Human Antimicrobial Peptide Hepcidin 25-Induced Apoptosis in Candida albicans


Abstract
1. Introduction
2. Materials and Methods
2.1. Peptides and Regents
2.2. C. albicans Strain and Growth Conditions
2.3. Calcein Leakage Assay
2.4. C. albicans Killing Assay
2.5. Analysis of Apoptotic Markers
2.6. Cytosolic ROS Assay
2.7. Detection of Mitochondrial Membrane Potential (ΔΨm)
2.8. Metacaspase Activity Assay
2.9. Statistical Analysis
3. Results
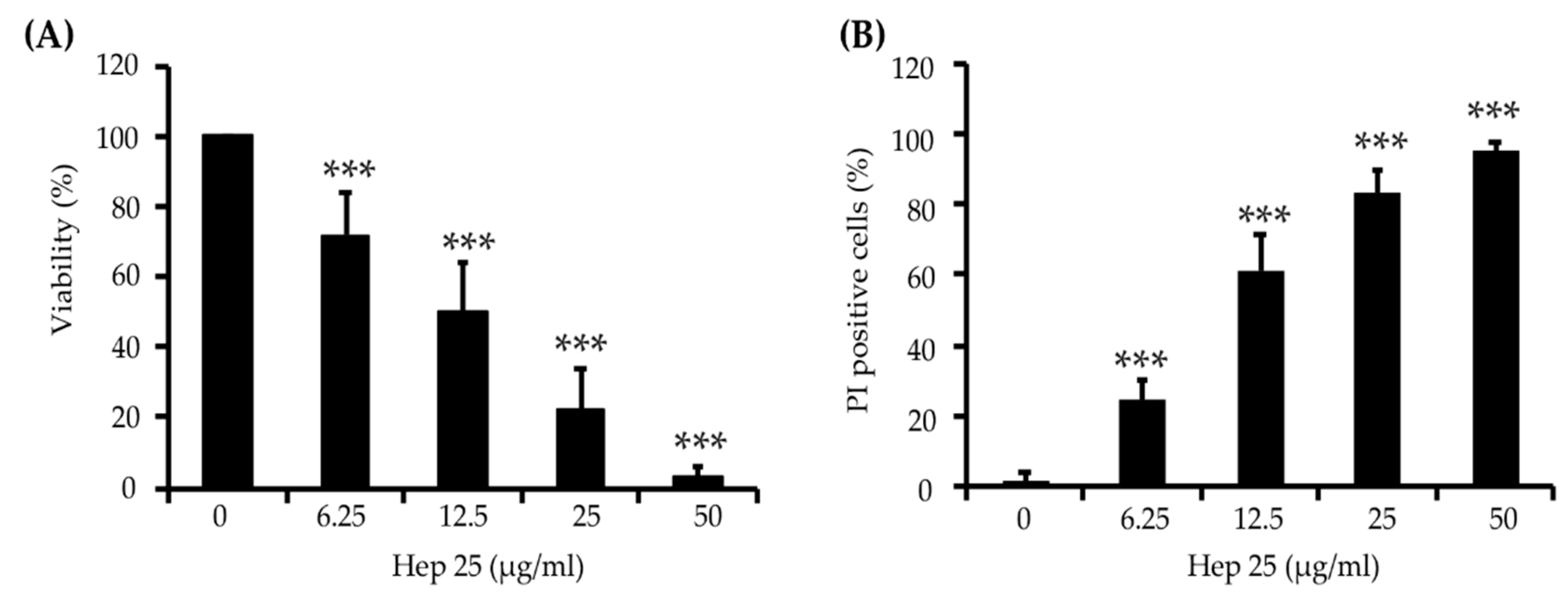
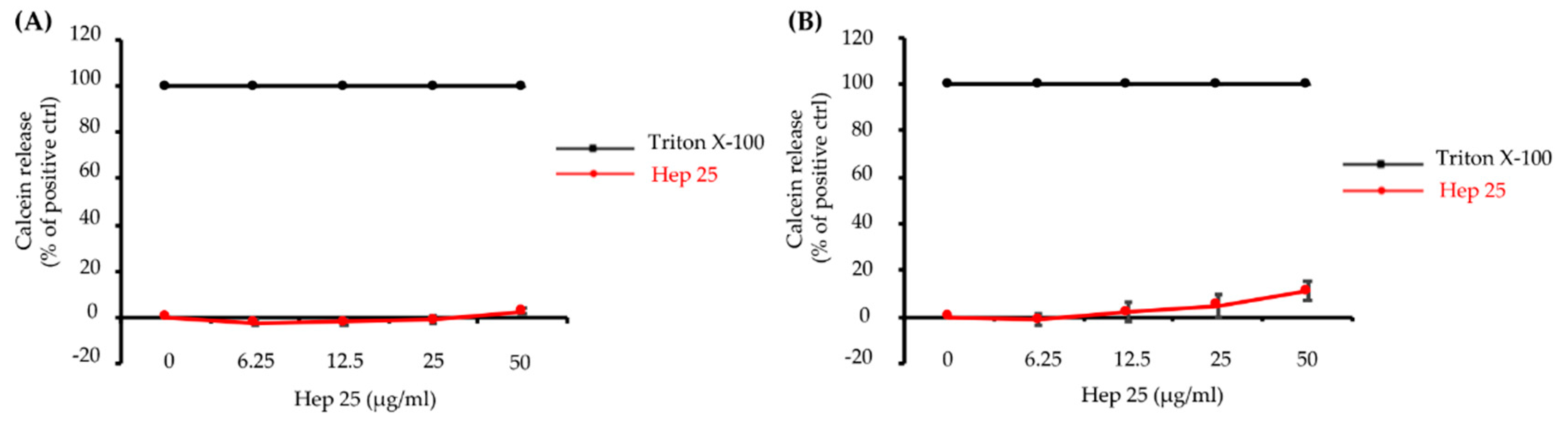
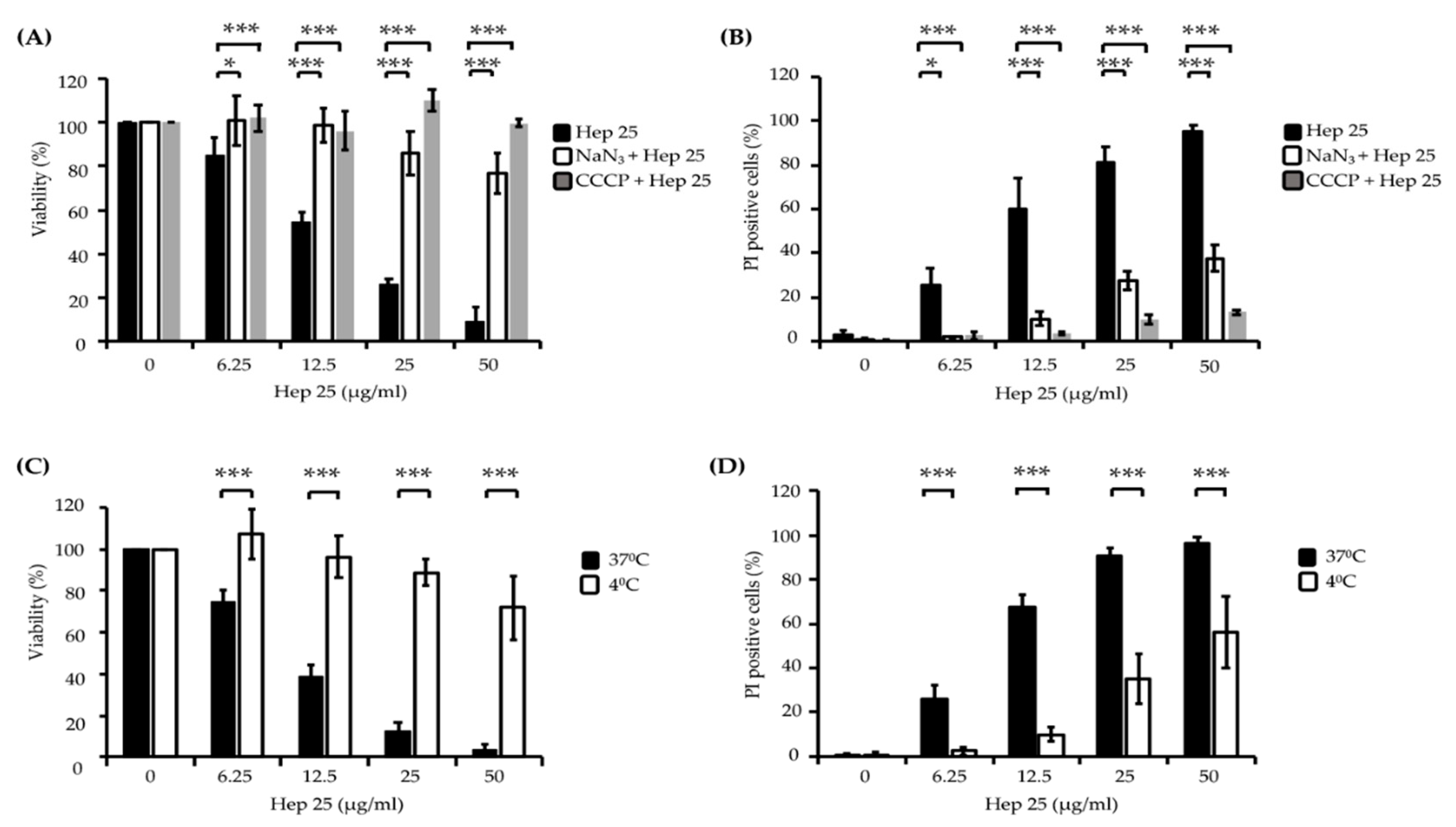
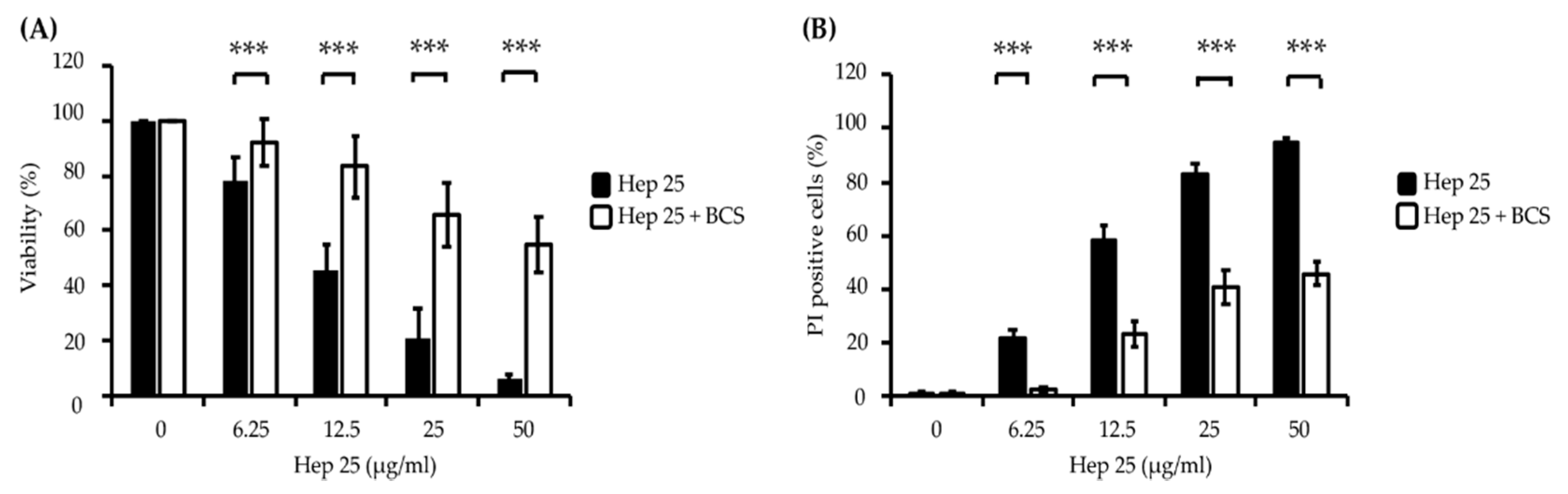
3.1. Hep 25 Shows Potent Candidacidal Activity in an Energy- and Temperature-Dependent Manner Without Causing Membrane Disruption
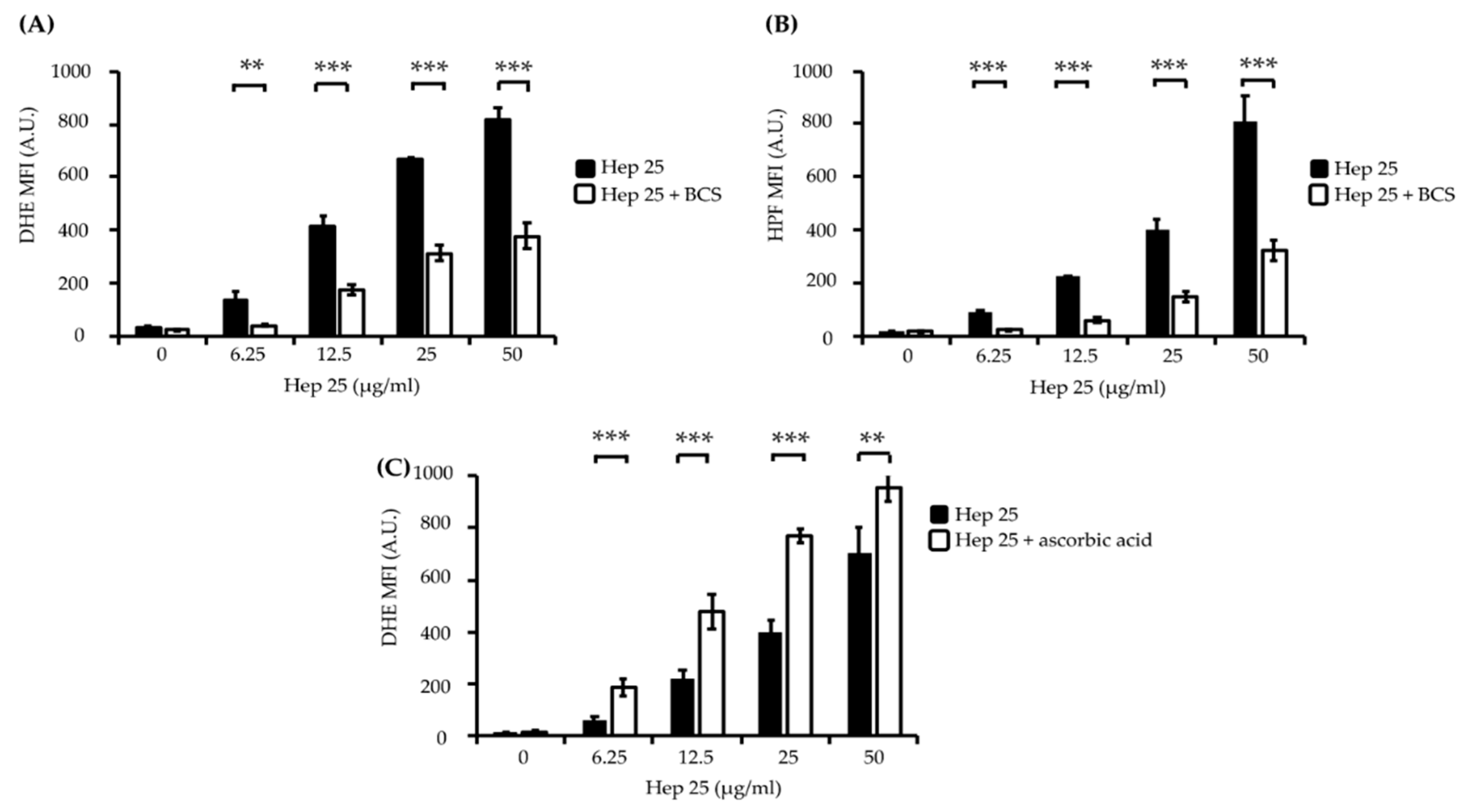
3.2. The ATUCN Motif of Hep 25 Is Involved in ROS Production and Correlates with the Activity of Hep 25
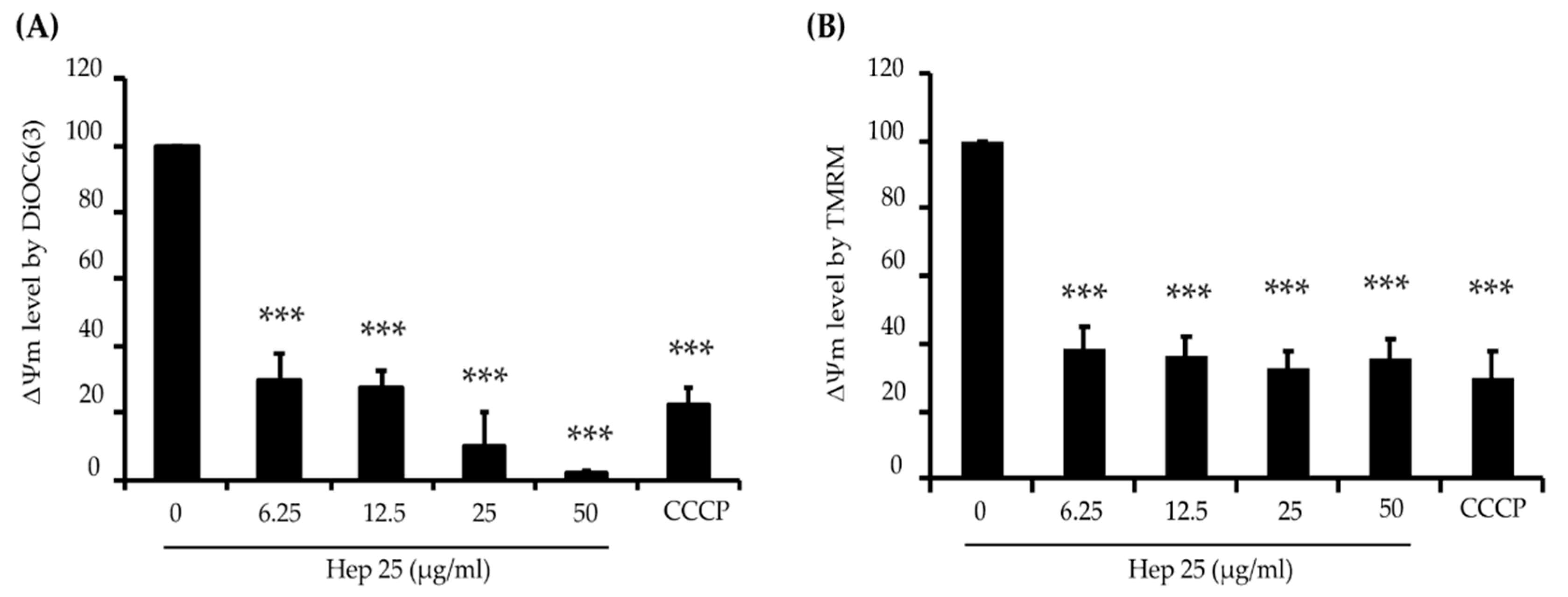
3.3. Hep 25 Induces Mitochondrial Depolarization in C. albicans
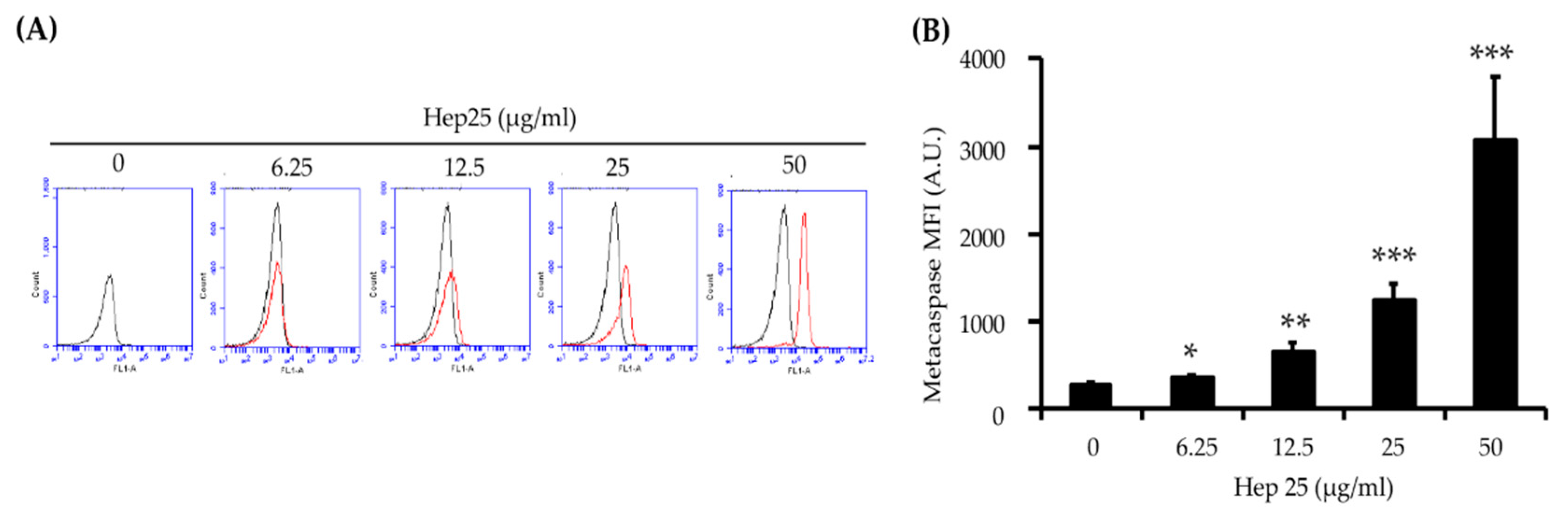
3.4. Hep 25 Triggers Metacaspase Activation
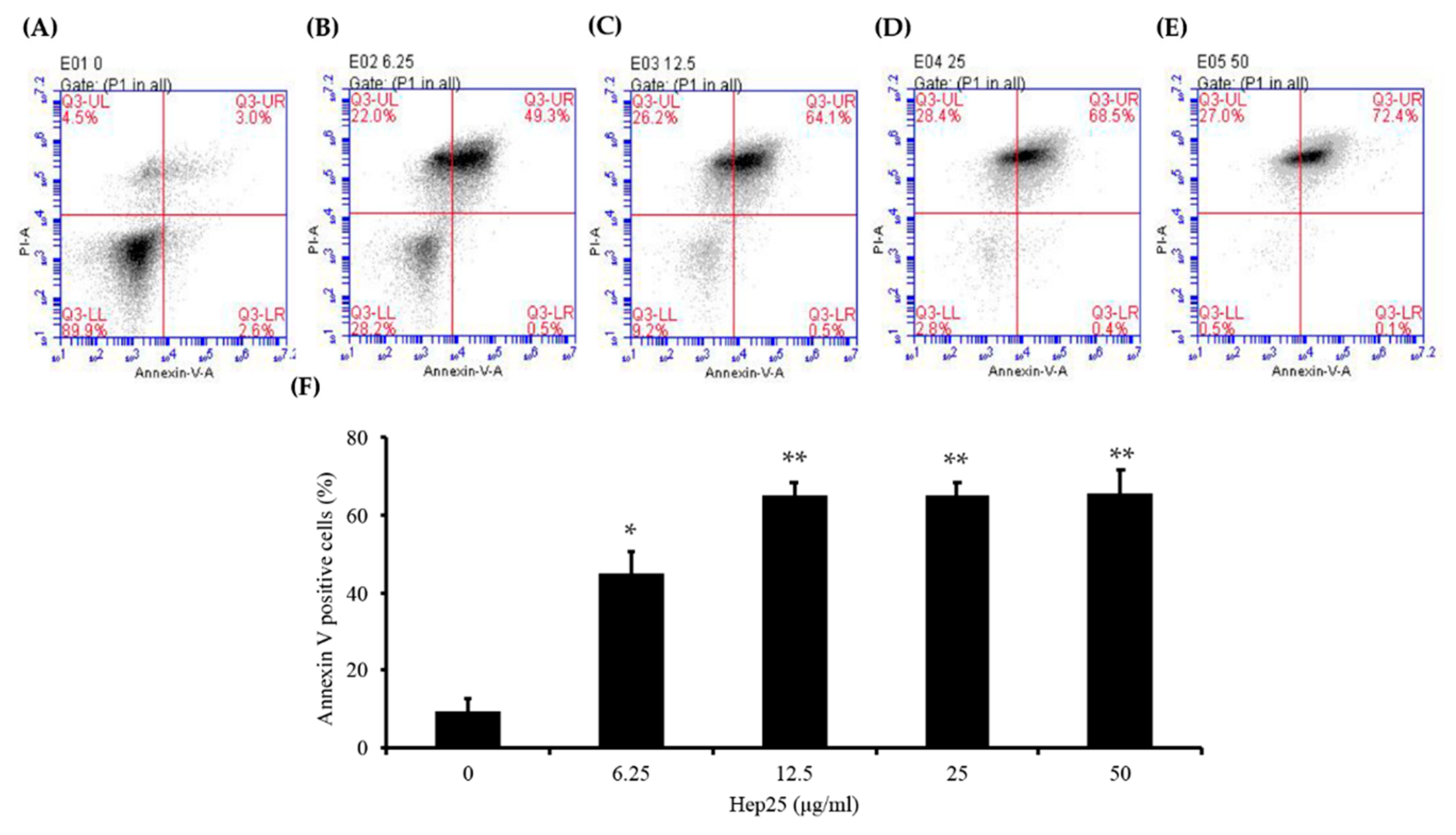
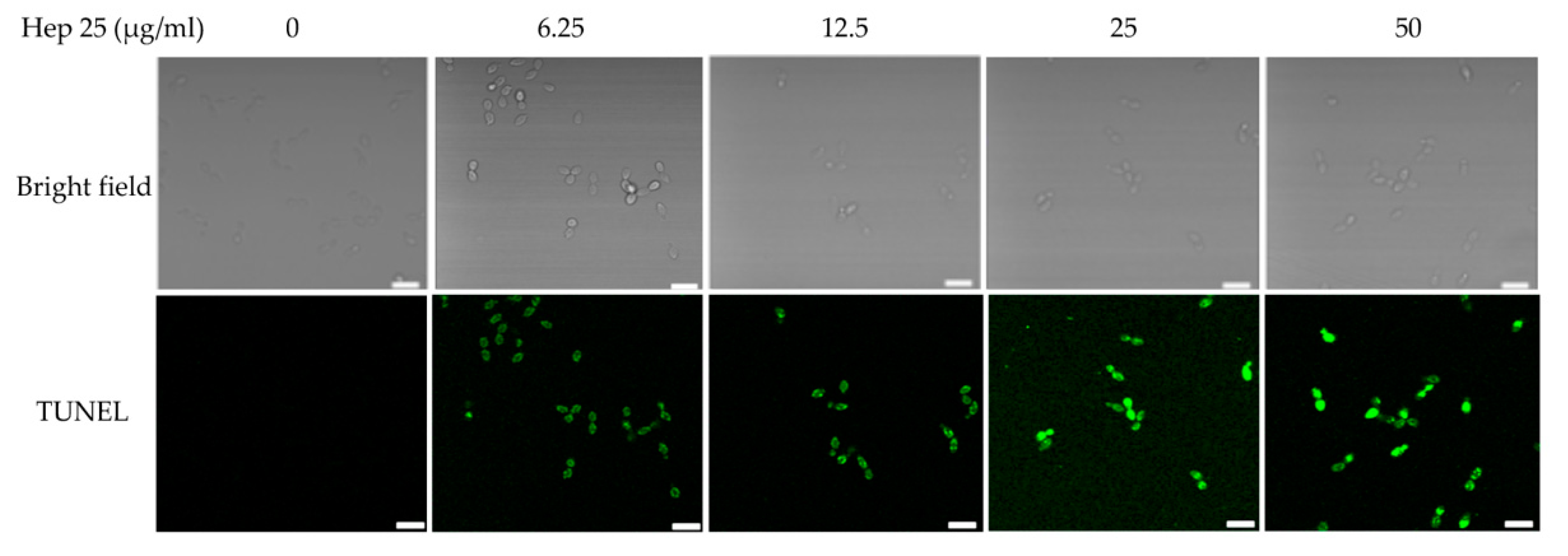
3.5. Phosphatidylserine was Externally Exposed in C. albicans Treated with Hep 25
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lopez-Medina, E.; Koh, A.Y. The complexities of bacterial-fungal interactions in the mammalian gastrointestinal tract. Microb. Cell 2016, 3, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Neville, B.A.; d’Enfert, C.; Bougnoux, M.E. Candida albicans commensalism in the gastrointestinal tract. Fems Yeast Res. 2015, 15. [Google Scholar] [CrossRef]
- Enoch, D.A.; Ludlam, H.A.; Brown, N.M. Invasive fungal infections: A review of epidemiology and management options. J. Med. Microbiol. 2006, 55, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, C.A. Fungal infections. Proc. Am. Thorac. Soc. 2006, 3, 35–40. [Google Scholar] [CrossRef]
- Swidergall, M.; Ernst, J.F. Interplay between Candida albicans and the antimicrobial peptide armory. Eukaryot. Cell 2014, 13, 950–957. [Google Scholar] [CrossRef]
- Boto, A.; Perez de la Lastra, J.M.; Gonzalez, C.C. The Road from host-defense peptides to a new generation of antimicrobial drugs. Molecules 2018, 23, 311. [Google Scholar] [CrossRef]
- Kumar, P.; Kizhakkedathu, J.N.; Straus, S.K. Antimicrobial peptides: Diversity, mechanism of action and strategies to improve the activity and biocompatibility in vivo. Biomolecules 2018, 8, 4. [Google Scholar] [CrossRef]
- Hancock, R.E.W.; Sahl, H.-G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef]
