Skin Microbiome Analysis for Forensic Human Identification: What Do We Know So Far?

 , ,
, ,  and
and 
{kind=link}
Abstract
1. Introduction
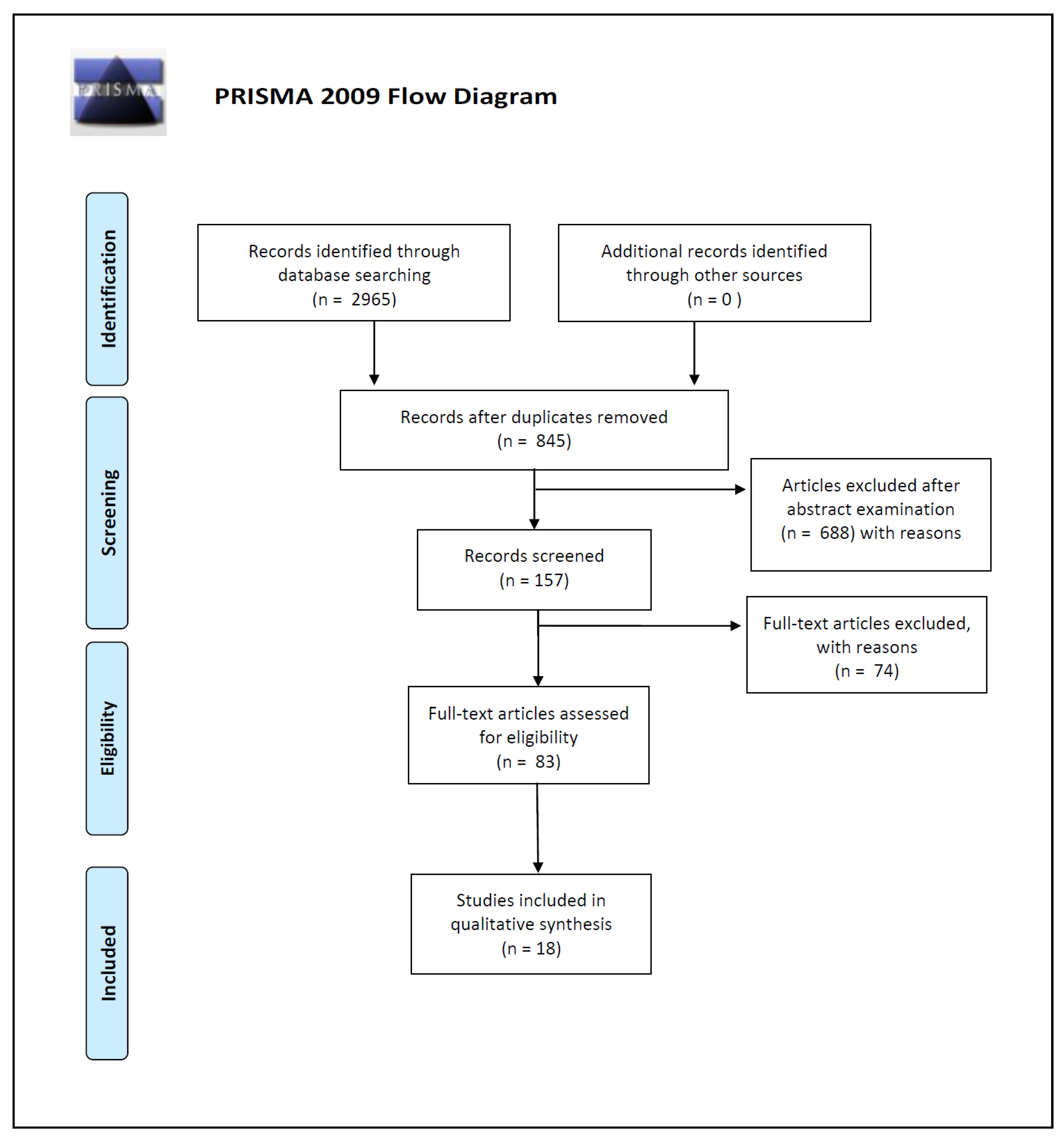
2. Materials and Methods
3. Results
3.1. The Search for the Best Identification Methodologies
3.1.1. The Amplicon Sequencing Approach
3.1.2. Shotgun Metagenomics Approach
3.2. Indirect Identification: the Possibility to Link People to Objects and Individuals by Skin Microbial Transferring
3.2.1. Keyboards and Computer Mice
- -
- the first one (the “keyboard study”) aimed to compare bacterial communities on the keys of three personal computers to the communities yielded by the fingertips of keyboard owners;
- -
- the second one (the “storage study”) concerned the evaluation of long-term temporal stability of bacteria yielded by skin on swabs, which were stored at -20 °C or left under typical indoor environmental conditions (about 20 °C) for up to 14 days;
- -
- the last study (the “computer mouse study”) analyzed the possibility of linking objects to specific individuals by comparing bacterial communities found on their computer mice against a database filled with information derived from the hand of the owner and from more than 270 hands that never touched the mouse.
