Genome-Scale Metabolic Network Reconstruction and In Silico Analysis of Hexanoic acid Producing Megasphaera elsdenii




Abstract
1. Introduction
2. Materials and Methods
2.1. Genome-Scale Metabolic Model Reconstruction
2.2. Biomass Composition
2.3. Constraint-Based Flux Analysis
2.4. Gene Essentiality Analysis
2.5. Flux Ratio Analysis
3. Results and Discussion
3.1. Reconstruction of M. elsdenii Metabolic Network Model
3.2. Network Characteristics of iME375 and Its Comparison with C. acetobutylicum and E. coli
3.3. Model Validation
3.4. Gene Essntiality Analysis
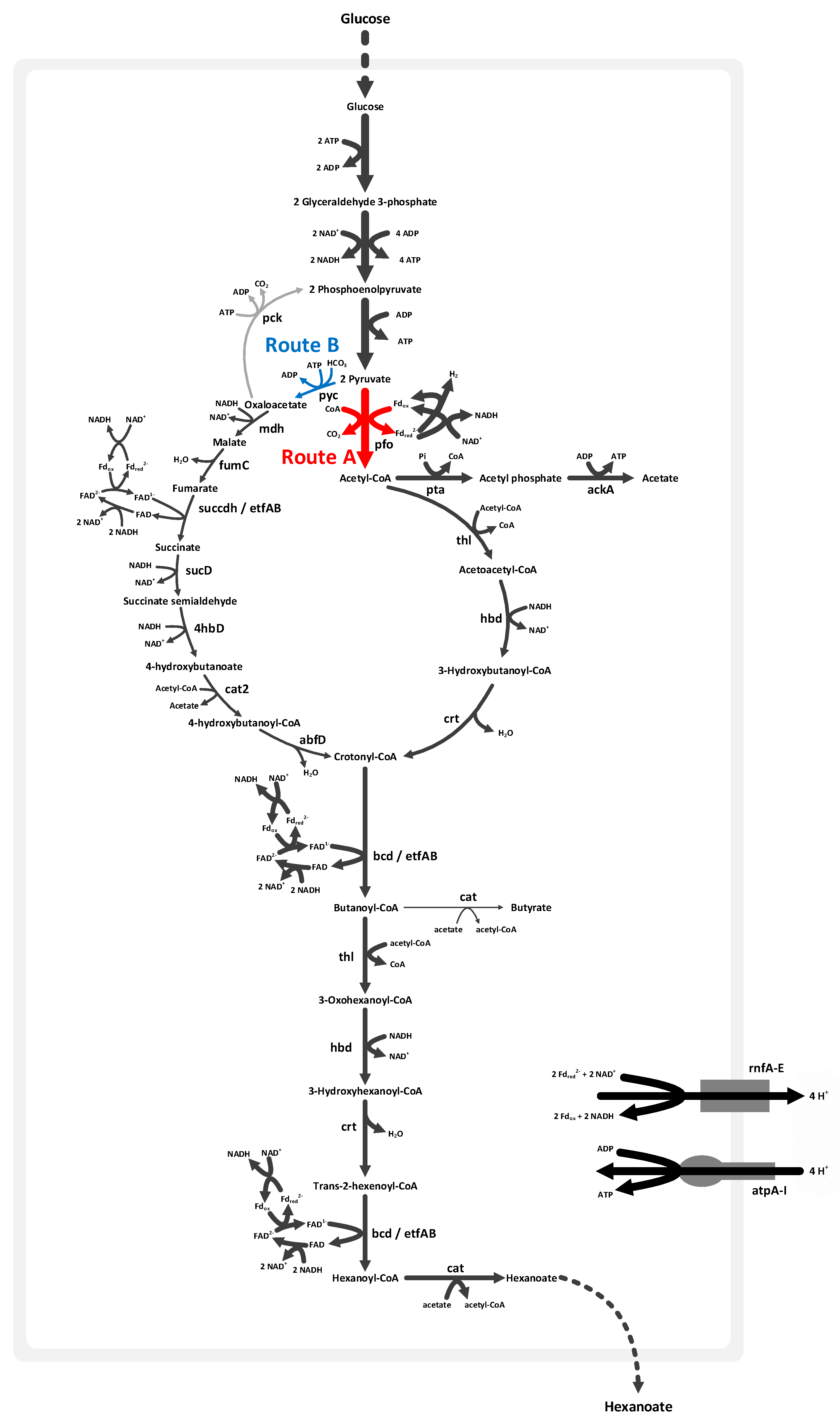
3.5. Characteristics of Volatile Fatty Acid Synthetic Metabolism
3.6. Flux Ratio Analysis for Hexanoic Acid Production
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Choi, K.; Jeon, B.S.; Kim, B.C.; Oh, M.K.; Um, Y.; Sang, B.I. In situ biphasic extractive fermentation for hexanoic acid production from sucrose by Megasphaera elsdenii NCIMB 702410. Appl. Biochem. Biotechnol. 2013, 171, 1094–1107. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Atiyeh, H.K.; Stevenson, B.S.; Tanner, R.S.; Wilkins, M.R.; Huhnke, R.L. Mixed culture syngas fermentation and conversion of carboxylic acids into alcohols. Bioresour. Technol. 2014, 152, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Milne, C.B.; Eddy, J.A.; Raju, R.; Ardekani, S.; Kim, P.-J.; Senger, R.S.; Jin, Y.-S.; Blaschek, H.P.; Price, N.D. Metabolic network reconstruction and genome-scale model of butanol-producing strain Clostridium beijerinckii NCIMB 8052. BMC Syst. Biol. 2011, 5, 130. [Google Scholar] [CrossRef] [PubMed]
- Šalić, A.; Pindrić, K.; Zelić, B. Bioproduction of food additives hexanal and hexanoic acid in a microreactor. Appl. Biochem. Biotechnol. 2013, 171, 2273–2284. [Google Scholar] [CrossRef] [PubMed]
- Scherf, U.; Söhling, B.; Gottschalk, G.; Linder, D.; Buckel, W. Succinate-ethanol fermentation in Clostridium kluyveri: Purification and characterisation of 4-hydroxybutyryl-CoA dehydratase/vinylacetyl-CoA Δ3-Δ2-isomerase. Arch. Microbiol. 1994, 161, 239–245. [Google Scholar] [CrossRef]
- Genthner, B.R.; Davis, C.L.; Bryant, M.P. Features of rumen and sewage sludge strains of Eubacterium limosum, a methanol- and H2-CO2-utilizing species. Appl. Environ. Microbiol. 1981, 42, 12–19. [Google Scholar] [CrossRef]
- Kohlmiller, E.F.; Gest, H. A comparative study of the light and dark fermentations of organic acids by Rhodospirillum rubrum. J. Bacteriol. 1951, 61, 269–282. [Google Scholar] [CrossRef]
- Lanjekar, V.B.; Marathe, N.P.; Venkata Ramana, V.; Shouche, Y.S.; Ranade, D.R. Megasphaera indica sp. nov., an obligate anaerobic bacteria isolated from human faeces. Int. J. Syst. Evol. Microbiol. 2014, 64, 2250–2256. [Google Scholar] [CrossRef]
- Roddick, F.A.; Britz, M.L. Production of hexanoic acid by free and immobilised cells of Megasphaera elsdenii: Influence of in-situ product removal using ion exchange resin. J. Chem. Technol. Biotechnol. 1997, 69, 383–391. [Google Scholar] [CrossRef]
