Importance of Defluviitalea raffinosedens for Hydrolytic Biomass Degradation in Co-Culture with Hungateiclostridium thermocellum

 ,
,  , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Availability
2.2. Reactor Operation and Sampling
2.3. Organisms
2.4. Growth Conditions Used for D. raffinosedens 249c-K6 and H. thermocellum DSM 1237T
2.5. Determination of Cellulose Hydrolyzing Activities by the Anthrone Assay of H. thermocellum Alone or in Co-Culture with D. raffinosedens
2.6. Determination of Volatile Acids and Alcohols with Gas Chromatography
2.7. Enzymatic Digestion of Cellulose Biomass and Determination of Enzymatic Activity
2.8. Species Identification, Sequencing, and Annotation of D. raffinosedens, Isolate 249c-K6
2.9. Prevalence and Activity of D. raffinosedens in Mesophilic and Thermophilic Biogas Fermenters Applying 16S rRNA Gene Amplicon Sequencing
2.10. Occurrence of D. raffinosedens Sequences in Biogas-Producing Microbial Communities Deduced from 16S rDNA and Reverse Transcribed 16S rRNA Amplicons of this Study or from Publicly Available Metagenome Data
3. Results and Discussion
3.1. Isolation of D. raffinosedens from Cellulolytic Mixed Cultures Isolated from Biogas Fermenters
3.2. Genome Sequence Analysis of the Strain D. raffinosedens 249c-K6
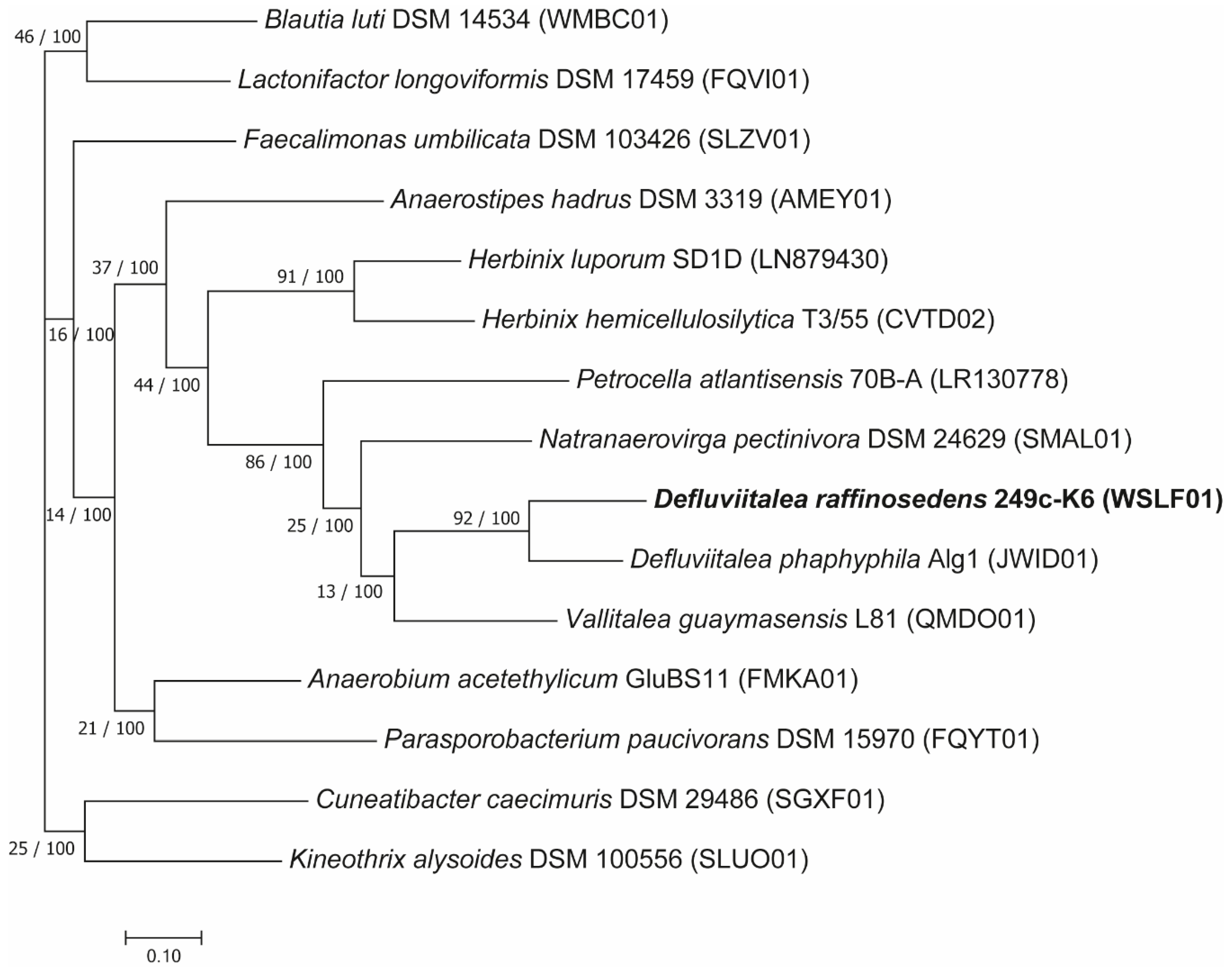
3.2.1. Phylogenetic Classification of D. raffinosedens 249c-K6 in Relation to Members of the Genus Defluviitalea
3.2.2. D. raffinosedens 249c-K6 Metabolic Pathways Predicted by Means of KEGG
3.2.3. D. raffinosedens 249c-K6 Genes Predicted to be Involved in Carbohydrate Utilization
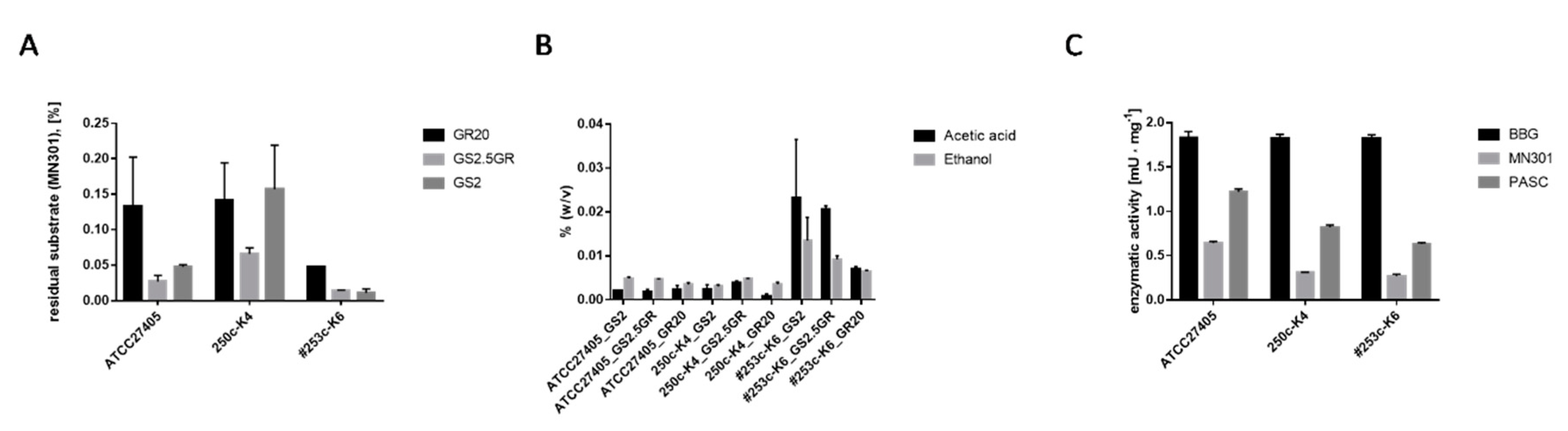
3.3. Physiological Comparison of H. thermocellum as Pure Culture and in Co-Culture with D. raffinosedens
- Cultures: ATCC27405T and 250c-K4, H. thermocellum alone; 253c-K6, H. thermocellum and D. raffinosedens in co-culture. Co-cultures are highlighted with ‘#’.
