Identification of Nitrogen Fixation Genes in Lactococcus Isolated from Maize Using Population Genomics and Machine Learning



Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Isolation, DNA Extraction and Whole Genome Sequencing
2.2. Whole Genome Assembly, Comparison and Taxonomic Analysis
2.3. Pangenome Analysis
2.4. Identification of BNF Associated Genes by Pan-GWAS Analysis
2.5. Identification of Important Genes Using Random Forests
2.6. Functional Annotation of Identified Protein Coding Sequences
2.7. Materials Availability
3. Results
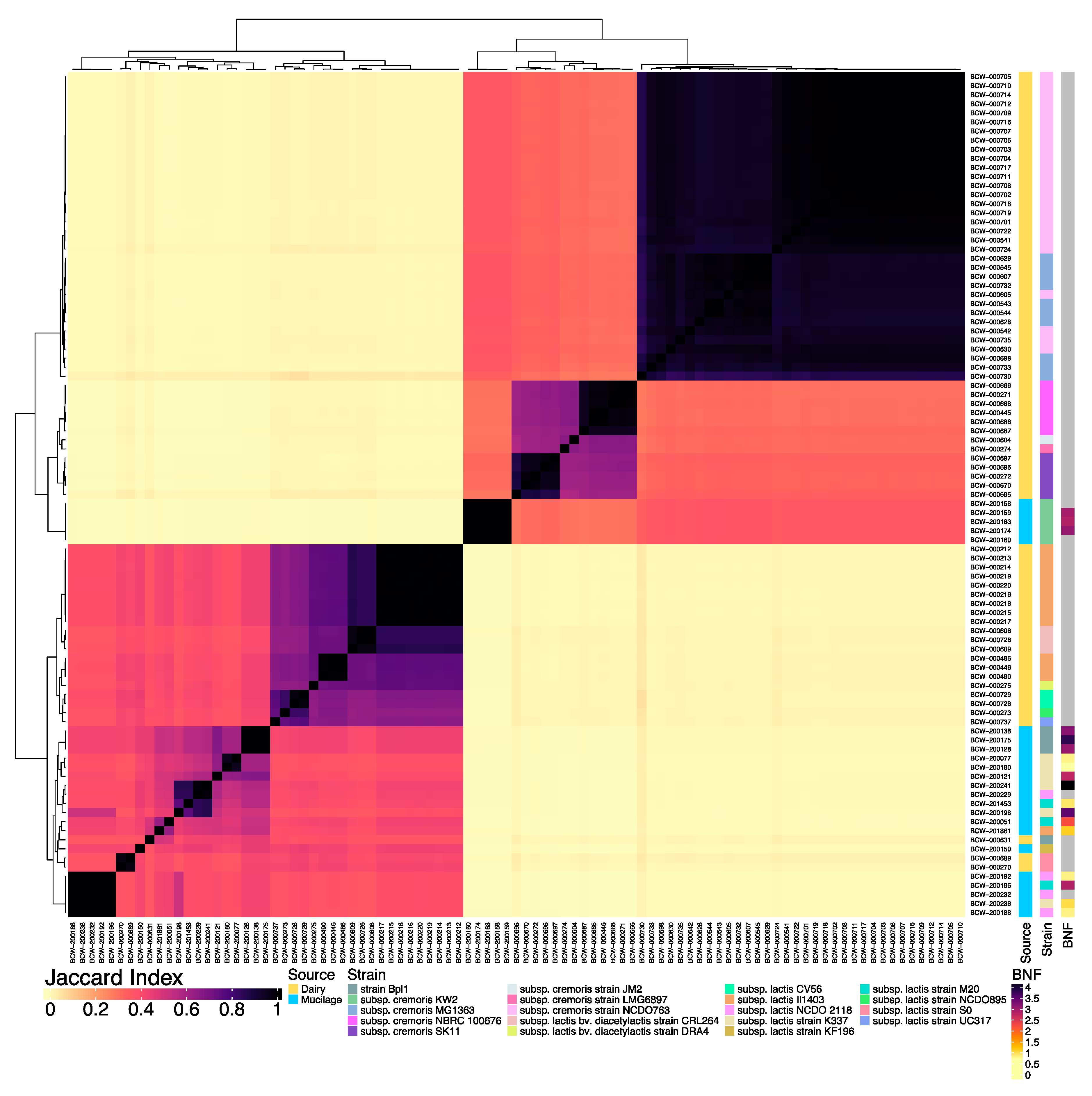
3.1. Comparison of WGS Assemblies Revealed Genomic Distance between Mucilage and Dairy Isolates
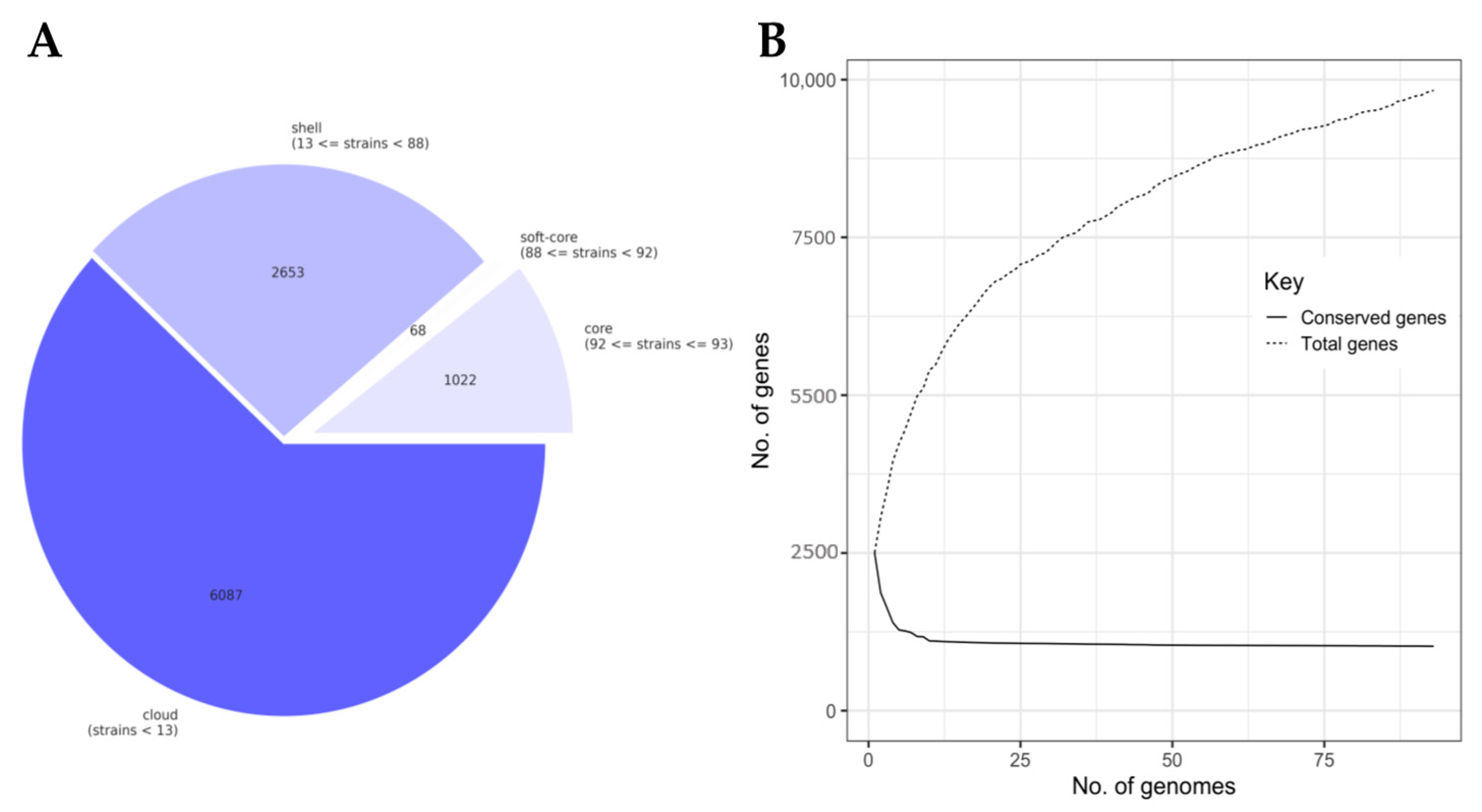
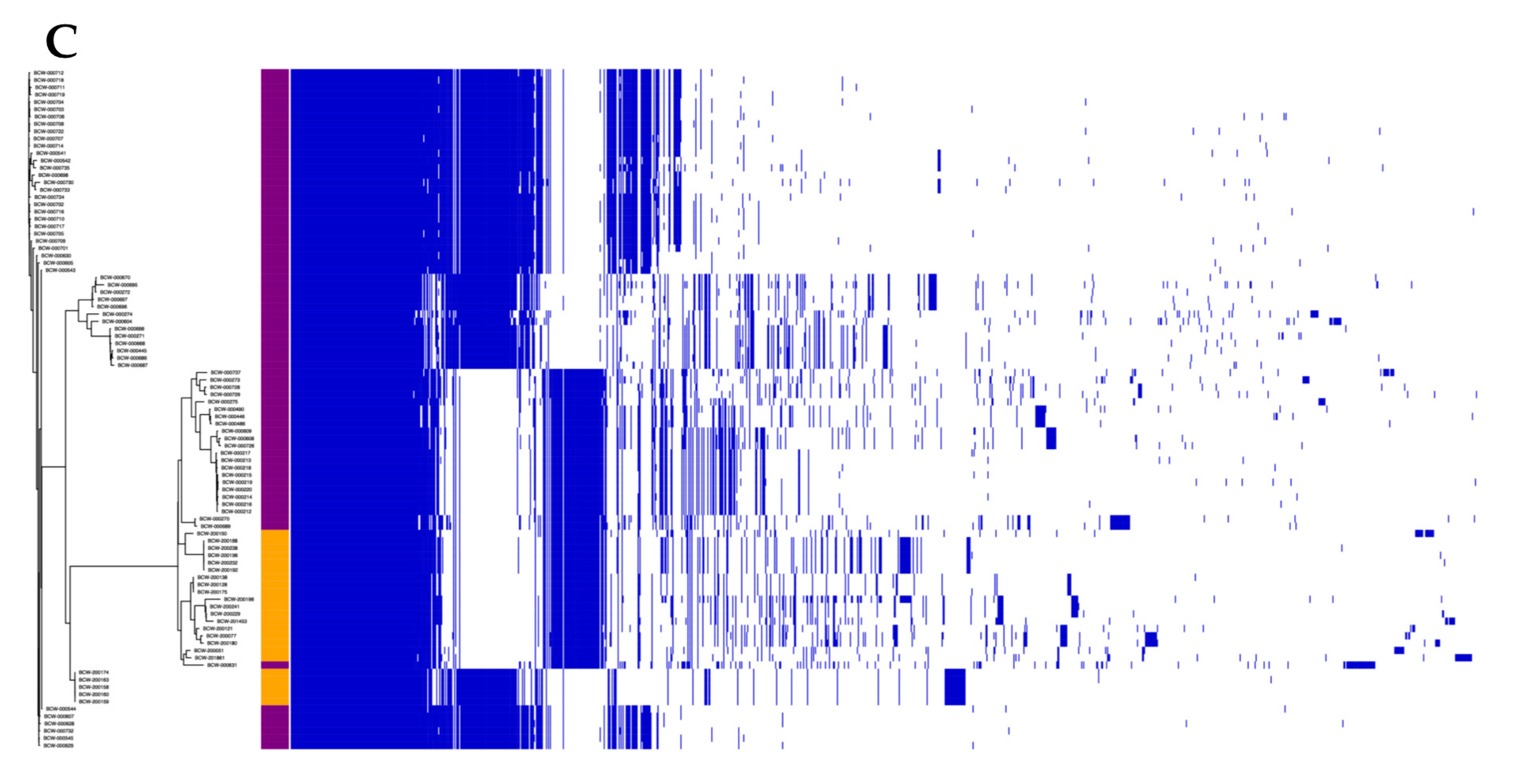
3.2. The Lactococcal Pangenome Revealed a Unique Genotype in Mucilage Isolates
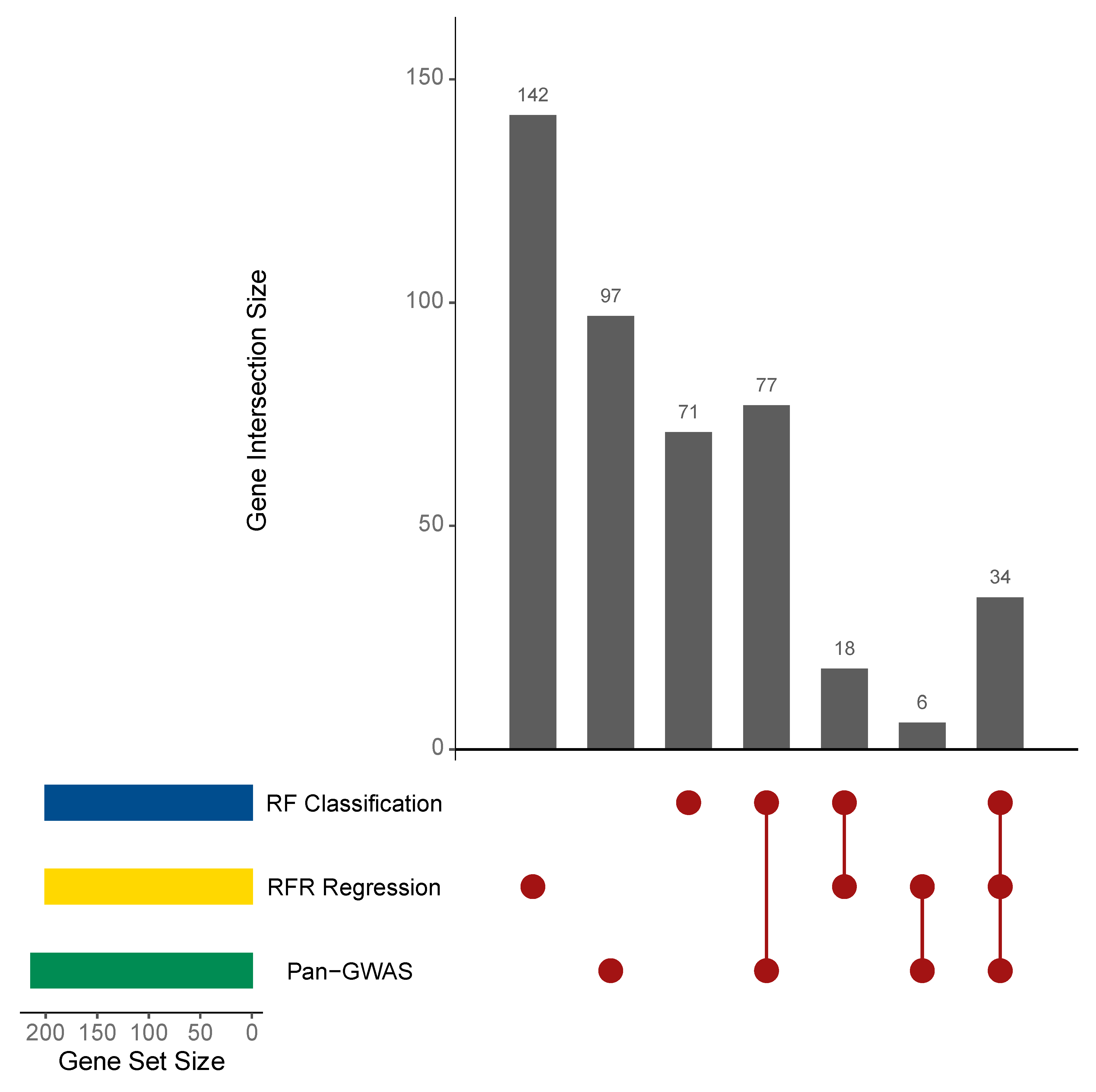
3.3. Statistical and Machine Learning Analysis Identified L. lactis Genes Associated with BNF
3.4. Domain Annotations Elucidated Predicted Functions of Genes Associated with the BNF Trait
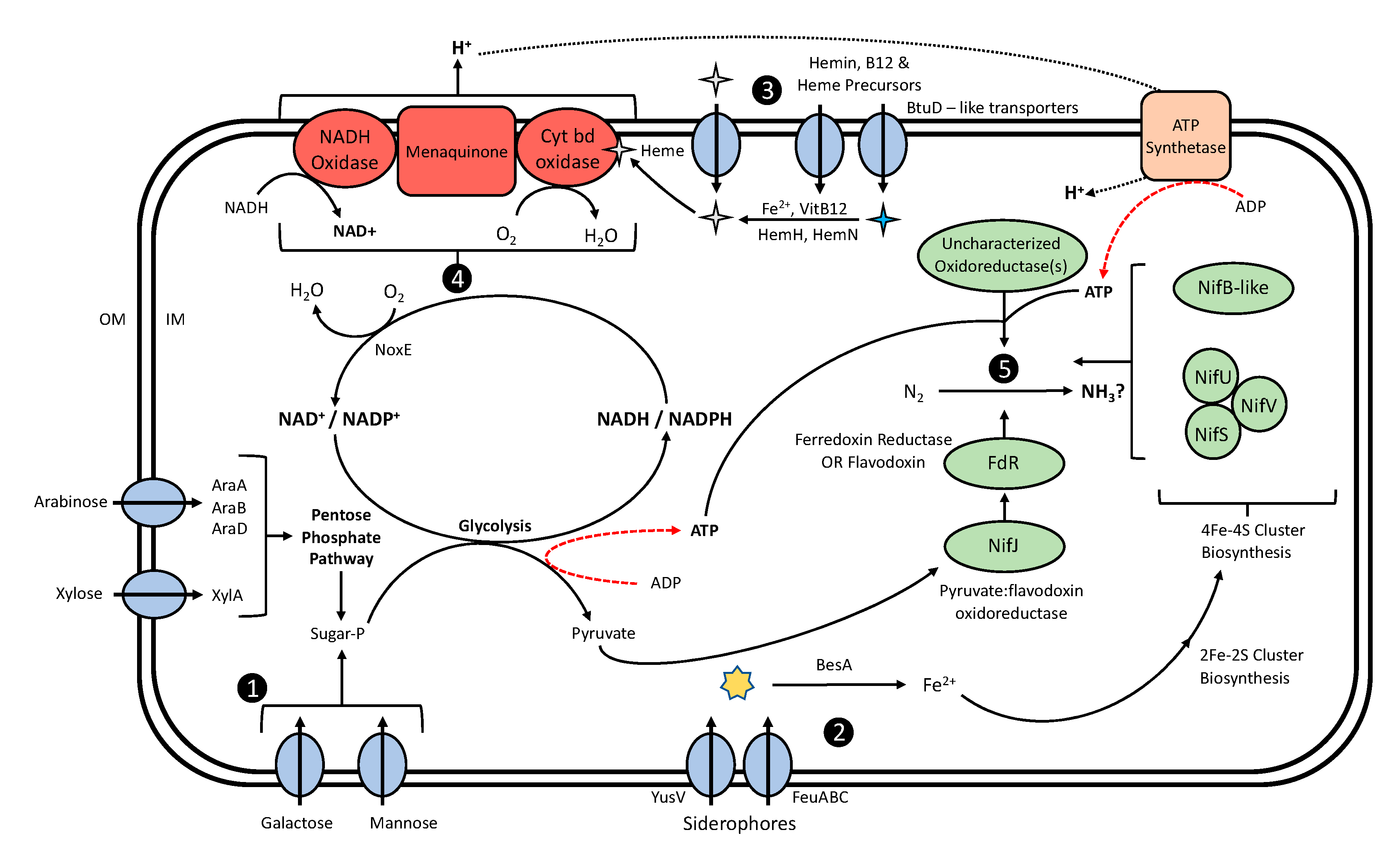
4. Discussion
4.1. L. lactis Isolates from Mucilage Have Genomes Distinguished from Dairy Isolates
4.2. Mucilage Lactococci Exhibit Genomic Potential to Create a Suitable Environment for BNF
4.2.1. Mucilage Lactococci Exhibit the Potential to Utilize Mucilage Carbohydrates as Energy for BNF
4.2.2. Diazotrophic L. lactis Genomes Are Enriched with Iron Accumulation Genes
4.2.3. L. lactis Is Capable of Molecular O2 Depletion
4.2.4. Diazotrophic Lactococci Possess Genes with Domains Analogous to Nitrogenase Proteins
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wessels, S.; Axelsson, L.; