; Alidjinou, E.K.; Moutairou, K.; Yessoufou, A.; Hober, D. Enteroviral Pathogenesis of Type 1 Diabetes: The Role of Natural Killer Cells. Microorganisms 2020, 8, 989. https://doi.org/10.3390/microorganisms8070989
Nekoua MP, Dechaumes A, Sane F, Alidjinou EK, Moutairou K, Yessoufou A, Hober D. Enteroviral Pathogenesis of Type 1 Diabetes: The Role of Natural Killer Cells. Microorganisms. 2020; 8(7):989. https://doi.org/10.3390/microorganisms8070989
Chicago/Turabian StyleNekoua, Magloire Pandoua, Arthur Dechaumes, Famara Sane, Enagnon Kazali Alidjinou, Kabirou Moutairou, Akadiri Yessoufou, and Didier Hober. 2020. "Enteroviral Pathogenesis of Type 1 Diabetes: The Role of Natural Killer Cells" Microorganisms 8, no. 7: 989. https://doi.org/10.3390/microorganisms8070989
APA StyleNekoua, M. P., Dechaumes, A., Sane, F., Alidjinou, E. K., Moutairou, K., Yessoufou, A., & Hober, D. (2020). Enteroviral Pathogenesis of Type 1 Diabetes: The Role of Natural Killer Cells. Microorganisms, 8(7), 989. https://doi.org/10.3390/microorganisms8070989