Comparative Genomic Analysis Confirms Five Genetic Populations of the Select Agent, Rathayibacter toxicus

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Genome Sequences
2.2. Genome Content
2.3. Average Nucleotide Identity
2.4. In Silico DNA-DNA Hybridization
2.5. Pangenome Analysis
2.6. Identification of Prophage and Phage Remnants
2.7. Secondary Metabolite and Biosynthetic Gene Cluster Analysis
2.8. Horizontal Gene Transfer
2.9. Individual Gene, Gene Cluster, Multi-Locus and Whole-Genome Sequence Analyses
3. Results and Discussion
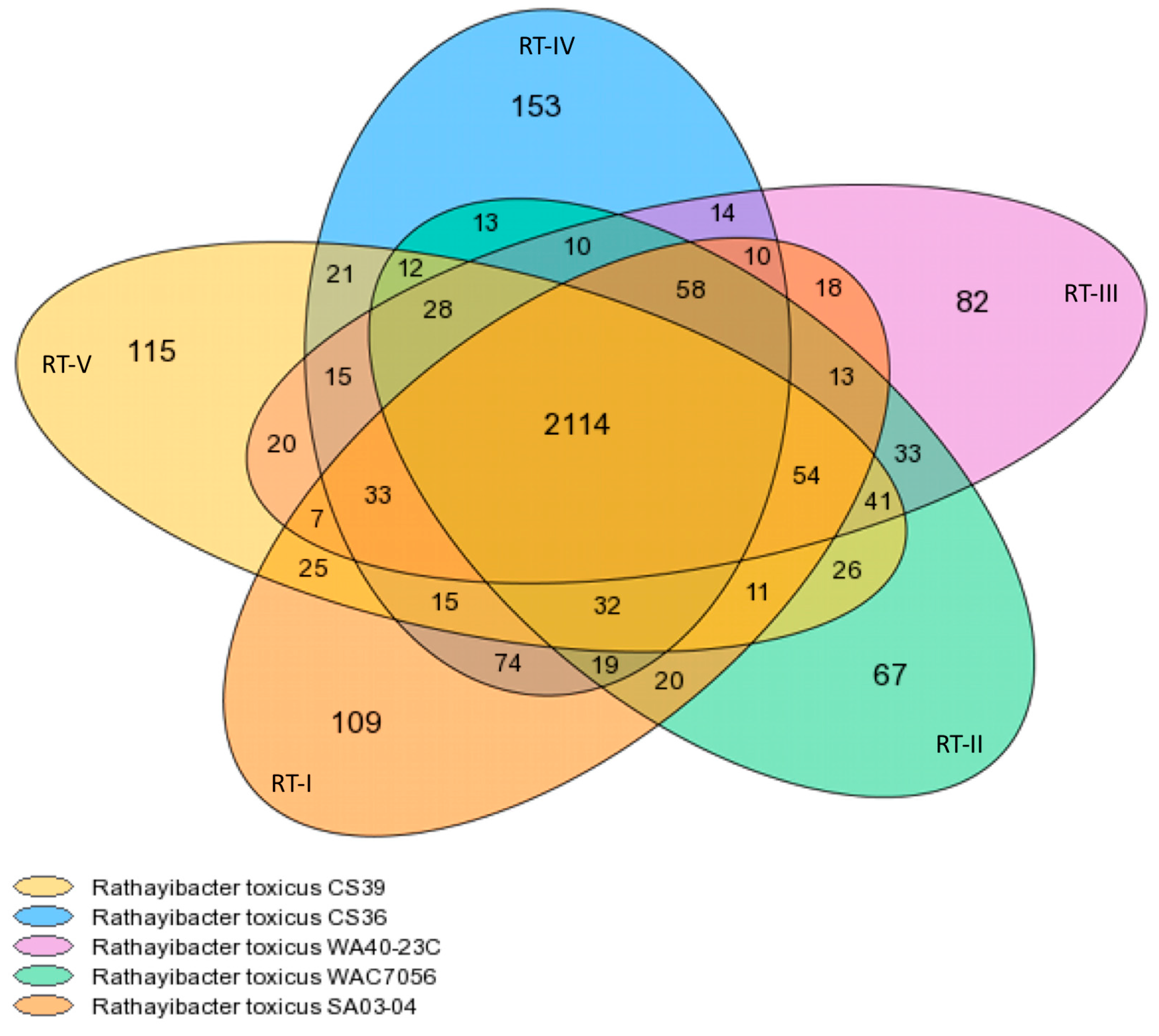
3.1. Genome Content and Organization
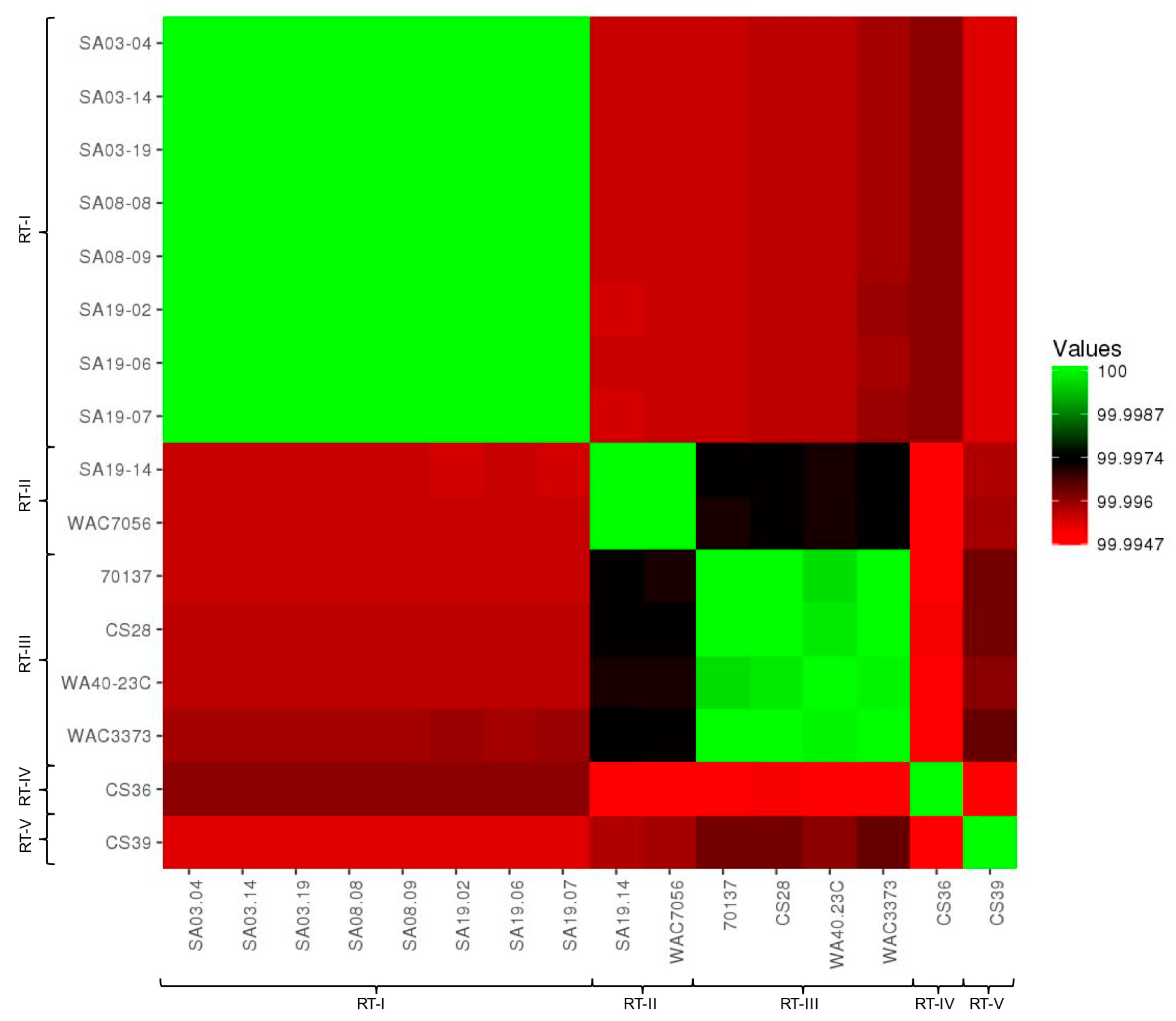
3.2. Average Nucleotide Identity and Digital DNA-DNA Hybridization
3.3. Phylogenetic Analyses Using Single Gene, Multi-Locus and Whole-Genome Sequence
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zgurskaya, H.I.; Evtushenko, L.I.; Akimov, V.N.; Kalakoutskii, L.V. Rathayibacter gen. nov., including the species Rathayibacter rathayi comb. nov., Rathayibacter tritici comb. nov., Rathayibacter iranicus comb. nov., and six strains from annual grasses. Int. J. Syst. Bacteriol. 1993, 43, 143–149. [Google Scholar] [CrossRef]
- Schroeder, B.K.; Schneider, W.L.; Luster, D.G.; Sechler, A.; Murray, T.D. Rathayibacter agropyri (non O’Gara, 1916) comb. nov., nom. rev., isolated from western wheatgrass (Pascopyrum smithii). Int. J. Syst. Evol. Microbiol. 2018, 68, 1519–1525. [Google Scholar] [CrossRef] [PubMed]
- Murray, T.D. Isolation of Corynebacterium agropyri from 30- to 40-year-old herbarium specimens of Agropyron species. Plant Dis. 1986, 70, 378–380. [Google Scholar] [CrossRef]
- Dorofeeva, L.V.; Evtushenko, L.I.; Krausova, V.I.; Karpov, A.V.; Subbotin, S.A.; Tiedje, J.M. Rathayibacter caricis sp. nov. and Rathayibacter festucae sp. nov., isolated from the phyllosphere of Carex sp. and the leaf gall induced by the nematode Anguina graminis on Festuca rubra L., respectively. Int. J. Syst. Evol. Microbiol. 2002, 52, 1917–1923. [Google Scholar]
- Dorofeeva, L.V.; Starodumova, I.P.; Krauzova, V.I.; Prisyazhnaya, N.V.; Vinokurova, N.G.; Lysanskaya, V.Y.; Tarlachkov, S.V.; Evtushenko, L.I. Rathayibacter oskolensis sp. nov., a novel actinobacterium from Androsace koso-poljanskii Ovcz.(Primulaceae) endemic to the Central Russian Upland. Int. J. Syst. Evol. Microbiol. 2018, 68, 1442–1447. [Google Scholar] [CrossRef]
- Vasilenko, O.V.; Starodumova, I.P.; Tarlachkov, S.V.; Dorofeeva, L.V.; Avtukh, A.N.; Evtushenko, L.I. Draft genome sequence of “Rathayibacter tanaceti” strain VKM Ac-2596 isolated from Tanacetum vulgare infested by a foliar nematode. Genome Announc. 2016, 4, e00512–e00516. [Google Scholar] [CrossRef]
- Riley, I.T.; Ophel, K.M. Clavibacter toxicus sp. nov., the bacterium responsible for annual ryegrass toxicity in Australia. Int. J. Syst. Bacteriol. 1992, 42, 64–68. [Google Scholar] [CrossRef]
- Sasaki, J.; Chijimatsu, M.; Suzuki, K.-I. Taxonomic significance of 2,4-diaminobutyric acid isomers in the cell wall peptidoglycan of actinomycetes and reclassification of Clavibacter toxicus as Rathayibacter toxicus comb. nov. Int. J. Syst. Bacteriol. 1998, 48, 403–410. [Google Scholar] [CrossRef]
- McKay, A.C.; Ophel, K.M. Toxigenic Clavibacter/Anguina associations infecting grass seedheads. Annu. Rev. Phytopathol. 1993, 31, 151–167. [Google Scholar] [CrossRef]
- Riley, I.T.; McKay, A.C. Inoculation of Lolium rigidum with Clavibacter sp., the bacterium responsible for toxicity of annual ryegrass. J. Appl. Bacteriol. 1991, 71, 302–306. [Google Scholar] [CrossRef]
- Bertozzi, T.; McKay, A.C. Incidence on Polypogon monspeliensis of Clavibacter toxicus and Anguina sp., the organisms associated with ‘flood plain stagger’ in South Australia. Aust. J. Exp. Agric. 1995, 35, 567–569. [Google Scholar] [CrossRef]
- McKay, A.C.; Ophel, K.M.; Reardon, T.B.; Gooden, J.M. Livestock deaths associated with Clavibacter toxicus/Auguina sp. infection in seedheads of Agrostis avenacea and Polypogon monspeliensis. Plant Dis. 1993, 77, 635–641. [Google Scholar] [CrossRef]
- Riley, I.T. Anguina tritici is a potential vector of Clavibacter toxicus. Australas. Plant Pathol. 1992, 21, 147–148. [Google Scholar] [CrossRef]
- Riley, I.T.; McKay, A.C. Invasion of some grasses by Anguina funesta (Nematoda, Anguinidae) juveniles. Nematologica 1991, 37, 447–454. [Google Scholar] [CrossRef]
- Riley, I.T.; McKay, A.C. Specificity of the adhesion of some plant pathogenic micro-organisms to the cuticle of nematodes in the genus Anguina (Nematoda: Anguinidae). Nematologica 1990, 36, 90–103. [Google Scholar] [CrossRef]