- (A) All cultures were incubated as biological duplicates in three different media (GS2, GS2.5GR and GR20) with a total volume of 50 mL and 0.2% (w/v) MN301. Residual MN301 after cultivation was determined by Anthrone assay. Data were normalized to an incubation period of two days. D. raffinosedens alone did not grow on MN301 as sole carbon source and is therefore not included in the figure (residual substrate 100%).
- (B) Metabolite production was compared using three different cultivation media (GS2, GS2.5GR, and GR20) in biological duplicates. Data were normalized to an incubation period of two days at 55 °C. One hundred microliters culture supernatant were measured in a total volume of 500 μL with 50 μL of 0.5% (v/v) 1-propanol as internal standard for quantification.
- (C) All digests were performed as biological duplicates in a total volume of 200 μL with 0.5% (w/v) substrate in a water bath at 55 °C for 30 h. Protein concentration within the batch was 0.1 μg·μL−1. DNSA assay was performed in technical triplicates. Enzymatic activity was calculated with Equation (1):Equation 1: Calculation of the specific enzymatic activity via DNSA assay.
- U is units (μmol × min−1); mglu is the mass of glucose (μg) calculated by means of glucose standard curve; Mglu is the molar mass of glucose (μg × μmol−1); t is the incubation period (min); Ve is the volume of enzyme within the batch (µL); and ce is the concentration of enzyme (μg × μL−1).
3.4. Importance of the Genus Defluviitalea in Microbial Communities of Biogas Fermenters
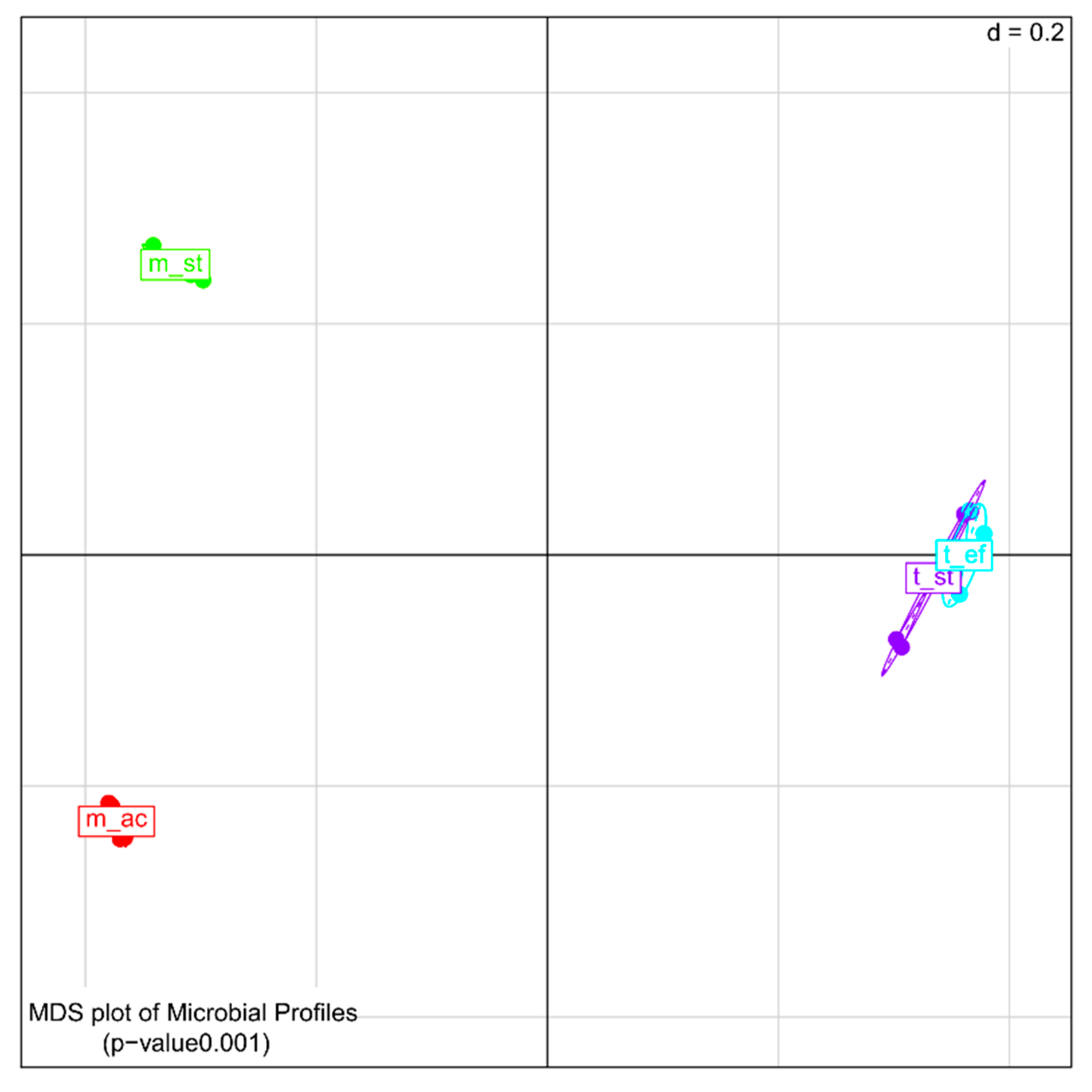
3.4.1. Comparison of Microbial Community Members’ Abundance and Activity as Deduced by 16S rDNA and Reverse Transcribed 16S rRNA Amplicon Sequencing
3.4.2. The Occurrence of Defluviitalea Genus in Biogas-Producing Plants as Deduced from Publicly Available Metagenome Data
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Weiland, P. Biogas production: Current state and perspectives. Appl. Microbiol. Biotechnol. 2010, 85, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Lebuhn, M.; Munk, B.; Effenberger, M. Agricultural biogas production in Germany-from practice to microbiology basics. Energy Sustain. Soc. 2014, 4, 10. [Google Scholar] [CrossRef]
- Li, K.; Liu, R.; Sun, C. A review of methane production from agricultural residues in China. Renew. Sustain. Energy Rev. 2016, 54, 857–865. [Google Scholar] [CrossRef]
- Zverlov, V.V.; Koeck, D.E.; Schwarz, W.H. The role of cellulose-hydrolyzing bacteria in the production of biogas from plant biomass. In Microorganisms in Biorefineries; Springer: Berlin/Heidelberg, Germany, 2015; pp. 335–361. [Google Scholar]
- Evans, P.N.; Boyd, J.A.; Leu, A.O.; Woodcroft, B.J.; Parks, D.H.; Hugenholtz, P.; Tyson, G.W. An evolving view of methane metabolism in the Archaea. Nat. Rev. Microbiol. 2019, 17, 219–232. [Google Scholar] [CrossRef]