- Lee, J.; Yun, H.; Feist, A.M.; Palsson, B.; Lee, S.Y. Genome-scale reconstruction and in silico analysis of the Clostridium acetobutylicum ATCC 824 metabolic network. Appl. Microbiol. Biotechnol. 2008, 80, 849–862. [Google Scholar] [CrossRef]
- Marx, H.; Graf, A.B.; Tatto, N.E.; Thallinger, G.G.; Mattanovich, D.; Sauer, M. Genome sequence of the ruminal bacterium Megasphaera elsdenii. J. Bacteriol. 2011, 193, 5578–5579. [Google Scholar] [CrossRef] [PubMed]
- Thiele, I.; Palsson, B.Ø. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 2010, 5, 93–121. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Hattori, M.; Aoki-Kinoshita, K.; Itoh, M.; Kawashima, S.; Katayama, T.; Araki, M.; Hirakawa, M. From genomics to chemical genomics: New developments in KEGG. Nucleic Acids Res. 2006, 34, D354–D357. [Google Scholar] [CrossRef] [PubMed]
- Caspi, R.; Billington, R.; Ferrer, L.; Foerster, H.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Mueller, L.A.; et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2016, 44, D471–D480. [Google Scholar] [CrossRef]
- Wallace, R.J. Catabolism of amino acids by Megasphaera elsdenii LC1. Appl. Environ. Microbiol. 1986, 51, 1141–1143. [Google Scholar] [CrossRef] [PubMed]
- Johnston, N.C.; Goldfine, H. Effects of growth temperature on fatty acid and alk-1-enyl group composition of Veillonella parvula and Megasphaera elsdenii phospholipids. J. Bacteriol. 1982, 149, 567–575. [Google Scholar] [CrossRef]
- Feist, A.M.; Henry, C.S.; Reed, J.L.; Krummenacker, M.; Joyce, A.R.; Karp, P.D.; Broadbelt, L.J.; Hatzimanikatis, V.; Palsson, B.Ø. A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol. Syst. Biol. 2007, 3, 121. [Google Scholar] [CrossRef]
- Orth, J.D.; Thiele, I.; Palsson, B.Ø. What is Flux Balance Analysis? Nat. Biotechnol. 2010, 28, 245–248. [Google Scholar] [CrossRef]
- Raman, K.; Chandra, N. Flux balance analysis of biological systems: Applications and challenges. Brief. Bioinform. 2009, 10, 435–449. [Google Scholar] [CrossRef]
- Schellenberger, J.; Que, R.; Fleming, R.M.T.; Thiele, I.; Orth, J.D.; Feist, A.M.; Zielinski, D.C.; Bordbar, A.; Lewis, N.E.; Rahmanian, S.; et al. Quantitative prediction of cellular metabolism with constraint-based models: The COBRA Toolbox v2.0. Nat. Protoc. 2011, 6, 1290–1307. [Google Scholar] [CrossRef]
- Hino, T.; Kuroda, S. Presence of lactate dehydrogenase and lactate racemase in Megasphaera elsdenii grown on glucose or lactate. Appl. Environ. Microbiol. 1993, 59, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, R.; Altman, E.; Eitemana, M.A. Lactate and acrylate metabolism by Megasphaera elsdenii under batch and steady-state conditions. Appl. Environ. Microbiol. 2012, 78, 8564–8570. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A. Regulation of Volatile Fatty Acid Synthesis in Megasphaera elsdenii and Hexanoic Acid Utilisation by Pseudomonas putida. Ph.D. Thesis, Victoria University, Footscray, Australia, 2006. [Google Scholar]