- Zhang, L.J.; Gallo, R.L. Antimicrobial peptides. Curr. Biol. 2016, 26, R14–R19. [Google Scholar] [CrossRef]
- Wu, Q.; Patocka, J.; Kuca, K. Insect antimicrobial peptides, a mini review. Toxins 2018, 10, 461. [Google Scholar] [CrossRef]
- Mahlapuu, M.; Hakansson, J.; Ringstad, L.; Bjorn, C. Antimicrobial peptides: An emerging category of therapeutic agents. Front. Cell Infect. Microbiol. 2016, 6, 194. [Google Scholar] [CrossRef]
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef]
- Wiig, M.E.; Dahlin, L.B.; Friden, J.; Hagberg, L.; Larsen, S.E.; Wiklund, K.; Mahlapuu, M. PXL01 in sodium hyaluronate for improvement of hand recovery after flexor tendon repair surgery: Randomized controlled trial. PLoS ONE 2014, 9, e110735. [Google Scholar] [CrossRef]
- Gronberg, A.; Mahlapuu, M.; Stahle, M.; Whately-Smith, C.; Rollman, O. Treatment with LL-37 is safe and effective in enhancing healing of hard-to-heal venous leg ulcers: A randomized, placebo-controlled clinical trial. Wound Repair Regen. 2014, 22, 613–621. [Google Scholar] [CrossRef]
- Park, C.H.; Valore, E.V.; Waring, A.J.; Ganz, T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J. Biol. Chem. 2001, 276, 7806–7810. [Google Scholar] [CrossRef] [PubMed]
- Boumaiza, M.; Abidi, S. Hepcidin cDNA evolution in vertebrates. Vitam. Horm. 2019, 110, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Oliveira-Filho, J.P.; Marques, J.A.; Cunha, P.H.J.; Medeiros, G.X.; Riet-Correa, F.; Machado, V.M.V.; Borges, A.S. Sequencing and expression analysis of hepcidin mRNA in donkey (Equus asinus) liver. Pesqui. Vet. Bras. 2012, 32, 1050–1054. [Google Scholar] [CrossRef]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [PubMed]
- Michels, K.; Nemeth, E.; Ganz, T.; Mehrad, B. Hepcidin and host defense against infectious diseases. PLoS Pathog. 2015, 11, e1004998. [Google Scholar] [CrossRef] [PubMed]
- Abbas, I.M.; Vranic, M.; Hoffmann, H.; El-Khatib, A.H.; Montes-Bayon, M.; Moller, H.M.; Weller, M.G. Investigations of the copper peptide hepcidin-25 by LC-MS/MS and NMR. Int. J. Mol. Sci. 2018, 19, 2271. [Google Scholar] [CrossRef] [PubMed]
- Verga Falzacappa, M.V.; Muckenthaler, M.U. Hepcidin: Iron-hormone and anti-microbial peptide. Gene 2005, 364, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Maisetta, G.; Petruzzelli, R.; Brancatisano, F.L.; Esin, S.; Vitali, A.; Campa, M.; Batoni, G. Antimicrobial activity of human hepcidin 20 and 25 against clinically relevant bacterial strains: Effect of copper and acidic pH. Peptides 2010, 31, 1995–2002. [Google Scholar] [CrossRef] [PubMed]