- Edgar, J.A.; Frahn, J.L.; Cockrum, P.A.; Anderton, N.; Jago, M.V.; Culvenor, C.C.J.; Jones, A.J.; Murray, K.; Shaw, K.J. Corynetoxins, causative agents of annual ryegrass toxicity; their identification as tunicamycin group antibiotics. J. Chem. Soc. Chem. Commun. 1982, 4, 222–224. [Google Scholar] [CrossRef]
- Finnie, J.W. Review of corynetoxins poisoning of livestock, a neurological disorder produced by a nematode-bacterium complex. Aust. Vet. J. 2006, 84, 271–277. [Google Scholar] [CrossRef]
- Eckardt, K. Tunicamycins, streptovirudins, and corynetoxins, a special subclass of nucleoside antibiotics. J. Nat. Prod. 1983, 46, 544–550. [Google Scholar] [CrossRef]
- Sechler, A.J.; Tancos, M.A.; Schneider, D.J.; King, J.G.; Fenessey, C.M.; Schroeder, B.K.; Murray, T.D.; Luster, D.G.; Schnieder, W.L.; Rogers, E.E. Whole genome sequence of two Rathayibacter toxicus strains reveals a tunicamycin biosynthetic cluster similar to Streptomyces chartreusis. PLoS ONE 2017, 12, e0183005. [Google Scholar] [CrossRef]
- Jago, M.V.; Payne, A.L.; Peterson, J.E.; Bagust, T.J. Inhibition of glycosylation by corynetoxin, the causative agent of annual ryegrass toxicity: A comparison with tunicamycin. Chem. Biol. Interact. 1983, 45, 223–234. [Google Scholar] [CrossRef]
- Murray, T.D.; Schroeder, B.K.; Schneider, W.L.; Luster, D.G.; Sechler, A.; Rogers, E.E.; Subbotin, S.A. Rathayibacter toxicus, other Rathayibacter species inducing bacterial head blight of grasses and the potential for livestock poisonings. Phytopathology 2017, 107, 804–815. [Google Scholar] [CrossRef] [PubMed]
- Allen, J. Annual ryegrass toxicity—An animal disease caused by toxins produced by a bacterial plant pathogen. Microbiol. Aust. 2012, 33, 18–21. [Google Scholar]
- Finnie, J.W. Plant-Associated Toxins: Agricultural, Phytochemical and Ecological Aspects; CAB International: Wallingford, Oxfordshire, UK, 1994. [Google Scholar]
- Jubb, K.V.F.; Huxtable, C.R.; Palmer, N. Pathology of Domestic Animals, 5th ed.; Elsevier: Edinburgh, UK, 2007. [Google Scholar]
- Grewar, J.D.; Allen, J.G.; Guthrie, A.J. Annual ryegrass toxicity in Thoroughbred horses in Ceres in the Western Cape Province, South Africa. J. S. Afr. Vet. Assoc. 2009, 80, 220–223. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S. Livestock poisonings: Exposing a familiar culprit. Rural Res. 1991, 153, 9–12. [Google Scholar]
- Schneider, D.J. First report of annual ryegrass toxicity in the Republic of South Africa. Onderstepoort J. Vet. Res. 1981, 48, 251–255. [Google Scholar]
- Davis, E.W., II. Phylogeny and Evolution of Gall-Associated Plant Pathogenic Bacteria; Oregon State University: Corvallis, OR, USA, 2017. [Google Scholar]
- Stack, J.P.; Busot, G.Y.; Arif, M. CRISPR/Cas sequence-based confirmation of an emergent population of Rathayibacter toxicus in South Australia. Phytopathology 2017, 107, S5.127. [Google Scholar]
- Davis, E.W., II; Tabima, J.F.; Weisberg, A.J.; Dantas Lopes, L.; Wiseman, M.S.; Wiseman, M.S.; Pupko, T.; Belcher, M.S.; Sechler, A.J.; Tancos, M.A.; et al. Evolution of the U.S. biological select agent Rathayibacter toxicus. mBio 2018, 9, e01280-18. [Google Scholar] [CrossRef] [PubMed]
- Agarkova, I.V.; Vidaver, A.K.; Postnikova, E.N.; Riley, I.T.; Schaad, N.W. Genetic characterization and diversity of Rathayibacter toxicus. Phytopathology 2006, 96, 1270–1277. [Google Scholar] [CrossRef]
- Johnston, M.S.; Sutherland, S.S.; Constantine, C.C.; Hampson, D.J. Genetic analysis of Clavibacter toxicus, the agent of annual ryegrass toxicity. Epidemiol. Infect. 1996, 117, 393–400. [Google Scholar] [CrossRef]
- Riley, I.T. Serological relationships between strains of coryneform bacteria responsible for annual ryegrass toxicity and other plant-pathogenic Corynebacteria. Int. J. Syst. Bacteriol. 1987, 35, 153–159. [Google Scholar] [CrossRef]
- Riley, I.T.; Reardon, T.B.; McKay, A.C. Genetic analysis of plant pathogenic bacteria in the genus Clavibacter using allozyme electrophoresis. J. Gen. Appl. Microbiol. 1988, 34, 3025–3030. [Google Scholar] [CrossRef][Green Version]
- Arif, M.; Busot, G.Y.; Mann, R.; Rodoni, B.; Liu, S.; Stack, J.P. Emergence of a new population of Rathayibacter toxicus: An ecologically complex, geographically isolated bacterium. PLoS ONE 2016, 11, e0156182. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yasuhara-Bell, J.; Stack, J.P. Panel of three loop-mediated isothermal amplification assays differentiates Rathayibacter toxicus populations RT-I, RT-II, RT-III, RT-IV and RT-V. J. Plant Pathol. 2019, 101, 707–717. [Google Scholar] [CrossRef]
- Benson, D.; Boguski, M.; Lipman, D.; Ostell, J. The National Center for Biotechnology Information. Genomics 1990, 6, 389–391. [Google Scholar] [CrossRef]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2013, 41, D36–D42. [Google Scholar] [CrossRef] [PubMed]