- Bremges, A.; Maus, I.; Belmann, P.; Eikmeyer, F.; Winkler, A.; Albersmeier, A.; Pühler, A.; Schlüter, A.; Sczyrba, A. Deeply sequenced metagenome and metatranscriptome of a biogas-producing microbial community from an agricultural production-scale biogas plant. Gigascience 2015, 4, 33. [Google Scholar] [CrossRef]
- Ortseifen, V.; Stolze, Y.; Maus, I.; Sczyrba, A.; Bremges, A.; Albaum, S.P.; Jaenicke, S.; Fracowiak, J.; Pühler, A.; Schlüter, A. An integrated metagenome and-proteome analysis of the microbial community residing in a biogas production plant. J. Biotechnol. 2016, 231, 268–279. [Google Scholar] [CrossRef]
- Campanaro, S.; Treu, L.; Kougias, P.G.; De Francisci, D.; Valle, G.; Angelidaki, I. Metagenomic analysis and functional characterization of the biogas microbiome using high throughput shotgun sequencing and a novel binning strategy. Biotechnol. Biofuels 2016, 9, 26. [Google Scholar] [CrossRef]
- Maus, I.; Koeck, D.E.; Cibis, K.G.; Hahnke, S.; Kim, Y.S.; Langer, T.; Kreubel, J.; Erhard, M.; Bremges, A.; Off, S.; et al. Unraveling the microbiome of a thermophilic biogas plant by metagenome and metatranscriptome analysis complemented by characterization of bacterial and archaeal isolates. Biotechnol. Biofuels 2016, 9, 171. [Google Scholar] [CrossRef]
- Hassa, J.; Maus, I.; Off, S.; Puhler, A.; Scherer, P.; Klocke, M.; Schluter, A. Metagenome, metatranscriptome, and metaproteome approaches unraveled compositions and functional relationships of microbial communities residing in biogas plants. Appl. Microbiol. Biotechnol. 2018, 102, 5045–5063. [Google Scholar] [CrossRef] [PubMed]
- Maus, I.; Bremges, A.; Stolze, Y.; Hahnke, S.; Cibis, K.G.; Koeck, D.E.; Kim, Y.S.; Kreubel, J.; Hassa, J.; Wibberg, D.; et al. Genomics and prevalence of bacterial and archaeal isolates from biogas-producing microbiomes. Biotechnol. Biofuels 2017, 10, 264. [Google Scholar] [CrossRef] [PubMed]
- Antoni, D.; Zverlov, V.V.; Schwarz, W.H. Biofuels from microbes. Appl. Microb. Biotechnol. 2007, 77, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Campanaro, S.; Treu, L.; Seshadri, R.; Ivanova, N.; Kougias, P.G.; Kyrpides, N.; Angelidaki, I. Metabolic dependencies govern microbial syntrophies during methanogenesis in an anaerobic digestion ecosystem. Microbiome 2020, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, M.; Carey, D.E.; Zitomer, D.H.; McNamara, P.J. Syntroph diversity and abundance in anaerobic digestion revealed through a comparative core microbiome approach. Appl. Microbiol. Biotechnol. 2019, 103, 6353–6367. [Google Scholar] [CrossRef] [PubMed]
- Levin, D.B.; Verbeke, T.J.; Munir, R.; Islam, R.; Ramachandran, U.; Lal, S.; Schellenberg, J.; Sparling, R. Omics approaches for designing biofuel producing cocultures for enhanced microbial conversion of lignocellulosic substrates. In Direct Microbial Conversion of Biomass to Advanced Biofuels; Elsevier: Amsterdam, The Netherlands, 2015; pp. 335–363. [Google Scholar]