- Krause, A.; Neitz, S.; Mägert, H.-J.; Schulz, A.; Forssmann, W.-G.; Schulz-Knappe, P.; Adermann, K. LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett. 2000, 480, 147–150. [Google Scholar] [CrossRef]
- Jiang, X.F.; Liu, Z.F.; Lin, A.F.; Xiang, L.X.; Shao, J.Z. Coordination of bactericidal and iron regulatory functions of Hepcidin in innate antimicrobial immunity in a zebrafish model. Sci. Rep. 2017, 7, 4265. [Google Scholar] [CrossRef]
- Aschi, M.; Bozzi, A.; Di Bartolomeo, R.; Petruzzelli, R. The role of disulfide bonds and N-terminus in the structural properties of hepcidins: Insights from molecular dynamics simulations. Biopolymers 2010, 93, 917–926. [Google Scholar] [CrossRef]
- Maisetta, G.; Vitali, A.; Scorciapino, M.A.; Rinaldi, A.C.; Petruzzelli, R.; Brancatisano, F.L.; Esin, S.; Stringaro, A.; Colone, M.; Luzi, C.; et al. pH-dependent disruption of Escherichia coli ATCC 25922 and model membranes by the human antimicrobial peptides hepcidin 20 and 25. FEBS J. 2013, 280, 2842–2854. [Google Scholar] [CrossRef]
- Gillum, A.M.; Tsay, E.Y.; Kirsch, D.R. Isolation of the Candida albicans gene for orotidine-5′-phosphate decarboxylase by complementation of S. cerevisiae ura3 and E. coli pyrF mutations. Mol. Gen. Genet. 1984, 198, 179–182. [Google Scholar] [CrossRef]
- Lee, J.; Hwang, J.S.; Hwang, I.S.; Cho, J.; Lee, E.; Kim, Y.; Lee, D.G. Coprisin-induced antifungal effects in Candida albicans correlate with apoptotic mechanisms. Free Radic. Biol. Med. 2012, 52, 2302–2311. [Google Scholar] [CrossRef]
- Kwolek-Mirek, M.; Zadrag-Tecza, R. Comparison of methods used for assessing the viability and vitality of yeast cells. FEMS Yeast Res. 2014, 14, 1068–1079. [Google Scholar] [CrossRef]
- Madeo, F.; Frohlich, E.; Frohlich, K.U. A yeast mutant showing diagnostic markers of early and late apoptosis. J. Cell Biol. 1997, 139, 729–734. [Google Scholar] [CrossRef]
- Hao, B.; Cheng, S.; Clancy, C.J.; Nguyen, M.H. Caspofungin kills Candida albicans by causing both cellular apoptosis and necrosis. Antimicrob. Agents Chemoter. 2013, 57, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Phillips, A.J.; Sudbery, I.; Ramsdale, M. Apoptosis induced by environmental stresses and amphotericin B in Candida albicans. Proc. Natl. Acad. Sci. USA 2003, 100, 14327–14332. [Google Scholar] [CrossRef] [PubMed]
- Higuchi-Sanabria, R.; Charalel, J.K.; Viana, M.P.; Garcia, E.J.; Sing, C.N.; Koenigsberg, A.; Swayne, T.C.; Vevea, J.D.; Boldogh, I.R.; Rafelski, S.M.; et al. Mitochondrial anchorage and fusion contribute to mitochondrial inheritance and quality control in the budding yeast Saccharomyces cerevisiae. Mol. Biol. Cell 2016, 27, 776–787. [Google Scholar] [CrossRef] [PubMed]
- Creed, S.; McKenzie, M. Measurement of mitochondrial membrane potential with the fluorescent dye tetramethylrhodamine methyl ester (TMRM). Method Mol. Biol. 2019, 1928, 69–76. [Google Scholar] [CrossRef]
- Shirazi, F.; Kontoyiannis, D.P. Micafungin triggers caspase-dependent apoptosis in Candida albicans and Candida parapsilosis biofilms, including caspofungin non-susceptible isolates. Virulence 2015, 6, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Mochon, A.B.; Liu, H. The antimicrobial peptide histatin-5 causes a spatially restricted disruption on the Candida albicans surface, allowing rapid entry of the peptide into the cytoplasm. PLoS Pathog. 2008, 4, e1000190. [Google Scholar] [CrossRef]
- Du, H.; Puri, S.; McCall, A.; Norris, H.L.; Russo, T.; Edgerton, M. Human salivary protein histatin 5 has potent bactericidal activity against ESKAPE pathogens. Front. Cell. Infect. Microbiol. 2017, 7, 41. [Google Scholar] [CrossRef]
- Puri, S.; Edgerton, M. How does it kill? Understanding the candidacidal mechanism of salivary histatin 5. Eukaryot. Cell 2014, 13, 958–964. [Google Scholar] [CrossRef]
- Henriques, S.T.; Quintas, A.; Bagatolli, L.A.; Homblé, F.; Castanho, M.A. Energy-independent translocation of cell-penetrating peptides occurs without formation of pores. A biophysical study with pep-1. Mol. Membr. Biol. 2007, 24, 282–293. [Google Scholar] [CrossRef]
- Hayes, B.M.E.; Bleackley, M.R.; Anderson, M.A.; van der Weerden, N.L. The plant defensin NaD1 enters the cytoplasm of Candida albicans via endocytosis. J. Fungi 2018, 4, 20. [Google Scholar] [CrossRef]
- Munoz, A.; Marcos, J.F.; Read, N.D. Concentration-dependent mechanisms of cell penetration and killing by the de novo designed antifungal hexapeptide PAF26. Mol. Microbiol. 2012, 85, 89–106. [Google Scholar] [CrossRef] [PubMed]
- Jiao, C.Y.; Delaroche, D.; Burlina, F.; Alves, I.D.; Chassaing, G.; Sagan, S. Translocation and endocytosis for cell-penetrating peptide internalization. J. Biol. Chem. 2009, 284, 33957–33965. [Google Scholar] [CrossRef] [PubMed]
- El-Andaloussi, S.; Holm, T.; Langel, U. Cell-penetrating peptides: Mechanisms and applications. Curr. Pharm. Des. 2005, 11, 3597–3611. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Lee, D.G. Oxidative stress by antimicrobial peptide pleurocidin triggers apoptosis in Candida albicans. Biochimie 2011, 93, 1873–1879. [Google Scholar] [CrossRef]