- Bilofsky, H.S.; Christian, B. The GenBank® genetic sequence data bank. Nucleic Acids Res. 1988, 16, 1861–1863. [Google Scholar] [CrossRef]
- Benson, D.; Lipman, D.J.; Ostell, J. GenBank. Nucleic Acids Res. 1993, 21, 2963–2965. [Google Scholar] [CrossRef]
- Arif, M.; Busot, G.Y.; Mann, R.; Rodoni, B.; Liu, S.; Stack, J.P. Complete genome of select agent Rathayibacter toxicus isolate SA03-04 from South Australia. Phytopathology 2015, 105, S4.96. [Google Scholar]
- Chin, C.S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E.; et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Tamura, K.; Nei, M. MEGA: Molecular evolutionary genetics analysis software for microcomputers. Bioinformatics 1994, 10, 189–191. [Google Scholar] [CrossRef] [PubMed]
- Haft, D.H.; DiCuccio, M.; Badretdin, A.; Brover, V.; Chetvernin, V.; O’Neill, K.; Li, W.; Chitsaz, F.; Derbyshire, M.K.; Gonzales, N.R.; et al. RefSeq: An update on prokaryotic genome annotation and curation. Nucleic Acids Res. 2018, 46, D851–D860. [Google Scholar] [CrossRef] [PubMed]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2013, 42, D206–D214. [Google Scholar] [CrossRef]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2011, 40, D109–D114. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Abby, S.S.; Néron, B.; Ménager, H.; Touchon, M.; Rocha, E.P. MacSyFinder: A program to mine genomes for molecular systems with an application to CRISPR-Cas systems. PLoS ONE 2014, 9, e110726. [Google Scholar] [CrossRef] [PubMed]
- Abby, S.S.; Rocha, E.P. Identification of protein secretion systems in bacterial genomes using MacSyFinder. In Bacterial Protein Secretion Systems: Methods and Protocols; Journet, L., Cascales, E., Eds.; Humana Press: New York, NY, USA, 2017; pp. 1–21. [Google Scholar]
- Afgan, E.; Baker, D.; Batut, B.; Van Den Beek, M.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [PubMed]
- Giardine, B.; Riemer, C.; Hardison, R.C.; Burhans, R.; Elnitski, L.; Shah, P.; Zhang, Y.; Blankenberg, D.; Albert, I.; Taylor, J.; et al. Galaxy: A platform for interactive large-scale genome analysis. Genome Res. 2005, 15, 1451–1455. [Google Scholar] [CrossRef]
- Mareuil, F.; Doppelt-Azeroual, O.; Ménager, H. A public galaxy platform at pasteur used as an execution engine for web services. F1000Research 2017, 6, 1030. [Google Scholar]
- Martínez-García, P.M.; Ramos, C.; Rodríguez-Palenzuela, P. T346Hunter: A novel web-based tool for the prediction of type III, type IV and type VI secretion systems in bacterial genomes. PLoS ONE 2015, 10, e0119317. [Google Scholar] [CrossRef]
- Bi, D.; Liu, L.; Tai, C.; Deng, Z.; Rajakumar, K.; Ou, H.Y. SecReT4: A web-based bacterial type IV secretion system resource. Nucleic Acids Res. 2012, 41, D660–D665. [Google Scholar] [CrossRef]
- Li, J.; Yao, Y.; Xu, H.H.; Hao, L.; Deng, Z.; Rajakumar, K.; Ou, H.Y. SecReT6: A web-based resource for type VI secretion systems found in bacteria. Environ. Microbiol. 2015, 17, 2196–2202. [Google Scholar] [CrossRef]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef]
- Laing, C.; Buchanan, C.; Taboada, E.N.; Zhang, Y.; Kropinski, A.; Villegas, A.; Thomas, J.E.; Gannon, V.P. Pan-genome sequence analysis using Panseq: An online tool for the rapid analysis of core and accessory genomic regions. BMC Bioinform. 2010, 11, 461. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-R, L.M.; Konstantinidis, K.T. The enveomics collection: A toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ Prepr. 2016, 4, e1900v1. [Google Scholar]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.H.; Ha, S.M.; Lim, J.M.; Kwon, S.J.; Chun, J. A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie van Leeuwenhoek 2017, 110, 1281–1286. [Google Scholar] [CrossRef]
- Lee, I.; Kim, Y.O.; Park, S.C.; Chun, J. OrthoANI: An improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 2016, 66, 1100–1103. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Ew, M.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Richter, M.; Rosselló-Móra, R.; Oliver Glöckner, F.; Peplies, J. JSpeciesWS: A web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 2015, 32, 929–931. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Delcher, A.L.; Kasif, S.; Fleischmann, R.D.; Peterson, J.; White, O.; Salzberg, S.L. Alignment of whole genomes. Nucleic Acids Res. 1999, 27, 2369–2376. [Google Scholar] [CrossRef]
- Delcher, A.L.; Phillippy, A.; Carlton, J.; Salzberg, S.L. Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res. 2002, 30, 2478–2483. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef] [PubMed]
- Bohlin, J.; Skjerve, E.; Ussery, D.W. Reliability and applications of statistical methods based on oligonulcleotide frequencies in bacterial and archaeal genomes. BMC Genomics 2008, 9, 104. [Google Scholar] [CrossRef] [PubMed]
- Pride, D.T.; Meinersmann, R.J.; Wassenaar, T.M.; Blaser, M.J. Evolutionary implications of microbial genome tetranucleotide frequency biases. Genome Res. 2003, 13, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Van Passel, M.W.J.; Kuramae, E.E.; Luyf, A.C.M.; Bart, A.; Boekhout, T. The reach of the genome signature in prokaryotes. BCM Evol. Biol. 2006, 6, 84. [Google Scholar] [CrossRef][Green Version]
- Teeling, H.; Meyerdierks, A.; Bauer, M.; Amann, R.; Glöckner, F.O. Application of tetranucleotide frequencies for the assignment of genomic fragments. Environ. Microbiol. 2004, 6, 938–947. [Google Scholar] [CrossRef]
- Babicki, S.; Arndt, D.; Marcu, A.; Liang, Y.; Grant, J.R.; Maciejewski, A.; Wishart, D.S. Heatmapper: Web-enabled heat mapping for all. Nucleic Acids Res. 2016, 44, W147–W153. [Google Scholar] [CrossRef]
- Auch, A.F.; Jan, M.; Klenk, H.P.; Göker, M. Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand. Genom. Sci. 2010, 2, 117. [Google Scholar] [CrossRef]
- Auch, A.F.; Klenk, H.P.; Göker, M. Standard operating procedure for calculating genome-to-genome distances based on high-scoring segment pairs. Stand. Genom. Sci. 2010, 2, 142. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef]
- Nelder, J.A.; Wedderburn, R.W.M. Generalized linear models. J. R. Stat. Soc. 1972, 135, 370–384. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M.; Klenk, H.-P. Taxonomic use of DNA G + C content and DNA-DNA hybridization in the genomic age. Int. J. Syst. Evol. Microbiol. 2014, 64, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Vallenet, D.; Calteau, A.; Cruveiller, S.; Gachet, M.; Lajus, A.; Josso, A.; Mercier, J.; Renaux, A.; Rollin, J.; Rouy, Z.; et al. MicroScope in 2017: An expanding and evolving integrated resource for community expertise of microbial genomes. Nucleic Acids Res. 2016, 45, D517–D528. [Google Scholar] [CrossRef] [PubMed]
- Miele, V.; Penel, S.; Duret, L. Ultra-fast sequence clustering from similarity networks with SiLiX. BMC Bioinform. 2011, 12, 116. [Google Scholar] [CrossRef] [PubMed]
- Sousa, A.L.; Maués, D.; Lobato, A.; Franco, E.F.; Pinheiro, K.; Araújo, F.; Pantoja, Y.; Costa da Silva, A.L.; Morais, J.; Ramos, R.T.J. PhageWeb—Web interface for rapid identification and characterization of prophages in bacterial enomes. Front. Genet. 2018, 9, 644. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A fast phage search tool. Nucleic Acids Res. 2011, 39, W347–W352. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef]
- Lima-Mendez, G.; Van Helden, J.; Toussaint, A.; Leplae, R. Prophinder: A computational tool for prophage prediction in prokaryotic genomes. Bioinformatics 2008, 24, 863–865. [Google Scholar] [CrossRef]
- Roux, S.; Enault, F.; Hurwitz, B.L.; Sullivan, M.B. VirSorter: Mining viral signal from microbial genomic data. PeerJ 2015, 3, e985. [Google Scholar] [CrossRef]
- Deng, W.; Nickle, D.C.; Learn, G.H.; Maust, B.; Mullins, J.I. ViroBLAST: A stand-alone BLAST web server for flexible queries of multiple databases and user’s datasets. Bioinformatics 2007, 23, 2334–2336. [Google Scholar] [CrossRef]
- Russell, D.A.; Hatfull, G.F. PhagesDB: The actinobacteriophage database. Bioinformatics 2016, 33, 784–786. [Google Scholar] [CrossRef] [PubMed]
- Weber, T.; Blin, K.; Duddela, S.; Krug, D.; Kim, H.U.; Bruccoleri, R.; Lee, S.Y.; Fischbach, M.A.; Müller, R.; Wohlleben, W.; et al. AntiSMASH 3.0––A comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015, 43, W237–W243. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Medema, M.H.; Kazempour, D.; Fischbach, M.A.; Breitling, R.; Takano, E.; Weber, T. antiSMASH 2.0—A versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res. 2013, 41, W204–W212. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Wolf, T.; Chevrette, M.G.; Lu, X.; Schwalen, C.J.; Kautsar, S.A.; Suarez Duran, H.G.; De Los Santos, E.L.; Kim, H.U.; Nave, M.; et al. antiSMASH 4.0—Improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Res. 2017, 45, W36–W41. [Google Scholar] [CrossRef] [PubMed]
- Medema, M.H.; Blin, K.; Cimermancic, P.; de Jager, V.; Zakrzewski, P.; Fischbach, M.A.; Weber, T.; Takano, E.; Breitling, R. antiSMASH: Rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011, 39, W339–W346. [Google Scholar] [CrossRef]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Group, S.F.U.R.C.; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef]
- Langille, M.G.I.; Brinkman, F.S.L. IslandViewer: An integrated interface for computational identification and visualization of genomic islands. Bioinformatics 2009, 25, 664–665. [Google Scholar] [CrossRef]
- Langille, M.G.; Hsiao, W.W.; Brinkman, F.S. Evaluation of genomic island predictors using a comparative genomics approach. BMC Bioinform. 2008, 9, 329. [Google Scholar] [CrossRef]
- Bertelli, C.; Brinkman, F.S. Improved genomic island predictions with IslandPath-DIMOB. Bioinformatics 2018, 1, 7. [Google Scholar] [CrossRef]
- Hsiao, W.; Wan, I.; Jones, S.J.; Brinkman, F.S. IslandPath: Aiding detection of genomic islands in prokaryotes. Bioinformatics 2003, 19, 418–420. [Google Scholar] [CrossRef]
- Merkl, R. SIGI: Score-based identification of genomic islands. BMC Bioinform. 2004, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Waack, S.; Keller, O.; Asper, R.; Brodag, T.; Damm, C.; Fricke, W.F.; Surovcik, K.; Meinicke, P.; Merkl, R. Score-based prediction of genomic islands in prokaryotic genomes using hidden Markov models. BMC Bioinform. 2006, 7, 142. [Google Scholar] [CrossRef] [PubMed]
- Vernikos, G.S.; Parkhill, J. Interpolated variable order motifs for identification of horizontally acquired DNA: Revisiting the Salmonella pathogenicity islands. Bioinformatics 2006, 22, 2196–2203. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Bertels, F.; Silander, O.K.; Pachkov, M.; Rainey, P.B.; van Nimwegen, E. Automated reconstruction of whole-genome phylogenies from short-sequence reads. Mol. Biol. Evol. 2014, 31, 1077–1088. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar]
- Jukes, T.H.; Cantor, C.R. Evolution of protein molecules. In Mammalian Protein Metabolism; Munro, H.N., Ed.; Academic Press: New York, NY, USA, 1969; pp. 21–132. [Google Scholar]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Darling, A.E.; Jospin, G.; Lowe, E.; Matsen IV, F.A.; Bik, H.M.; Eisen, J.A. PhyloSift: Phylogenetic analysis of genomes and metagenomes. PeerJ 2013, 2, e243. [Google Scholar] [CrossRef]
- Kiełbasa, S.M.; Wan, R.; Sato, K.; Horton, P.; Frith, M.C. Adaptive seeds tame genomic sequence comparison. Genome Res. 2011, 21, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Matsen, F.A.; Kodner, R.B.; Armbrust, E.V. pplacer: Linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinform. 2010, 11, 538. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. ClustalW. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.I.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Henikoff, S.; Henikoff, J.G. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA 1992, 89, 10915–10919. [Google Scholar] [CrossRef]
- Galperin, M.Y.; Brenner, S.E. Using metabolic pathway databases for functional annotation. Trends Genet. 1998, 14, 332–333. [Google Scholar] [CrossRef]
- Natale, D.A.; Galperin, M.Y.; Tatusov, R.L.; Koonin, E.V. Using the COG database to improve gene recognition in complete genomes. Genetica 2000, 108, 9–17. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Natale, D.A.; Garkavtsev, I.V.; Tatusova, T.A.; Shankavaram, U.T.; Rao, B.S.; Kiryutin, B.; Galperin, M.Y.; Fedorova, N.D.; Koonin, E.V. The COG database: New developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2001, 29, 22–28. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; et al. The COG database: An updated version includes eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef]
- Bateman, A.; Birney, E.; Durbin, R.; Eddy, S.R.; Howe, K.L.; Sonnhammer, E.L. The Pfam protein families database. Nucleic Acids Res. 2000, 28, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2013, 42, D222–D230. [Google Scholar] [CrossRef]
- Punta, M.; Coggill, P.C.; Eberhardt, R.Y.; Mistry, J.; Tate, J.; Boursnell, C.; Pang, N.; Forslund, K.; Ceric, G.; Clements, J.; et al. The Pfam protein families database. Nucleic Acids Res. 2011, 40, D290–D301. [Google Scholar] [CrossRef] [PubMed]
- Haft, D.H.; Loftus, B.J.; Richardson, D.L.; Yang, F.; Eisen, J.A.; Paulsen, I.T.; White, O. TIGRFAMs: A protein family resource for the functional identification of proteins. Nucleic Acids Res. 2001, 29, 41–43. [Google Scholar] [CrossRef] [PubMed]
- Haft, D.H.; Selengut, J.D.; Richter, R.A.; Harkins, D.; Basu, M.K.; Beck, E. TIGRFAMs and genome properties in 2013. Nucleic Acids Res. 2012, 41, D387–D395. [Google Scholar] [CrossRef]
- Haft, D.H.; Selengut, J.D.; White, O. The TIGRFAMs database of protein families. Nucleic Acids Res. 2003, 31, 371–373. [Google Scholar] [CrossRef]
- Selengut, J.D.; Haft, D.H.; Davidsen, T.; Ganapathy, A.; Gwinn-Giglio, M.; Nelson, W.C.; Richter, A.R.; White, O. TIGRFAMs and Genome Properties: Tools for the assignment of molecular function and biological process in prokaryotic genomes. Nucleic Acids Res. 2006, 35, D260–D264. [Google Scholar] [CrossRef]
- Leplae, R.; Hebrant, A.; Wodak, S.J.; Toussaint, A. ACLAME: A CLAssification of Mobile genetic Elements. Nucleic Acids Res. 2004, 33, D45–D49. [Google Scholar] [CrossRef]
- Leplae, R.; Lima-Mendez, G.; Toussaint, A. ACLAME: A CLAssification of Mobile genetic Elements, update 2010. Nucleic Acids Res. 2009, 38, D57–D61. [Google Scholar] [CrossRef]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef]
- Delcher, A.L.; Harmon, D.; Kasif, S.; White, O.; Salzberg, S.L. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999, 27, 4636–4641. [Google Scholar] [CrossRef] [PubMed]
- Salzberg, S.; Delcher, A.L.; Kasif, S.; White, O. Microbial gene identification using interpolated Markov models. Nucleic Acids Res. 1998, 26, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Gomez, J.A.L.; Bhatty, M.; Christie, P.J. PrgK, a multidomain peptidoglycan hydrolase, is essential for conjugative transfer of the pheromone-responsive plasmid pCF10. J. Bacteriol. 2014, 196, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Bantwal, R.; Bannam, T.L.; Porter, C.J.; Quinsey, N.S.; Lyras, D.; Adams, V.; Rood, J.I. The peptidoglycan hydrolase TcpG is required for efficient conjugative transfer of pCW3 in Clostridium perfringens. Plasmid 2012, 67, 13–147. [Google Scholar] [CrossRef]
- Frost, L.S.; Leplae, R.; Summers, A.O.; Toussaint, A. Mobile genetic elements: The agents of open source evolution. Nat. Rev. Microbiol. 2005, 3, 722. [Google Scholar] [CrossRef]
- Guglielmini, J.; De La Cruz, F.; Rocha, E.P. Evolution of conjugation and type IV secretion systems. Mol. Biol. Evol. 2012, 30, 315–331. [Google Scholar] [CrossRef]
- Parsons, J.A.; Bannam, T.L.; Devenish, R.J.; Rood, J.I. TcpA, an FtsK/SpoIIIE homolog, is essential for transfer of the conjugative plasmid pCW3 in Clostridium perfringens. J. Bacteriol. 2007, 189, 7782–7790. [Google Scholar] [CrossRef]
- Steen, J.A.; Bannam, T.L.; Teng, W.L.; Devenish, R.J.; Rood, J.I. The putative coupling protein TcpA interacts with other pCW3-encoded proteins to form an essential part of the conjugation complex. J. Bacteriol. 2009, 191, 2926–2933. [Google Scholar] [CrossRef]
- Lang, S.; Gruber, K.; Mihajlovic, S.; Arnold, R.; Gruber, C.J.; Steinlechner, S.; Jehl, M.A.; Rattei, T.; Fröhlich, K.U.; Zechner, E.L. Molecular recognition determinants for type IV secretion of diverse families of conjugative relaxases. Mol. Microbiol. 2010, 78, 1539–1555. [Google Scholar] [CrossRef]
- Cascales, E.; Cambillau, C. Structural biology of type VI secretion systems. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 1102–1111. [Google Scholar] [CrossRef]
- Cascales, E.; Christie, P.J. Definition of a bacterial type IV secretion pathway for a DNA substrate. Science 2004, 304, 1170–1173. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, S.J.; Krishnamoorthy, V.; Cascales, E.; Christie, P.J. Agrobacterium tumefaciens VirB6 domains direct the ordered export of a DNA substrate through a type IV secretion System. J. Mol. Biol. 2004, 341, 961–977. [Google Scholar] [CrossRef] [PubMed]
- Mary, C.; Fouillen, A.; Bessette, B.; Nanci, A.; Baron, C. Interaction via the N terminus of the type IV secretion system (T4SS) protein VirB6 with VirB10 is required for VirB2 and VirB5 incorporation into T-pili and for T4SS function. J. Biol. Chem. 2018, 293, 13415–13426. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Rosselló-Mora, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [PubMed]
- Figueras, M.J.; Beaz-Hidalgo, R.; Hossain, M.J.; Liles, M.R. Taxonomic affiliation of new genomes should be verified using average nucleotide identity and multilocus phylogenetic analysis. Genome Announc. 2014, 2, e00927-14. [Google Scholar] [CrossRef] [PubMed]
- Gascuel, O. BIONJ: An improved version of the NJ algorithm based on a simple model of sequence data. Mol. Biol. Evol. 1997, 14, 685–695. [Google Scholar] [CrossRef]
- Nei, M. Average distance method (UPGMA). In Molecular evolutionary genetics; Columbia University Press: New York, NY, USA, 1987; pp. 293–298. [Google Scholar]
- Konstantinidis, K.T.; Tiedje, J.M. Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. USA 2005, 102, 2567–2572. [Google Scholar] [CrossRef]
- Wu, D.; Jospin, G.; Eisen, J.A. Systematic identification of gene families for use as markers for phylogenetic and phylogeny-driven ecological studies of bacteria and archaea and their major subgroups. PLoS ONE 2013, 8, e77033. [Google Scholar] [CrossRef]
- Gevers, D.; Cohan, F.M.; Lawrence, J.G.; Spratt, B.G.; Coenye, T.; Feil, E.J.; Stackebrandt, E.; Van de Peer, Y.; Vandamme, P.; Thompson, F.L.; et al. Re-evaluating prokaryotic species. Nat. Rev. Microbiol. 2005, 3, 733. [Google Scholar] [CrossRef]