3.2.2. Cell Phones
3.2.3. Fabrics
3.2.4. Handled Objects in Crime Scenes
3.2.5. Direct and Indirect Contact between Individuals
3.3. Ecological Features: Microbiome’s Temporal and Spatial Stability
3.3.1. Microbiome and Identification of Living People
3.3.2. Microbiome and Identification Analysis after Death
3.3.3. Touch Microbiome
4. Discussion
- -
- Skin-microbiome profiling for human identification is a comparative analysis that requires the collection and analysis of skin samples and reference samples;
- -
- Human microbiome identifiability is deeply connected with microbiome structure, personalization, and stability;
- -
- To acquire forensic value, microbial identification should be reliable and reproducible, even after a large temporal interval;
- -
- The huge quantity of data that can be extracted from a microbiome analysis has to be characterized and classified to obtain information which is not only useful for identification, but which is also admissible as forensic evidence in court.
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Marchesi, J.R.; Ravel, J. The vocabulary of microbiome research: A proposal. Microbiome 2015, 3, 31. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The human microbiome project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Sender, R.; Fuchs, S.; Milo, R. Are We Really Vastly Outnumbered? Revisiting the Ratio of Bacterial to Host Cells in Humans. Cell 2016, 164, 337–340. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.A.; Blaser, M.J.; Caporaso, J.G.; Jansson, J.K.; Lynch, S.V.; Knight, R. Current understanding of the human microbiome. Nat. Med. 2018, 24, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Consortium, H.M.P. A framework for human microbiome research. Nature 2012, 486, 215–221. [Google Scholar]
- Budowle, B.; Schutzer, S.E.; Einseln, A.; Kelley, L.C.; Walsh, A.C.; Smith, J.A.; Marrone, B.L.; Robertson, J.; Campos, J. Public health. Building microbial forensics as a response to bioterrorism. Science 2003, 301, 1852–1853. [Google Scholar] [CrossRef]
- Schmedes, S.E.; Sajantila, A.; Budowle, B. Expansion of Microbial Forensics. J. Clin. Microbiol. 2016, 54, 1964–1974. [Google Scholar] [CrossRef]
- Oliveira, M.; Amorim, A. Microbial forensics: New breakthroughs and future prospects. Appl. Microbiol. Biotechnol. 2018, 102, 10377–10391. [Google Scholar] [CrossRef]
- Børsting, C.; Morling, N. Next generation sequencing and its applications in forensic genetics. Forensic Sci. Int. Genet. 2015, 18, 78–89. [Google Scholar]
- Kuiper, I. Microbial forensics: Next-generation sequencing as catalyst: The use of new sequencing technologies to analyze whole microbial communities could become a powerful tool for forensic and criminal investigations. EMBO Rep. 2016, 17, 1085–1087. [Google Scholar] [CrossRef]
- Cullen, C.M.; Aneja, K.K.; Beyhan, S.; Cho, C.E.; Woloszynek, S.; Convertino, M.; McCoy, S.J.; Zhang, Y.; Anderson, M.Z.; Alvarez-Ponce, D.; et al. Emerging Priorities for Microbiome Research. Front. Microbiol. 2020, 11, 136. [Google Scholar] [CrossRef]
- Williams, D.W.; Gibson, G. Classification of individuals and the potential to detect sexual contact using the microbiome of the pubic region. Forensic Sci. Int. Genet. 2019, 41, 177–187. [Google Scholar] [CrossRef]
- Bashan, A.; Gibson, T.E.; Friedman, J.; Carey, V.J.; Weiss, S.T.; Hohmann, E.L.; Liu, Y.Y. Universality of human microbial dynamics. Nature 2016, 534, 259–262. [Google Scholar] [CrossRef]
- Gao, Z.; Tseng, C.H.; Pei, Z.; Blaser, M.J. Molecular analysis of human forearm superficial skin bacterial biota. Proc. Natl. Acad. Sci. USA 2007, 104, 2927–2932. [Google Scholar] [CrossRef]