- Kim, S.; Lee, D.G. Role of calcium in reactive oxygen species-induced apoptosis in Candida albicans: An antifungal mechanism of antimicrobial peptide, PMAP-23. Free Radic. Res. 2019, 53, 8–17. [Google Scholar] [CrossRef]
- Libardo, M.D.; Cervantes, J.L.; Salazar, J.C.; Angeles-Boza, A.M. Improved bioactivity of antimicrobial peptides by addition of amino-terminal copper and nickel (ATCUN) binding motifs. ChemMedChem 2014, 9, 1892–1901. [Google Scholar] [CrossRef]
- Libardo, M.D.; Nagella, S.; Lugo, A.; Pierce, S.; Angeles-Boza, A.M. Copper-binding tripeptide motif increases potency of the antimicrobial peptide anoplin via reactive oxygen species generation. Biochem. Biophys. Res. Commun. 2015, 456, 446–451. [Google Scholar] [CrossRef]
- Santoro, A.; Walke, G.; Vileno, B.; Kulkarni, P.P.; Raibaut, L.; Faller, P. Low catalytic activity of the Cu(ii)-binding motif (Xxx-Zzz-His; ATCUN) in reactive oxygen species production and inhibition by the Cu(i)-chelator BCS. Chem. Commun. 2018, 54, 11945–11948. [Google Scholar] [CrossRef]
- Libardo, M.D.; Gorbatyuk, V.Y.; Angeles-Boza, A.M. Central Role of the copper-binding motif in the complex mechanism of action of ixosin: Enhancing oxidative damage and promoting synergy with ixosin B. ACS Infect. Dis. 2016, 2, 71–81. [Google Scholar] [CrossRef]
- Conklin, S.E.; Bridgman, E.C.; Su, Q.; Riggs-Gelasco, P.; Haas, K.L.; Franz, K.J. Specific histidine residues confer histatin peptides with copper-dependent activity against Candida albicans. Biochemistry 2017, 56, 4244–4255. [Google Scholar] [CrossRef]
- Pan, Y.; Schroeder, E.A.; Ocampo, A.; Barrientos, A.; Shadel, G.S. Regulation of yeast chronological life span by TORC1 via adaptive mitochondrial ROS signaling. Cell Metab. 2011, 13, 668–678. [Google Scholar] [CrossRef] [PubMed]
- Mokudai, T.; Kanno, T.; Niwano, Y. Postantifungal-like effect of sublethal treatment of Candida albicans with acid-electrolyzed water. Arch. Oral Biol. 2015, 60, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Sendzik, M.; Pushie, M.J.; Stefaniak, E.; Haas, K.L. Structure and affinity of Cu(I) bound to human serum albumin. Inorg. Chem. 2017, 56, 15057–15065. [Google Scholar] [CrossRef] [PubMed]
- Esmieu, C.; Guettas, D.; Conte-Daban, A.; Sabater, L.; Faller, P.; Hureau, C. Copper-targeting approaches in alzheimer’s disease: How to improve the fallouts obtained from in vitro studies. Inorg. Chem. 2019, 58, 13509–13527. [Google Scholar] [CrossRef] [PubMed]