- Stackebrandt, E.; Frederiksen, W.; Garrity, G.M.; Grimont, P.A.; Kämpfer, P.; Maiden, M.C.; Nesme, X.; Rosselló-Mora, R.; Swings, J.; Trüper, H.G.; et al. Report of the ad hoc committee for the re-evaluation of the species definition in bacteriology. Int. J. Syst. Evol. Microbiol. 2002, 52, 1043–1047. [Google Scholar]
- Jacques, M.-A.; Durand, K.; Ogreur, G.; Balidas, S.; Fricot, C.; Bonneau, S.; Quillévéré, A.; Audusseau, C.; Olivier, V.; Grimault, V.; et al. Phylogenetic analysis and polyphasic characterization of Clavibacter michiganensis strains isolated from tomato seeds reveal that non-pathgenic strains are distinct from C. michiganensis subsp. michiganensis. Appl. Environ. Microbiol. 2012, 78, 8388–8402. [Google Scholar] [CrossRef]
- Yasuhara-Bell, J.; Alvarez, A.M. Seed-associated subspecies of the genus Clavibacter are clearly distinguishable from Clavibacter michiganensis subsp. michiganensis. Int. J. Syst. Evol. Microbiol. 2015, 65, 811–826. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.J.; Gillaspie, A.G., Jr.; Vidaver, A.K.; Harris, R.W. Clavibacter: A new genus containing some phytopathogenic coryneform bacteria, including Clavibacter xyli subsp. xyli sp. nov., subsp. nov. and Clavibacter xyli subsp. cynodontis subsp. nov., pathogens that cause ratoon stunting disease of sugarcane and bermudagrass stunting disease. Int. J. Syst. Evol. Microbiol. 1984, 34, 107–117. [Google Scholar]
- Ophel, K.M.; Bird, A.F.; Kerr, A. Association of bacteriophage particles with toxin production by Clavibacter toxicus, the causal agent of annual ryegrass toxicity. Phytopathology 1993, 83, 676–681. [Google Scholar] [CrossRef]
- Kowalski, M.C.; Cahill, D.; Doran, T.J.; Colegate, S.M. Development and application of polymerase chain reaction-based assays for Rathayibacter toxicus and a bacteriophage associated with annual ryegrass (Lolium rigidum) toxicity. Aust. J. Exp. Agric. 2007, 47, 177–183. [Google Scholar] [CrossRef]
- Avonce, N.; Mendoza-Vargas, A.; Morett, E.; Iturriaga, G. Insights on the evolution of trehalose biosynthesis. BMC Evol. Biol. 2006, 6, 109. [Google Scholar] [CrossRef] [PubMed]
- Behrendt, U.; Ulrich, A.; Schumann, P.; Naumann, D.; Suzuki, K.I. Diversity of grass-associated Microbacteriaceae isolated from the phyllosphere and litter layer after mulching the sward; polyphasic characterization of Subtercola pratensis sp. nov., Curtobacterium herbarum sp. nov. and Plantibacter flavus gen. nov., sp. nov. Int. J. Syst. Evol. Microbiol. 2002, 52, 1441–1454. [Google Scholar] [PubMed]
- Evtushenko, L.I.; Dorofeeva, L.V.; Subbotin, S.A.; Cole, J.R.; Tiedje, J.M. Leifsonia poae gen. nov., sp. nov., isolated from nematode galls on Poa annua, and reclassification of ‘Corynebacterium aquaticum’ Leifson 1962 as Leifsonia aquatica (ex Leifson 1962) gen. nov., nom. rev., comb. nov and Clavibacter xyli Davis et al. 1984 with two subspecies as Leifsonia xyli (Davis et al. 1984) gen. nov., comb. nov. Int. J. Syst. Evol. Microbiol. 2000, 50, 371–380. [Google Scholar] [PubMed]
- Foerstner, K.U.; Von Mering, C.; Hooper, S.D.; Bork, P. Environments shape the nucleotide composition of genomes. EMBO Rep. 2005, 6, 1208–1213. [Google Scholar] [CrossRef]
- Hayward-Farmer, E.; Otto, S.P. The evolution of genomic base composition in bacteria. Evolution 2003, 57, 1783–1792. [Google Scholar] [CrossRef]
- Riley, I.T.; Schmitz, A.; de Silva, P. Anguina australis, a vector for Rathayibacter toxicus in Ehrharta longiflora. Australas. Plant Pathol. 2001, 30, 171–175. [Google Scholar]
- Riley, I.T.; Swart, A.; Postnikova, E.; Agarkova, I.; Vidaver, A.; Schaad, N. New association of a toxigenic Rathayibacter sp. and Anguina woodi in Ehrhata villosa var. villosa in South Africa. Phytopathology 2004, 94, S88. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Other ID | Group a | Host b | Year | Location c | Collector | GenBank Accession |
|---|---|---|---|---|---|---|---|
| SA03-04 | RT-I | ARG | 2014 | Corny Point, SA | M. Arif/J. Stack | CP037987 | |
| SA03-14 | RT-I | ARG | 2014 | Corny Point, SA | M. Arif/J. Stack | CP037986 | |
| SA03-19 | RT-I | ARG | 2014 | Corny Point, SA | M. Arif/J. Stack | CP037985 | |
| SA08-08 | RT-I | ARG | 2014 | Lake Sunday, SA | M. Arif/J. Stack | CP037984 | |
| SA08-09 | RT-I | ARG | 2014 | Lake Sunday, SA | M. Arif/J. Stack | CP037983 | |
| SA19-02 | RT-I | ARG | 2013 | Yorketown, SA | M. Arif/J. Stack | CP037982 | |
| SA19-06 | RT-I | ARG | 2013 | Yorketown, SA | M. Arif/J. Stack | CP037981 | |
| SA19-07 | RT-I | ARG | 2013 | Yorketown, SA | M. Arif/J. Stack | CP037980 | |
| SA08-07 | RT-II | ARG | 2013 | Lake Sunday, SA | M. Arif/J. Stack | N/A | |
| SA19-14 | RT-II | ARG | 2013 | Yorketown, SA | M. Arif/J. Stack | CP037979 | |
| WAC7056 (type strain) | SAC7056, CS14, ICMP 9525, JCM9669, NCPPB 3552, D84127, FH49, FH137 | RT-II | ARG | 1983 | Murray Bridge, SA | I.T. Riley | CP037977 |
