NetMet: A Network-Based Tool for Predicting Metabolic Capacities of Microbial Species and their Interactions



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Description of the Expansion Algorithm and its Implementation in the NetMet Tool
2.2. Single-Species Mode
2.3. Interaction Mode
3. Results
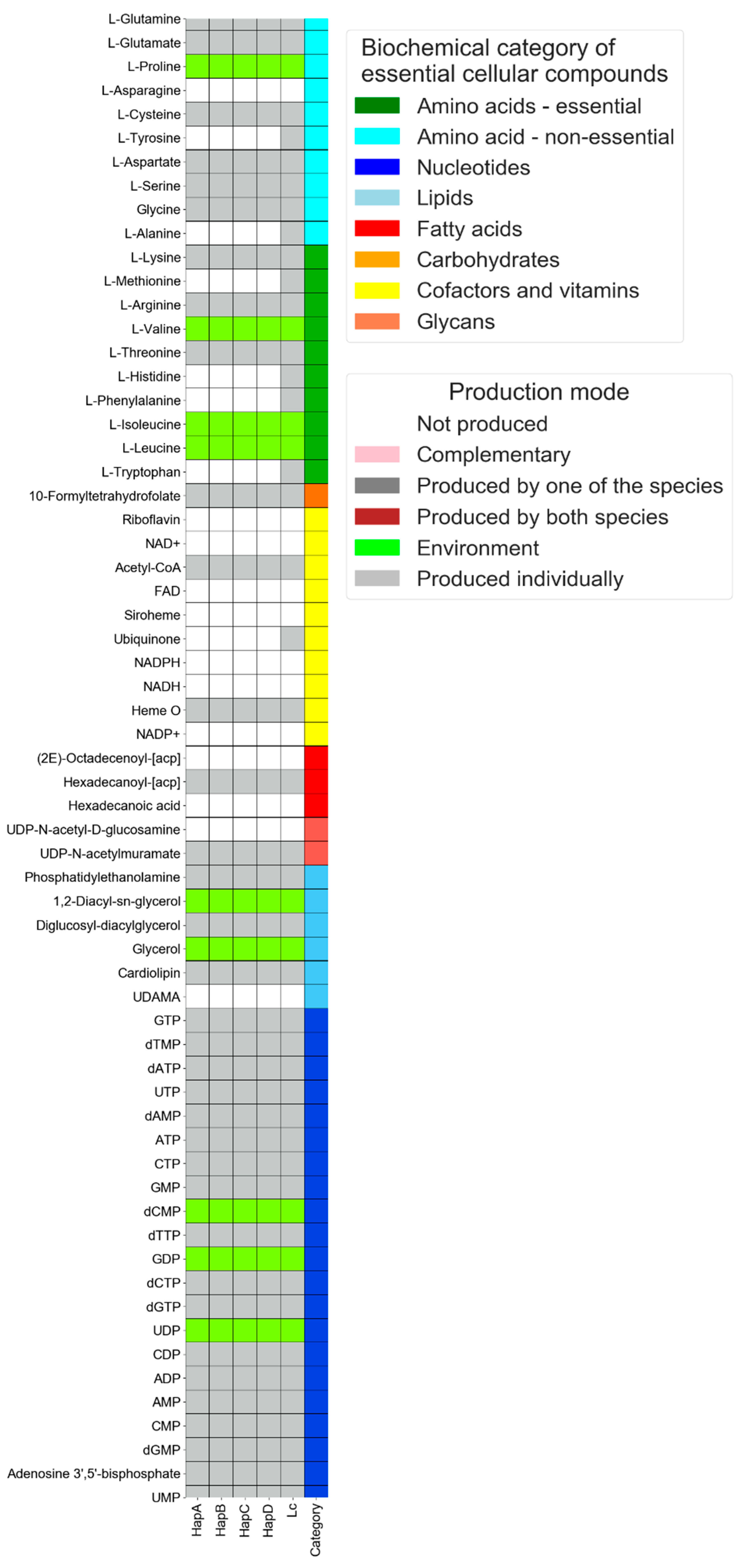
3.1. Single-Species Mode: Comparative Analysis of the Metabolic Capacities of Liberibacter Species That Have Undergone Genomic Reduction
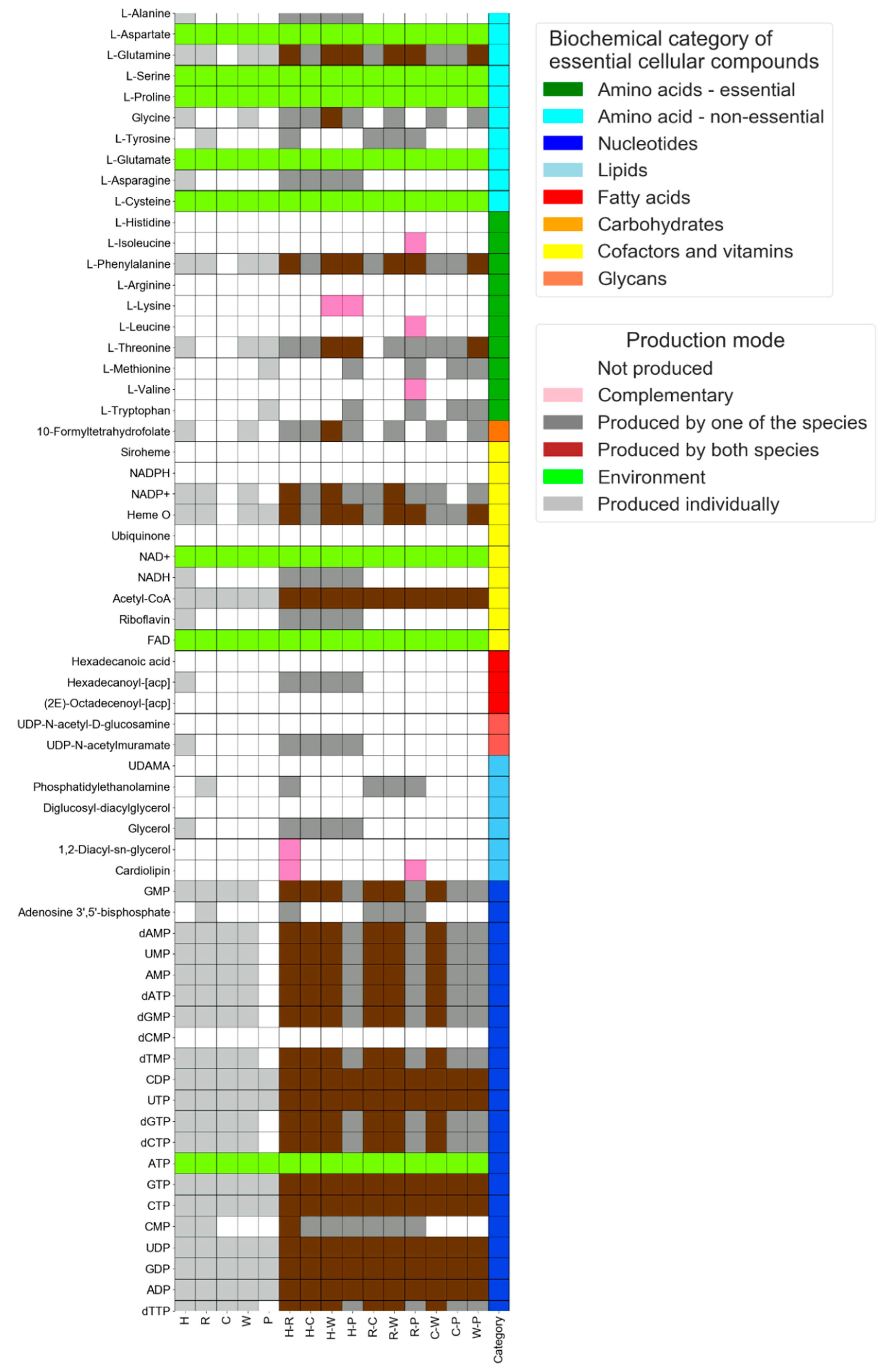
3.2. Interaction Mode: Delineating Exchange Interactions between Endosymbionts of Phloem-Feeding Whitefly Bemisia tabaci
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Muller, E.E.L.; Faust, K.; Widder, S.; Herold, M.; Martínez Arbas, S.; Wilmes, P. Using metabolic networks to resolve ecological properties of microbiomes. Curr. Op. Syst. Biol. 2018, 8, 73–80. [Google Scholar] [CrossRef]
- Kruse, K.A.I.; Ebenhöh, O. Comparing flux balance analysis to network expansion: Producibility, sustainability and the scope of compounds. Genome Inform. 2008, 20, 91–101. [Google Scholar]
- Ebenhöh, O.; Handorf, T.; Heinrich, R. Structural analysis of expanding metabolic networks. Genome Inform. 2004, 15, 35–45. [Google Scholar]
- Ebenhöh, O.; Handorf, T.; Heinrich, R. A Cross Species Comparison of Metabolic Network Functions. Genome Inform. 2005, 16, 203–213. [Google Scholar]
- Katsir, L.; Zhepu, R.; Santos Garcia, D.; Piasezky, A.; Jiang, J.; Sela, N.; Freilich, S.; Bahar, O. Genome analysis of haplotype D of Candidatus Liberibacter Solanacearum. Front. Microbiol. 2018, 9, 2933. [Google Scholar] [CrossRef] [PubMed]
- Freilich, S.; Kreimer, A.; Borenstein, E.; Yosef, N.; Sharan, R.; Gophna, U.; Ruppin, E. Metabolic-network-driven analysis of bacterial ecological strategies. Genome Biol. 2009, 10, R61. [Google Scholar] [CrossRef] [PubMed]
- Freilich, S.; Kreimer, A.; Borenstein, E.; Gophna, U.; Sharan, R.; Ruppin, E. Decoupling Environment-Dependent and Independent Genetic Robustness across Bacterial Species. PLoS Comput. Biol. 2010, 6, e1000690. [Google Scholar] [CrossRef] [PubMed]
- Handorf, T.; Ebenhöh, O.; Heinrich, R. Expanding Metabolic Networks: Scopes of Compounds, Robustness, and Evolution. J. Mol. Evol. 2005, 61, 498–512. [Google Scholar] [CrossRef] [PubMed]