- Costello, E.K.; Lauber, C.L.; Hamady, M.; Fierer, N.; Gordon, J.I.; Knight, R. Bacterial community variation in human body habitats across space and time. Science 2009, 26, 1694–1697. [Google Scholar] [CrossRef]
- Grice, E.A.; Kong, H.H.; Conlan, S.; Deming, C.B.; Davis, J.; Young, A.C.; Bouffard, G.G.; Blakesley, R.W.; Murray, P.R.; Green, E.D.; et al. Topographical and temporal diversity of the human skin microbiome. Science 2009, 324, 1190–1192. [Google Scholar] [CrossRef]
- Fitz-Gibbon, S.; Tomida, S.; Chiu, B.H.; Nguyen, L.; Du, C.; Liu, M.; Elashoff, D.; Erfe, M.C.; Loncaric, A.; Kim, J.; et al. Propionibacterium acnes strain populations in the human skin microbiome associated with acne. J. Investig. Dermatol. 2013, 133, 2152–2160. [Google Scholar] [CrossRef]
- Metcalf, J.L.; Xu, Z.Z.; Bouslimani, A.; Dorrestein, P.; Carter, D.O.; Knight, R. Microbiome Tools for Forensic Science. Trends Biotechnol. 2017, 35, 814–823. [Google Scholar] [CrossRef]
- Song, S.J.; Lauber, C.; Costello, E.K.; Lozupone, C.A.; Humphrey, G.; Berg-Lyons, D.; Caporaso, J.G.; Knights, D.; Clemente, J.C.; Nakielny, S.; et al. Cohabiting family members share microbiota with one another and with their dogs. eLife 2013, 2, e00458. [Google Scholar] [CrossRef]
- Misic, A.M.; Davis, M.F.; Tyldsley, A.S.; Hodkinson, B.P.; Tolomeo, P.; Hu, B.; Nachamkin, I.; Lautenbach, E.; Morris, D.O.; Grice, E.A. The shared microbiota of humans and companion animals as evaluated from Staphylococcus carriage sites. Microbiome 2015, 3, 2. [Google Scholar] [CrossRef]
- Casarin, R.C.; Barbagallo, A.; Meulman, T.; Santos, V.R.; Sallum, E.A.; Nociti, F.H.; Duarte, P.M.; Casati, M.Z.; Gonçalves, R.B. Subgingival biodiversity in subjects with uncontrolled type-2 diabetes and chronic periodontitis. J. Periodontal. Res. 2013, 48, 30–36. [Google Scholar] [CrossRef]
- Camelo-Castillo, A.J.; Mira, A.; Pico, A.; Nibali, L.; Henderson, B.; Donos, N.; Tomás, I. Subgingival microbiota in health compared to periodontitis and the influence of smoking. Front. Microbiol. 2015, 6, 119. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef]
- Fujiyoshi, S.; Tanaka, D.; Maruyama, F. Transmission of Airborne Bacteria across Built Environments and Its Measurement Standards: A Review. Front. Microbiol. 2017, 8, 2336. [Google Scholar] [CrossRef]
- Prussin, A.J.; Marr, L.C. Sources of airborne microorganisms in the built environment. Microbiome 2015, 3, 78. [Google Scholar] [CrossRef]
- Flores, G.E.; Bates, S.T.; Caporaso, J.G.; Lauber, C.L.; Leff, J.W.; Knight, R.; Fierer, N. Diversity, distribution and sources of bacteria in residential kitchens. Environ. Microbiol. 2013, 15, 588–596. [Google Scholar] [CrossRef]
- Chase, J.; Fouquier, J.; Zare, M.; Sonderegger, D.L.; Knight, R.; Kelley, S.T.; Siegel, J.; Caporaso, J.G. Geography and Location Are the Primary Drivers of Office Microbiome Composition. mSystems 2016, 1. [Google Scholar] [CrossRef] [PubMed]
- Kembel, S.W.; Jones, E.; Kline, J.; Northcutt, D.; Stenson, J.; Womack, A.M.; Bohannan, B.J.; Brown, G.Z.; Green, J.L. Architectural design influences the diversity and structure of the built environment microbiome. ISME J. 2012, 6, 1469–1479. [Google Scholar] [CrossRef]
- Meadow, J.F.; Altrichter, A.E.; Kembel, S.W.; Moriyama, M.; O’Connor, T.K.; Womack, A.M.; Brown, G.Z.; Green, J.L.; Bohannan, B.J. Bacterial communities on classroom surfaces vary with human contact. Microbiome 2014, 2, 7. [Google Scholar] [CrossRef]