- Zhang, X.; Tu, B.; Dai, L.R.; Lawson, P.A.; Zheng, Z.Z.; Liu, L.Y.; Deng, Y.; Zhang, H.; Cheng, L. Petroclostridium xylanilyticum gen. nov., sp. nov., a xylan-degrading bacterium isolated from an oilfield, and reclassification of clostridial cluster III members into four novel genera in a new Hungateiclostridiaceae fam. nov. Int. J. Syst. Evol. Microbiol. 2018, 68, 3197–3211. [Google Scholar] [CrossRef]
- Cavedon, K.; Canale-Parola, E. Physiological interactions between a mesophilic cellulolytic Clostridium and a non-cellulolytic bacterium. FEMS Microbiol. Lett. 1992, 86, 237–245. [Google Scholar] [CrossRef][Green Version]
- He, Q.; Hemme, C.L.; Jiang, H.; He, Z.; Zhou, J. Mechanisms of enhanced cellulosic bioethanol fermentation by co-cultivation of Clostridium and Thermoanaerobacter spp. Bioresour. Technol. 2011, 102, 9586–9592. [Google Scholar] [CrossRef]
- Kato, S.; Haruta, S.; Cui, Z.J.; Ishii, M.; Igarashi, Y. Stable coexistence of five bacterial strains as a cellulose-degrading community. Appl. Environ. Microbiol. 2005, 71, 7099–7106. [Google Scholar] [CrossRef]
- Rettenmaier, R.; Duerr, C.; Neuhaus, K.; Liebl, W.; Zverlov, V.V. Comparison of sampling techniques and different media for the enrichment and isolation of cellulolytic organisms from biogas fermenters. Syst. Appl. Microbiol. 2019, 42, 481–487. [Google Scholar] [CrossRef]
- Johnson, E.A.; Madia, A.; Demain, A.L. Chemically defined minimal medium for growth of the anaerobic cellulolytic thermophile Clostridium thermocellum. Appl. Environ. Microbiol. 1981, 41, 1060. [Google Scholar] [CrossRef]
- Koeck, D.E.; Ludwig, W.; Wanner, G.; Zverlov, V.V.; Liebl, W.; Schwarz, W.H. Herbinix hemicellulosilytica gen. nov., sp. nov., a thermophilic cellulose-degrading bacterium isolated from a thermophilic biogas reactor. Int. J. Syst. Evol. Microbiol. 2015, 65, 2365–2371. [Google Scholar] [CrossRef]
- Miller, G.L. Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal. Chem. 1959, 31, 426–428. [Google Scholar] [CrossRef]
- Koeck, D.E.; Zverlov, V.V.; Liebl, W.; Schwarz, W.H. Comparative genotyping of Clostridium thermocellum strains isolated from biogas plants: Genetic markers and characterization of cellulolytic potential. Syst. Appl. Microbiol. 2014, 37, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Huptas, C.; Scherer, S.; Wenning, M. Optimized Illumina PCR-free library preparation for bacterial whole genome sequencing and analysis of factors influencing de novo assembly. BMC Res. Notes 2016, 9, 269. [Google Scholar] [CrossRef] [PubMed]
- Nurk, S.; Bankevich, A.; Antipov, D.; Gurevich, A.A.; Korobeynikov, A.; Lapidus, A.; Prjibelski, A.D.; Pyshkin, A.; Sirotkin, A.; Sirotkin, Y.; et al. Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J. Comput. Biol. 2013, 20, 714–737. [Google Scholar] [CrossRef]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y. KEGG Mapper for inferring cellular functions from protein sequences. Protein Sci. 2020, 29, 28–35. [Google Scholar] [CrossRef]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59. [Google Scholar] [CrossRef]
- Petersen, T.N.; Brunak, S.; Von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785. [Google Scholar] [CrossRef] [PubMed]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2013, 42, D490–D495. [Google Scholar] [CrossRef] [PubMed]
- Na, S.-I.; Kim, Y.O.; Yoon, S.-H.; Ha, S.-m.; Baek, I.; Chun, J. UBCG: Up-to-date bacterial core gene set and pipeline for phylogenomic tree reconstruction. J. Microbiol. 2018, 56, 280–285. [Google Scholar] [CrossRef]
- Richter, M.; Rosselló-Móra, R.; Oliver Glöckner, F.; Peplies, J. JSpeciesWS: A web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 2016, 32, 929–931. [Google Scholar] [CrossRef] [PubMed]
- Lebuhn, M.; Effenberger, M.; Gronauer, A.; Wilderer, P.; Wuertz, S. Using quantitative real-time PCR to determine the hygienic status of cattle manure. Water Sci. Technol. 2003, 48, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Lebuhn, M.; Hanreich, A.; Klocke, M.; Schlüter, A.; Bauer, C.; Pérez, C.M. Towards molecular biomarkers for biogas production from lignocellulose-rich substrates. Anaerobe 2014, 29, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Engel, K.; Sauer, J.; Jünemann, S.; Winkler, A.; Wibberg, D.; Kalinowski, J.; Tauch, A.; Caspers, B.A. Individual-and species-specific skin microbiomes in three different estrildid finch species revealed by 16S amplicon sequencing. Microb. Ecol. 2018, 76, 518–529. [Google Scholar] [CrossRef] [PubMed]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Joshi, N.; Fass, J. Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool for FastQ Files. 2011. Available online: https://github.com/najoshi/sickle (accessed on 14 December 2019).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, E.P.; Kolbe, D.L.; Eddy, S.R. Infernal 1.0: Inference of RNA alignments. Bioinformatics 2009, 25, 1335–1337. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Lagkouvardos, I.; Fischer, S.; Kumar, N.; Clavel, T. Rhea: A transparent and modular R pipeline for microbial profiling based on 16S rRNA gene amplicons. PeerJ 2017, 5, e2836. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Yoon, S.-H.; Ha, S.-M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. 2009. The sequence alignment/map format and samtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Rodriguez-R, L.M.; Gunturu, S.; Harvey, W.T.; Rosselló-Mora, R.; Tiedje, J.M.; Cole, J.R.; Konstantinidis, K.T. The Microbial Genomes Atlas (MiGA) webserver: Taxonomic and gene diversity analysis of Archaea and Bacteria at the whole genome level. Nucleic Acids Res. 2018, 46, W282–W288. [Google Scholar] [CrossRef]
- Campanaro, S.; Treu, L.; Rodriguez-R, L.M.; Kovalovszki, A.; Ziels, R.M.; Maus, I.; Zhu, X.; Kougias, P.G.; Basile, A.; Luo, G. New insights from the biogas microbiome by comprehensive genome-resolved metagenomics of nearly 1600 species originating from multiple anaerobic digesters. Biotechnol. Biofuels 2020, 13, 1–18. [Google Scholar] [CrossRef]
- Ma, S.; Huang, Y.; Wang, C.; Fan, H.; Dai, L.; Zhou, Z.; Liu, X.; Deng, Y. Defluviitalea raffinosedens sp. nov., a thermophilic, anaerobic, saccharolytic bacterium isolated from an anaerobic batch digester treating animal manure and rice straw. Int. J. Syst. Evol. Microbiol. 2017, 67, 1607. [Google Scholar] [CrossRef] [PubMed]
- Koeck, D.; Koellmeier, T.; Zverlov, V.; Liebl, W.; Schwarz, W. Differences in biomass degradation between newly isolated environmental strains of Clostridium thermocellum and heterogeneity in the size of the cellulosomal scaffoldin. Syst. Appl. Microbiol. 2015, 38, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Jabari, L.; Gannoun, H.; Cayol, J.L.; Hamdi, M.; Fauque, G.; Ollivier, B.; Fardeau, M.L. Characterization of Defluviitalea saccharophila gen. nov., sp. nov., a thermophilic bacterium isolated from an upflow anaerobic filter treating abattoir wastewaters, and proposal of Defluviitaleaceae fam. nov. Int. J. Syst. Evol. Microbiol. 2012, 62, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.Q.; Wang, B.; Lu, M.; Li, F.L. Defluviitalea phaphyphila sp. nov., a Novel Thermophilic Bacterium That Degrades Brown Algae. Appl. Environ. Microbiol. 2016, 82, 868–877. [Google Scholar] [CrossRef]
- Konstantinidis, K.T.; Tiedje, J.M. Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. USA 2005, 102, 2567–2572. [Google Scholar] [CrossRef]
- Tindall, B.J.; Rosselló-Móra, R.; Busse, H.-J.; Ludwig, W.; Kämpfer, P. Notes on the characterization of prokaryote strains for taxonomic purposes. Int. J. Syst. Evol. Microbiol. 2010, 60, 249–266. [Google Scholar] [CrossRef]
- Koendjbiharie, J.G.; Wiersma, K.; van Kranenburg, R. Investigating the central metabolism of Clostridium thermosuccinogenes. Appl. Environ. Microbiol. 2018, 84, e00363-18. [Google Scholar] [CrossRef]
- Ng, T.K.; Ben-Bassat, A.; Zeikus, J. Ethanol production by thermophilic bacteria: Fermentation of cellulosic substrates by cocultures of Clostridium thermocellum and Clostridium thermohydrosulfuricum. Appl. Environ. Microbiol. 1981, 41, 1337–1343. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Lynd, L.R. Cellulose utilization by Clostridium thermocellum: Bioenergetics and hydrolysis product assimilation. Proc. Natl. Acad. Sci. USA 2005, 102, 7321–7325. [Google Scholar] [CrossRef]