- Zomorrodi, A.R.; Segrè, D. Synthetic ecology of microbes: Mathematical models and applications. J. Mol. Biol. 2016, 428, 837–861. [Google Scholar] [CrossRef]
- Klitgord, N.; Segrè, D. Ecosystems biology of microbial metabolism. Curr. Opin. Biotech. 2011, 22, 541–546. [Google Scholar] [CrossRef]
- Widder, S.; Allen, R.J.; Pfeiffer, T.; Curtis, T.P.; Wiuf, C.; Sloan, W.T.; Cordero, O.X.; Brown, S.P.; Momeni, B.; Shou, W.; et al. Challenges in microbial ecology: Building predictive understanding of community function and dynamics. ISME J. 2016, 10, 2557–2568. [Google Scholar] [CrossRef]
- Borenstein, E.; Feldman, M.W. Topological signatures of species interactions in metabolic networks. J. Comput. Biol. 2009, 16, 191–200. [Google Scholar] [CrossRef]
- Christian, N.; Handorf, T.; Ebenhöh, O. Metabolic synergy: Increasing biosynthetic capabilities by network cooperation. Genome Inform. 2007, 18, 320–329. [Google Scholar]
- Duan, G.; Christian, N.; Schwachtje, J.; Walther, D.; Ebenhöh, O. The metabolic interplay between plants and phytopathogens. Metabolites 2013, 3, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Freilich, S.; Kreimer, A.; Meilijson, I.; Gophna, U.; Sharan, R.; Ruppin, E. The large-scale organization of the bacterial network of ecological co-occurrence interactions. Nucleic Acids Res. 2010, 38, 3857–3868. [Google Scholar] [CrossRef]
- Levy, R.; Borenstein, E. Metabolic modeling of species interaction in the human microbiome elucidates community-level assembly rules. Proc. Natl. Acad. Sci. USA 2013, 110, 12804–12809. [Google Scholar] [CrossRef] [PubMed]
- Ofaim, S.; Ofek-Lalzar, M.; Sela, N.; Jinag, J.; Kashi, Y.; Minz, D.; Freilich, S. Analysis of microbial functions in the rhizosphere using a metabolic-network based framework for metagenomics interpretation. Front. Microbiol. 2017, 8, 1606. [Google Scholar] [CrossRef] [PubMed]
- Opatovsky, I.; Santos-Garcia, D.; Ruan, Z.; Lahav, T.; Ofaim, S.; Mouton, L.; Barbe, V.; Jiang, J.; Zchori-Fein, E.; Freilich, S. Modeling trophic dependencies and exchanges among insects’ bacterial symbionts in a host-simulated environment. BMC Genom. 2018, 19, 402. [Google Scholar] [CrossRef] [PubMed]