- Guilloreau, L.; Combalbert, S.; Sournia-Saquet, A.; Mazarguil, H.; Faller, P. Redox chemistry of copper-amyloid-beta: The generation of hydroxyl radical in the presence of ascorbate is linked to redox-potentials and aggregation state. Chembiochem 2007, 8, 1317–1325. [Google Scholar] [CrossRef] [PubMed]
- Perrone, G.G.; Tan, S.X.; Dawes, I.W. Reactive oxygen species and yeast apoptosis. Biochim. Biophys. Acta 2008, 1783, 1354–1368. [Google Scholar] [CrossRef]
- Farrugia, G.; Balzan, R. Oxidative stress and programmed cell death in yeast. Front. Oncol. 2012, 2, 64. [Google Scholar] [CrossRef]
- Carmona-Gutierrez, D.; Eisenberg, T.; Buttner, S.; Meisinger, C.; Kroemer, G.; Madeo, F. Apoptosis in yeast: Triggers, pathways, subroutines. Cell Death Differ. 2010, 17, 763–773. [Google Scholar] [CrossRef]
- Perry, S.W.; Norman, J.P.; Barbieri, J.; Brown, E.B.; Gelbard, H.A. Mitochondrial membrane potential probes and the proton gradient: A practical usage guide. BioTechniques 2011, 50, 98–115. [Google Scholar] [CrossRef]
- Krysko, D.V.; Roels, F.; Leybaert, L.; D’Herde, K. Mitochondrial transmembrane potential changes support the concept of mitochondrial heterogeneity during apoptosis. J. Histochem. Cytochem. 2001, 49, 1277–1284. [Google Scholar] [CrossRef]
- Eisenberg, T.; Buttner, S.; Kroemer, G.; Madeo, F. The mitochondrial pathway in yeast apoptosis. Apoptosis Int. J. Program. Cell Death 2007, 12, 1011–1023. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.A.V.; Megeney, L.A. Evolution of caspase-mediated cell death and differentiation: Twins separated at birth. Cell Death Differ. 2017, 24, 1359. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Lee, D.G. Reactive oxygen species modulate itraconazole-induced apoptosis via mitochondrial disruption in Candida albicans. Free Radic. Res. 2018, 52, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Seong, M.; Lee, D.G. Reactive oxygen species-independent apoptotic pathway by gold nanoparticles in Candida albicans. Microbiol. Res. 2018, 207, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.; Zhang, J.; Yu, L.; Wang, C.; Yang, Y.; Rong, X.; Xu, K.; Chu, M. Antifungal Activity of Coumarin Against Candida albicans Is Related to Apoptosis. Front. Cell. Infect. Microbiol. 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Herker, E.; Jungwirth, H.; Lehmann, K.A.; Maldener, C.; Fröhlich, K.U.; Wissing, S.; Büttner, S.; Fehr, M.; Sigrist, S.; Madeo, F. Chronological aging leads to apoptosis in yeast. J. Cell Biol. 2004, 164, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.R.; Gupta, V.K.; Deeba, F.; Bajpai, R.; Pandey, V.; Naqvi, A.H.; Upreti, D.K.; Gathergood, N.; Jiang, Y.; El Enshasy, H.A.; et al. Non-toxic and ultra-small biosilver nanoclusters trigger apoptotic cell death in fluconazole-resistant Candida albicans via Ras signaling. Biomolecules 2019, 9, 47. [Google Scholar] [CrossRef]