- Luongo, J.C.; Barberán, A.; Hacker-Cary, R.; Morgan, E.E.; Miller, S.L.; Fierer, N. Microbial analyses of airborne dust collected from dormitory rooms predict the sex of occupants. Indoor Air 2017, 27, 338–344. [Google Scholar] [CrossRef]
- Richardson, M.; Gottel, N.; Gilbert, J.A.; Lax, S. Microbial Similarity between Students in a Common Dormitory Environment Reveals the Forensic Potential of Individual Microbial Signatures. mBio 2019, 10, e01054-19. [Google Scholar] [CrossRef] [PubMed]
- Flores, G.E.; Bates, S.T.; Knights, D.; Lauber, C.L.; Stombaugh, J.; Knight, R.; Fierer, N. Microbial biogeography of public restroom surfaces. PLoS ONE 2011, 6, e28132. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.R.; Grimes, T.L.; Datta, S. Unraveling bacterial fingerprints of city subways from microbiome 16S gene profiles. Biol. Direct 2018, 13, 10. [Google Scholar] [CrossRef]
- Budowle, B.; Schmedes, S.E.; Wendt, F.R. Increasing the reach of forensic genetics with massively parallel sequencing. Forensic Sci. Med. Pathol. 2017, 13, 342–349. [Google Scholar] [CrossRef]
- Fierer, N.; Lauber, C.L.; Zhou, N.; McDonald, D.; Costello, E.K.; Knight, R. Forensic identification using skin bacterial communities. Proc. Natl. Acad. Sci. USA 2010, 107, 6477–6481. [Google Scholar] [CrossRef]
- Lax, S.; Smith, D.P.; Hampton-Marcell, J.; Owens, S.M.; Handley, K.M.; Scott, N.M.; Gibbons, S.M.; Larsen, P.B.; Shogan, D.; Weiss, S.; et al. Longitudinal analysis of microbial interaction between humans and the indoor environment. Science 2014, 345, 1048–1052. [Google Scholar] [CrossRef]
- Lax, S.; Hampton-Marcell, J.T.; Gibbons, S.M.; Colares, G.B.; Smith, D.; Eisen, J.A.; Gilbert, J.A. Forensic analysis of the microbiome of phones and shoes. Microbiome 2015, 3, 21. [Google Scholar] [CrossRef]
- Phan, K.; Barash, M.; Spindler, X.; Gunn, P.; Roux, C. Retrieving forensic information about the donor through bacterial profiling. Int. J. Legal. Med. 2020, 134, 21–29. [Google Scholar] [CrossRef]
- Kreft, J.U. Editorial: The microbiome as a source of new enterprises and job creation. Microb. Biotechnol. 2018, 11, 145–148. [Google Scholar] [CrossRef]
- Assenmacher, D.M.; Fields, S.D.; Crupper, S.S. Comparison of Commercial Kits for Recovery and Analysis of Bacterial DNA from Fingerprints. J. Forensic Sci. 2020, 27. [Google Scholar] [CrossRef]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; Prisma Group. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA Statement. Open Med. 2009, 3, e1000097. [Google Scholar]
- Knight, R.; Vrbanac, A.; Taylor, B.C.; Aksenov, A.; Callewaert, C.; Debelius, J.; Gonzalez, A.; Kosciolek, T.; McCall, L.I.; McDonald, D.; et al. Best practices for analysing microbiomes. Nat. Rev. Microbiol. 2018, 16, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Morgan, X.C.; Huttenhower, C. Chapter 12: Human microbiome analysis. PLoS Comput. Biol. 2012, 8, e1002808. [Google Scholar] [CrossRef]
- Galloway-Peña, J.; Hanson, B. Tools for Analysis of the Microbiome. Dig. Dis. Sci. 2020, 65, 674–685. [Google Scholar] [CrossRef]
- Franzosa, E.A.; Huang, K.; Meadow, J.F.; Gevers, D.; Lemon, K.P.; Bohannan, B.J.; Huttenhower, C. Identifying personal microbiomes using metagenomic codes. Proc. Natl. Acad. Sci. USA 2015, 112, E2930–E2938. [Google Scholar] [CrossRef]
- Schmedes, S.E.; Woerner, A.E.; Budowle, B. Forensic Human Identification Using Skin Microbiomes. Appl. Environ. Microbiol. 2017, 83. [Google Scholar] [CrossRef]
- Oh, J.; Byrd, A.L.; Park, M.; Kong, H.H.; Segre, J.A. N.C.S. Program. Temporal Stability of the Human Skin Microbiome. Cell 2016, 165, 854–866. [Google Scholar] [CrossRef]
- Schmedes, S.E.; Woerner, A.E.; Novroski, N.M.M.; Wendt, F.R.; King, J.L.; Stephens, K.M.; Budowle, B. Targeted sequencing of clade-specific markers from skin microbiomes for forensic human identification. Forensic Sci. Int. Genet. 2018, 32, 50–61. [Google Scholar] [CrossRef]
- Truong, D.T.; Franzosa, E.A.; Tickle, T.L.; Scholz, M.; Weingart, G.; Pasolli, E.; Tett, A.; Huttenhower, C.; Segata, N. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods 2015, 12, 902–903. [Google Scholar] [CrossRef]
- Woerner, A.E.; Novroski, N.M.M.; Wendt, F.R.; Ambers, A.; Wiley, R.; Schmedes, S.E.; Budowle, B. Forensic human identification with targeted microbiome markers using nearest neighbor classification. Forensic Sci. Int. Genet. 2019, 38, 130–139. [Google Scholar] [CrossRef]
- Meadow, J.F.; Altrichter, A.E.; Green, J.L. Mobile phones carry the personal microbiome of their owners. PeerJ 2014, 2, e447. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Woo, S.K.; Lee, S.M.; Eom, Y.B. Forensic analysis using microbial community between skin bacteria and fabrics. Toxicol. Environ. Health Sci. 2016, 8, 263–270. [Google Scholar] [CrossRef]
- Kodama, W.A.; Xu, Z.; Metcalf, J.L.; Song, S.J.; Harrison, N.; Knight, R.; Carter, D.O.; Happy, C.B. Trace Evidence Potential in Postmortem Skin Microbiomes: From Death Scene to Morgue. J. Forensic Sci. 2019, 64, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Neckovic, A.; van Oorschot, R.A.H.; Szkuta, B.; Durdle, A. Investigation of direct and indirect transfer of microbiomes between individuals. Forensic Sci. Int. Genet. 2020, 45, 102212. [Google Scholar] [CrossRef] [PubMed]
- Flores, G.E.; Caporaso, J.G.; Henley, J.B.; Rideout, J.R.; Domogala, D.; Chase, J.; Leff, J.W.; Vázquez-Baeza, Y.; Gonzalez, A.; Knight, R.; et al. Temporal variability is a personalized feature of the human microbiome. Genome Biol. 2014, 15, 531. [Google Scholar] [CrossRef]
- Bay, L.; Barnes, C.J.; Fritz, B.G.; Thorsen, J.; Restrup, M.E.M.; Rasmussen, L.; Sørensen, J.K.; Hesselvig, A.B.; Odgaard, A.; Hansen, A.J.; et al. Universal Dermal Microbiome in Human Skin. mBio 2020, 11, e02945–e19. [Google Scholar] [CrossRef]
- Pechal, J.L.; Schmidt, C.J.; Jordan, H.R.; Benbow, M.E. A large-scale survey of the postmortem human microbiome, and its potential to provide insight into the living health condition. Sci. Rep. 2018, 8, 5724. [Google Scholar] [CrossRef]
- Wilkins, D.; Leung, M.H.; Lee, P.K. Microbiota fingerprints lose individually identifying features over time. Microbiome 2017, 5, 1. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tozzo, P.; D’Angiolella, G.; Brun, P.; Castagliuolo, I.; Gino, S.; Caenazzo, L. Skin Microbiome Analysis for Forensic Human Identification: What Do We Know So Far? Microorganisms 2020, 8, 873. https://doi.org/10.3390/microorganisms8060873
Tozzo P, D’Angiolella G, Brun P, Castagliuolo I, Gino S, Caenazzo L. Skin Microbiome Analysis for Forensic Human Identification: What Do We Know So Far? Microorganisms. 2020; 8(6):873. https://doi.org/10.3390/microorganisms8060873
Chicago/Turabian StyleTozzo, Pamela, Gabriella D’Angiolella, Paola Brun, Ignazio Castagliuolo, Sarah Gino, and Luciana Caenazzo. 2020. "Skin Microbiome Analysis for Forensic Human Identification: What Do We Know So Far?" Microorganisms 8, no. 6: 873. https://doi.org/10.3390/microorganisms8060873
APA StyleTozzo, P., D’Angiolella, G., Brun, P., Castagliuolo, I., Gino, S., & Caenazzo, L. (2020). Skin Microbiome Analysis for Forensic Human Identification: What Do We Know So Far? Microorganisms, 8(6), 873. https://doi.org/10.3390/microorganisms8060873