- Kreimer, A.; Doron-Faigenboim, A.; Borenstein, E.; Freilich, S. NetCmpt: A network-based tool for calculating the metabolic competition between bacterial species. Bioinformatics 2012, 28, 2195–2197. [Google Scholar] [CrossRef] [PubMed]
- Levy, R.; Carr, R.; Kreimer, A.; Freilich, S.; Borenstein, E. NetCooperate: A network-based tool for inferring host-microbe and microbe-microbe cooperation. BMC Bioinform. 2015, 16, 164. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Caspi, R.; Billington, R.; Ferrer, L.; Foerster, H.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Mueller, L.A.; et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2015, 44, D471–D480. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, V.M.; Chen, I.M.A.; Palaniappan, K.; Chu, K.; Szeto, E.; Pillay, M.; Ratner, A.; Huang, J.; Woyke, T.; Huntemann, M.; et al. IMG 4 version of the integrated microbial genomes comparative analysis system. Nucleic Acids Res. 2013, 42, D560–D567. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef]
- Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019, 28, 1947–1951. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Furumichi, M.; Morishima, K.; Tanabe, M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2019, 47, D590–D595. [Google Scholar] [CrossRef]
- Blimkie, T.; Lee, A.H.-Y.; Hancock, R.E.W. MetaBridge: An Integrative Multi-Omics Tool for Metabolite-Enzyme Mapping. Curr. Protoc. Bioinform. 2020, 70, e98. [Google Scholar] [CrossRef]
- Burton, E.G.; Sakami, W. The formation of methionine from the monoglutamate form of methyltetrahydrofolate by higher plants. Biochem. Biophys. Res. Commun. 1969, 36, 228–234. [Google Scholar] [CrossRef]
- Guest, J.R.; Friedman, S.; Foster, M.A.; Tejerina, G.; Woods, D.D. Transfer of the methyl group from N5-methyltetrahydrofolates to homocysteine in Escherichia coli. Biochem. J. 1964, 92, 497–504. [Google Scholar] [CrossRef]
- Lin, H.; Lou, B.; Glynn, J.M.; Doddapaneni, H.; Civerolo, E.L.; Chen, C.; Duan, Y.; Zhou, L.; Vahling, C.M. The complete genome sequence of ‘Candidatus Liberibacter solanacearum’, the bacterium associated with potato zebra chip disease. PLoS ONE 2011, 6, e19135. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.M.; Johnson, C.P.; Lu, A.Y.; Frampton, R.A.; Sullivan, K.L.; Fiers, M.W.E.J.; Crowhurst, R.N.; Pitman, A.R.; Scott, I.A.W.; Wen, A.; et al. Genomes of ‘Candidatus Liberibacter solanacearum’ haplotype A from New Zealand and the United States suggest significant genome plasticity in the species. Phytopathology 2015, 105, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Haapalainen, M.; Schott, T.; Thompson, S.M.; Smith, G.R.; Nissinen, A.I.; Pirhonen, M. Genomic sequence of ‘Candidatus Liberibacter solanacearum’ haplotype C and its comparison with haplotype A and B genomes. PLoS ONE 2017, 12, e0171531. [Google Scholar] [CrossRef] [PubMed]
- Hartung, J.S.; Shao, J.; Kuykendall, L.D. Comparison of the ‘Ca. Liberibacter asiaticus’ genome adapted for an intracellular lifestyle with other members of the rhizobiales. PLoS ONE 2011, 6, e23289. [Google Scholar] [CrossRef]
- Bennett, G.M.; Moran, N.A. Heritable symbiosis: The advantages and perils of an evolutionary rabbit hole. Proc. Natl. Acad. Sci. USA 2015, 112, 10169–10176. [Google Scholar] [CrossRef]
- Fagen, J.R.; Leonard, M.T.; Coyle, J.F.; McCullough, C.M.; Davis-Richardson, A.G.; Davis, M.J.; Triplett, E.W. Liberibacter crescens gen. nov., sp. nov., the first cultured member of the genus Liberibacter. Int. J. Syst. Evol. Microbiol 2014, 64, 2461–2466. [Google Scholar] [CrossRef]
- Carr, R.; Borenstein, E. NetSeed: A network-based reverse-ecology tool for calculating the metabolic interface of an organism with its environment. Bioinformatics 2012, 28, 734–735. [Google Scholar] [CrossRef]
- Ankrah, N.Y.D.; Luan, J.; Douglas, A.E. Cooperative metabolism in a three-partner insect-bacterial symbiosis revealed by metabolic modeling. J. Bacteriol. 2017, 199, e00872-00816. [Google Scholar] [CrossRef]
- Luan, J.-B.; Chen, W.; Hasegawa, D.K.; Simmons, A.M.; Wintermantel, W.M.; Ling, K.-S.; Fei, Z.; Liu, S.-S.; Douglas, A.E. Metabolic Coevolution in the Bacterial Symbiosis of Whiteflies and Related Plant Sap-Feeding Insects. Genome Biol. Evol. 2015, 7, 2635–2647. [Google Scholar] [CrossRef]
- Santos-Garcia, D.; Juravel, K.; Freilich, S.; Zchori-Fein, E.; Latorre, A.; Moya, A.; Morin, S.; Silva, F.J. To B or Not to B: Comparative Genomics Suggests Arsenophonus as a Source of B Vitamins in Whiteflies. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef]