| 70137 | WSM447, CS30, FH145, WAC3396 | RT-III | Oat | 1980 | Gnowangerup, WA | D. Chatel | CP010848 |
| CS28 | ICMP 6307, FH138 | RT-III | ARG | 1978 | WA | I.T. Riley | CP037990 |
| WA40-23C | RT-III | ARG | 2015 | WA | M. Arif/J. Stack | CP037978 | |
| WAC3373 | WSM194 | RT-III | PG | 1978 | Gnowangerup, WA | D. Chatel | CP013292 |
| CS36 | SE-1 | RT-IV | PBG | 1990 | Gongolgon, NSW | A. McKay | CP037989 |
| CS38 | NSW-11 | RT-IV | ABG | 1990 | Lucindale, SA | A. McKay | N/A |
| CS39 | NSW-16 | RT-V | ABG | 1990 | Lucindale, SA | A. McKay | CP037988 |
| Strain | Population | Pan CDS | Core CDS | Core CDS % | Variable CDS | Variable CDS % | Strain Specific CDS | Strain Specific CDS % |
|---|---|---|---|---|---|---|---|---|
| SA03-04 | RT-I | 2666 | 2157 | 80.908 | 509 | 19.092 | 111 | 4.164 |
| WAC7056 | RT-II | 2605 | 2160 | 82.917 | 445 | 17.083 | 67 | 2.572 |
| WA40-23C | RT-III | 2592 | 2153 | 83.063 | 439 | 16.937 | 82 | 3.164 |
| CS36 | RT-IV | 2694 | 2182 | 80.995 | 512 | 19.005 | 154 | 5.716 |
| CS39 | RT-V | 2620 | 2160 | 82.443 | 460 | 17.557 | 115 | 4.389 |
| Region a | RT-I | RT-II | RT-III | RT-IV | RT-V | Size (bp) | Notes |
|---|---|---|---|---|---|---|---|
| RT-I-1 | x | 799 | In ASC-2; ASC-2 longer in RT-V than all others but missing RT-I-1 | ||||
| RT-I-2 | x | 945 | |||||
| RT-I-3 | x | 749 | In ASC-4 | ||||
| RT-I-4 | x | 1661 | In ASC-6, SA03-04 RGP-1; CS36 RGP-1 | ||||
| RT-I-5 | x | x | 3604 | In ASC-6, SA03-04 RGP-1; CS36 RGP-1 | |||
| RT-I-6 | x | x | 19,556 | Partly in ASC-13; SA03-04 RGP-2; CS36 RGP-2; LAMP primers designed here [36] | |||
| RT-I-7 | x | 1791 | Between TreY-TreZ and ASC-14; LAMP primers designed here [36] | ||||
| RT-I-8 | x | x | x | 1679 | In ASC-16; In RT-IV, RT-I-8 partially at end of ASC-16 and also after ASC-20 | ||
| RT-I-9 | x | 1023 | |||||
| RT-I-10 | x | 940 | |||||
| RT-II-1 | x | 604 | LAMP primers designed here [36] | ||||
| RT-II-2 | x | 607 | In ASC-4 | ||||
| RT-II-3 | x | x | x | 4863 | In ASC-15; LAMP primers designed here [36] | ||
| RT-II-4 | x | x | x | 4449 | In ASC-15 | ||
| RT-II-5 | x | x | 1522 | Slight overlap within WAC7056 RGP-3 and CS39 RGP-2 | |||
| RT-II-6 | x | x | 1371 | ||||
| RT-III-1 | x | 8944 | In ASC-2; WA40-23C RGP-1 | ||||
| RT-III-2 | x | 1329 | In ASC-2; WA40-23C RGP-1; LAMP primers designed here [36] | ||||
| RT-III-3 | x | x | x | 1908 | |||
| RT-III-4 | x | x | x | 1738 | |||
| RT-III-5 | x | x | x | x | 1521 | In ASC-14; SA03-04 RGP-3; WA40-23C RGP-1; CS36 RGP-3; CS39 RGP-1 | |
| RT-III-6 | x | x | x | x | 1053 | In ASC-14; SA03-04 RGP-3; WA40-23C RGP-1; CS36 RGP-3; CS39 RGP-1 | |
| RT-III-7 | x | x | x | x | 798 | In ASC-14; SA03-04 RGP-3; WA40-23C RGP-1; CS36 RGP-3; CS39 RGP-1; LAMP primers designed here [36] | |
| RT-III-8 | x | 1100 | |||||
| RT-IV-1 | x | 1218 | |||||
| RT-IV-2 | x | 1120 | |||||
| RT-IV-3 | x | 1263 | In ASC-16; CS36 RGP-4 | ||||
| RT-IV-4 | x | 889 | CS36 RGP-4 | ||||
| RT-IV-5 | x | 1270 | |||||
| RT-IV-6 | x | 1470 | |||||
| RT-IV-7 | x | 1734 | In ASC-17 | ||||
| RT-IV-8 | x | 2731 | In ASC-17 | ||||
| RT-IV-9 | x | 1430 | |||||
| RT-IV-10 | x | 5882 | CS36 RGP-6; LAMP primers designed here [36] | ||||
| RT-V-1 | x | 1725 | |||||
| RT-V-2 | x | 611 | In RT-V unique Thiopeptide Cluster (ASC-18); CS39 RGP-3 | ||||
| RT-V-3 | x | 8451 | In RT-V unique Thiopeptide Cluster (ASC-18); CS39 RGP-3; LAMP primers designed here [36] | ||||
| RT-V-4 | x | 10,447 | In RT-V unique Thiopeptide Cluster (ASC-18); CS39 RGP-3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yasuhara-Bell, J.; Arif, M.; Busot, G.Y.; Mann, R.; Rodoni, B.; Stack, J.P. Comparative Genomic Analysis Confirms Five Genetic Populations of the Select Agent, Rathayibacter toxicus. Microorganisms 2020, 8, 366. https://doi.org/10.3390/microorganisms8030366
Yasuhara-Bell J, Arif M, Busot GY, Mann R, Rodoni B, Stack JP. Comparative Genomic Analysis Confirms Five Genetic Populations of the Select Agent, Rathayibacter toxicus. Microorganisms. 2020; 8(3):366. https://doi.org/10.3390/microorganisms8030366
Chicago/Turabian StyleYasuhara-Bell, Jarred, Mohammad Arif, Grethel Y. Busot, Rachel Mann, Brendan Rodoni, and James P. Stack. 2020. "Comparative Genomic Analysis Confirms Five Genetic Populations of the Select Agent, Rathayibacter toxicus" Microorganisms 8, no. 3: 366. https://doi.org/10.3390/microorganisms8030366
APA StyleYasuhara-Bell, J., Arif, M., Busot, G. Y., Mann, R., Rodoni, B., & Stack, J. P. (2020). Comparative Genomic Analysis Confirms Five Genetic Populations of the Select Agent, Rathayibacter toxicus. Microorganisms, 8(3), 366. https://doi.org/10.3390/microorganisms8030366