- Hwang, J.H.; Choi, H.; Kim, A.R.; Yun, J.W.; Yu, R.; Woo, E.R.; Lee, D.G. Hibicuslide C-induced cell death in Candida albicans involves apoptosis mechanism. J. Appl. Microbiol. 2014, 117, 1400–1411. [Google Scholar] [CrossRef]
- Tselepis, C.; Ford, S.J.; McKie, A.T.; Vogel, W.; Zoller, H.; Simpson, R.J.; Diaz Castro, J.; Iqbal, T.H.; Ward, D.G. Characterization of the transition-metal-binding properties of hepcidin. Biochem. J. 2010, 427, 289–296. [Google Scholar] [CrossRef]
- Rózga, M.; Sokołowska, M.; Protas, A.M.; Bal, W. Human serum albumin coordinates Cu(II) at its N-terminal binding site with 1 pM affinity. J. Biol. Inorg. Chem. 2007, 12, 913–918. [Google Scholar] [CrossRef]
- Bal, W.; Jeżowska-Bojczuk, M.; Kasprzak, K.S. Binding of nickel(II) and copper(II) to the N-terminal sequence of human protamine HP2. Chem. Res. Toxicol. 1997, 10, 906–914. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Sharma, N.K.; Prasad, R.; Singh, U.P. DNA cleavage study using copper (II)-GlyAibHis: A tripeptide complex based on ATCUN peptide motifs. Protein Pept. Lett. 2008, 15, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Agbale, C.M.; Cardoso, M.H.; Galyuon, I.K.; Franco, O.L. Designing metallodrugs with nuclease and protease activity. Met. Integr. Biomet. Sci. 2016, 8, 1159–1169. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, P.; Bossak, K.; Stefaniak, E.; Hureau, C.; Raibaut, L.; Bal, W.; Faller, P. N-terminal Cu-binding motifs (Xxx-Zzz-His, Xxx-His) and their derivatives: Chemistry, biology and medicinal applications. Chem. A Eur. J. 2018, 24, 8029–8041. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Lee, D.G. The antimicrobial peptide arenicin-1 promotes generation of reactive oxygen species and induction of apoptosis. Biochim. Biophys. Acta 2011, 1810, 1246–1251. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Dang, W.; Xie, J.; Zhu, R.; Sun, M.; Jia, F.; Zhao, Y.; An, X.; Qiu, S.; Li, X.; et al. Antimicrobial peptide protonectin disturbs the membrane integrity and induces ROS production in yeast cells. Biochim. Biophys. Acta 2015, 1848, 2365–2373. [Google Scholar] [CrossRef]
- Hwang, B.; Hwang, J.S.; Lee, J.; Kim, J.K.; Kim, S.R.; Kim, Y.; Lee, D.G. Induction of yeast apoptosis by an antimicrobial peptide, Papiliocin. Biochem. Biophys. Res. Commun. 2011, 408, 89–93. [Google Scholar] [CrossRef]