- Zchori-Fein, E.; Lahav, T.; Freilich, S. Variations in the identity and complexity of endosymbiont combinations in whitefly hosts. Front. Microbiol. 2014, 5. [Google Scholar] [CrossRef]
- Heinken, A.; Thiele, I. Systems biology of host–microbe metabolomics. WIREs Syst. Biol. Med. 2015, 7, 195–219. [Google Scholar] [CrossRef] [PubMed]
- Charitou, T.; Bryan, K.; Lynn, D.J. Using biological networks to integrate, visualize and analyze genomics data. Genet. Sel. Evol. 2016, 48, 27. [Google Scholar] [CrossRef]
- Xu, X.; Zarecki, R.; Medina, S.; Ofaim, S.; Liu, X.; Chen, C.; Hu, S.; Brom, D.; Gat, D.; Porob, S.; et al. Modeling microbial communities from atrazine contaminated soils promotes the development of biostimulation solutions. ISME J. 2019, 13, 494–508. [Google Scholar] [CrossRef] [PubMed]
- Faust, K. Microbial consortium design benefits from metabolic modeling. Trends Biotechnol. 2019, 37, 123–125. [Google Scholar] [CrossRef] [PubMed]
- Freilich, S.; Zarecki, R.; Eilam, O.; Segal, E.S.; Henry, C.S.; Kupiec, M.; Gophna, U.; Sharan, R.; Ruppin, E. Competitive and cooperative metabolic interactions in bacterial communities. Nat. Commun. 2011, 2, 589. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tal, O.; Selvaraj, G.; Medina, S.; Ofaim, S.; Freilich, S. NetMet: A Network-Based Tool for Predicting Metabolic Capacities of Microbial Species and their Interactions. Microorganisms 2020, 8, 840. https://doi.org/10.3390/microorganisms8060840
Tal O, Selvaraj G, Medina S, Ofaim S, Freilich S. NetMet: A Network-Based Tool for Predicting Metabolic Capacities of Microbial Species and their Interactions. Microorganisms. 2020; 8(6):840. https://doi.org/10.3390/microorganisms8060840
Chicago/Turabian StyleTal, Ofir, Gopinath Selvaraj, Shlomit Medina, Shany Ofaim, and Shiri Freilich. 2020. "NetMet: A Network-Based Tool for Predicting Metabolic Capacities of Microbial Species and their Interactions" Microorganisms 8, no. 6: 840. https://doi.org/10.3390/microorganisms8060840
APA StyleTal, O., Selvaraj, G., Medina, S., Ofaim, S., & Freilich, S. (2020). NetMet: A Network-Based Tool for Predicting Metabolic Capacities of Microbial Species and their Interactions. Microorganisms, 8(6), 840. https://doi.org/10.3390/microorganisms8060840