- Lee, S.-H.; Kang, H.-J.; Lee, Y.H.; Lee, T.J.; Han, K.; Choi, Y.; Park, H.-D. Monitoring bacterial community structure and variability in time scale in full-scale anaerobic digesters. J. Environ. Monit. 2012, 14, 1893–1905. [Google Scholar] [CrossRef]
- Sundberg, C.; Al-Soud, W.A.; Larsson, M.; Alm, E.; Yekta, S.S.; Svensson, B.H.; Sørensen, S.J.; Karlsson, A. 454 pyrosequencing analyses of bacterial and archaeal richness in 21 full-scale biogas digesters. FEMS Microbiol. Ecol. 2013, 85, 612–626. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Updating the 97% identity threshold for 16S ribosomal RNA OTUs. Bioinformatics 2018, 34, 2371–2375. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.S.; Spakowicz, D.J.; Hong, B.Y.; Petersen, L.M.; Demkowicz, P.; Chen, L.; Leopold, S.R.; Hanson, B.M.; Agresta, H.O.; Gerstein, M.; et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat. Commun. 2019, 10, 5029. [Google Scholar] [CrossRef] [PubMed]
- Kouzuma, A.; Tsutsumi, M.; Ishii, S.I.; Ueno, Y.; Abe, T.; Watanabe, K. Non-autotrophic methanogens dominate in anaerobic digesters. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Maus, I.; Klocke, M.; Derenkó, J.; Stolze, Y.; Beckstette, M.; Jost, C.; Wibberg, D.; Blom, J.; Henke, C.; Willenbücher, K. Impact of process temperature and organic loading rate on cellulolytic/hydrolytic biofilm microbiomes during biomethanation of ryegrass silage revealed by genome-centered metagenomics and metatranscriptomics. Environ. Microbiome 2020, 15, 1–21. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rettenmaier, R.; Schneider, M.; Munk, B.; Lebuhn, M.; Jünemann, S.; Sczyrba, A.; Maus, I.; Zverlov, V.; Liebl, W. Importance of Defluviitalea raffinosedens for Hydrolytic Biomass Degradation in Co-Culture with Hungateiclostridium thermocellum. Microorganisms 2020, 8, 915. https://doi.org/10.3390/microorganisms8060915
Rettenmaier R, Schneider M, Munk B, Lebuhn M, Jünemann S, Sczyrba A, Maus I, Zverlov V, Liebl W. Importance of Defluviitalea raffinosedens for Hydrolytic Biomass Degradation in Co-Culture with Hungateiclostridium thermocellum. Microorganisms. 2020; 8(6):915. https://doi.org/10.3390/microorganisms8060915
Chicago/Turabian StyleRettenmaier, Regina, Martina Schneider, Bernhard Munk, Michael Lebuhn, Sebastian Jünemann, Alexander Sczyrba, Irena Maus, Vladimir Zverlov, and Wolfgang Liebl. 2020. "Importance of Defluviitalea raffinosedens for Hydrolytic Biomass Degradation in Co-Culture with Hungateiclostridium thermocellum" Microorganisms 8, no. 6: 915. https://doi.org/10.3390/microorganisms8060915
APA StyleRettenmaier, R., Schneider, M., Munk, B., Lebuhn, M., Jünemann, S., Sczyrba, A., Maus, I., Zverlov, V., & Liebl, W. (2020). Importance of Defluviitalea raffinosedens for Hydrolytic Biomass Degradation in Co-Culture with Hungateiclostridium thermocellum. Microorganisms, 8(6), 915. https://doi.org/10.3390/microorganisms8060915