- Lee, J.; Lee, D.G. Melittin triggers apoptosis in Candida albicans through the reactive oxygen species-mediated mitochondria/caspase-dependent pathway. FEMS Microbiol. Lett. 2014, 355, 36–42. [Google Scholar] [CrossRef]
- Libardo, M.D.; Paul, T.J.; Prabhakar, R.; Angeles-Boza, A.M. Hybrid peptide ATCUN-sh-Buforin: Influence of the ATCUN charge and stereochemistry on antimicrobial activity. Biochimie 2015, 113, 143–155. [Google Scholar] [CrossRef]
- Madeo, F.; Fröhlich, E.; Ligr, M.; Grey, M.; Sigrist, S.J.; Wolf, D.H.; Fröhlich, K.U. Oxygen stress: A regulator of apoptosis in yeast. J. Cell Biol. 1999, 145, 757–767. [Google Scholar] [CrossRef]
- Li, R.; Zhang, R.; Yang, Y.; Wang, X.; Yi, Y.; Fan, P.; Liu, Z.; Chen, C.; Chang, J. CGA-N12, a peptide derived from chromogranin A, promotes apoptosis of Candida tropicalis by attenuating mitochondrial functions. Biochem. J. 2018, 475, 1385–1396. [Google Scholar] [CrossRef] [PubMed]
- Canton, M.; Caffieri, S.; Dall’Acqua, F.; Di Lisa, F. PUVA-induced apoptosis involves mitochondrial dysfunction caused by the opening of the permeability transition pore. FEBS Lett. 2002, 522, 168–172. [Google Scholar] [CrossRef]
- Tsiatsiani, L.; Van Breusegem, F.; Gallois, P.; Zavialov, A.; Lam, E.; Bozhkov, P.V. Metacaspases. Cell Death Differ. 2011, 18, 1279–1288. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.S.; Lee, J.; Hwang, J.H.; Kim, K.J.; Lee, D.G. Silver nanoparticles induce apoptotic cell death in Candida albicans through the increase of hydroxyl radicals. FEBS J. 2012, 279, 1327–1338. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Amino Acid Sequence | Disulfide Bonds | Molecular Mass (Da) | Net Charge a | Hydrophobicity a |
|---|---|---|---|---|---|
| Hep 25 | DTHFPICIFCCGCCHRSKCGMCCKT | 7–23, 10–13, 11–19,14–22 | 2789.4 | 2 | 52% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, R.-C.; Lan, C.-Y. Human Antimicrobial Peptide Hepcidin 25-Induced Apoptosis in Candida albicans. Microorganisms 2020, 8, 585. https://doi.org/10.3390/microorganisms8040585
Chen R-C, Lan C-Y. Human Antimicrobial Peptide Hepcidin 25-Induced Apoptosis in Candida albicans. Microorganisms. 2020; 8(4):585. https://doi.org/10.3390/microorganisms8040585
Chicago/Turabian StyleChen, Ruei-Ching, and Chung-Yu Lan. 2020. "Human Antimicrobial Peptide Hepcidin 25-Induced Apoptosis in Candida albicans" Microorganisms 8, no. 4: 585. https://doi.org/10.3390/microorganisms8040585
APA StyleChen, R.-C., & Lan, C.-Y. (2020). Human Antimicrobial Peptide Hepcidin 25-Induced Apoptosis in Candida albicans. Microorganisms, 8(4), 585. https://doi.org/10.3390/microorganisms8040585