- Soto-Cruz, O.; Favela-Torres, E.; Saucedo-Castañeda, G. Modeling of growth, lactate consumption, and volatile fatty acid production by Megasphaera elsdenii cultivated in minimal and complex media. Biotechnol. Prog. 2002, 18, 193–200. [Google Scholar] [CrossRef]
- Schuster, S.; Pfeiffer, T.; Fell, D.A. Is maximization of molar yield in metabolic networks favoured by evolution? J. Theor. Biol. 2008, 252, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Hino, T.; Shimada, K.; Maruyama, T. Substrate preference in a strain of Megasphaera elsdenii, a ruminal bacterium, and its implications in propionate production and growth competition. Appl. Environ. Microbiol. 1994, 60, 1827–1831. [Google Scholar] [CrossRef] [PubMed]
- Hino, T.; Miyazaki, K.; Kuroda, S. Role of extracellular acetate in the fermentation of glucose by a ruminal bacterium, Megasphaera elsdenii. J. Gen. Appl. Microbiol. 1991, 37, 121–129. [Google Scholar] [CrossRef]
- Jeon, S.B.; Choi, O.; Um, Y.; Sang, B.-I. Production of medium-chain carboxylic acids by Megasphaera sp. MH with supplemental electron acceptors. Biotechnol. Biofuels 2016, 9, 129. [Google Scholar] [CrossRef]
- Amador-Noguez, D.; Feng, X.J.; Fan, J.; Roquet, N.; Rabitz, H.; Rabinowitz, J.D. Systems-level metabolic flux profiling elucidates a complete, bifurcated tricarboxylic acid cycle in Clostridium acetobutylicum. J. Bacteriol. 2010, 192, 4452–4461. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| M. Elsdenii CECT390 | M. Elsdenii ATCC17753 | M. Elsdenii ATCC25940 | ||||
|---|---|---|---|---|---|---|
| Exp 1 | Sim | Exp 2 | Sim | Exp 3 | Sim | |
| (mmol g−1DCW h−1) | ||||||
| Medium | Minimal with Lactate | Complex with Lactate | Complex with Glucose | |||
| Cell growth (h−1) | 0.06 | 0.06 | 0.19 | 0.21 | 0.03 | 0.03 |
| Lactate | −28.28 | −28.28 | −51.45 | −51.45 | ― | |
| Glucose | ― | ― | −2.62 | −2.62 | ||
| Acetate | 8.76 | 8.76 | 15.56 | 36 | ― | |
| Propionic acid | 17.61 | 16.7 | 3.37 | 3.71 | ― | |
| Butyric acid | 0.03 | 0.03 | 8.31 | 9.14 | 0.66 | 0.35 |
| Pentatnoic acid | 1.01 | 0.9 | ― | ― | ||
| Hexanoic acid | ― | ― | 1.8 | 1.5 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, N.-R.; Lee, C.H.; Lee, D.-Y.; Park, J.-B. Genome-Scale Metabolic Network Reconstruction and In Silico Analysis of Hexanoic acid Producing Megasphaera elsdenii. Microorganisms 2020, 8, 539. https://doi.org/10.3390/microorganisms8040539
Lee N-R, Lee CH, Lee D-Y, Park J-B. Genome-Scale Metabolic Network Reconstruction and In Silico Analysis of Hexanoic acid Producing Megasphaera elsdenii. Microorganisms. 2020; 8(4):539. https://doi.org/10.3390/microorganisms8040539
Chicago/Turabian StyleLee, Na-Rae, Choong Hwan Lee, Dong-Yup Lee, and Jin-Byung Park. 2020. "Genome-Scale Metabolic Network Reconstruction and In Silico Analysis of Hexanoic acid Producing Megasphaera elsdenii" Microorganisms 8, no. 4: 539. https://doi.org/10.3390/microorganisms8040539
APA StyleLee, N.-R., Lee, C. H., Lee, D.-Y., & Park, J.-B. (2020). Genome-Scale Metabolic Network Reconstruction and In Silico Analysis of Hexanoic acid Producing Megasphaera elsdenii. Microorganisms, 8(4), 539. https://doi.org/10.3390/microorganisms8040539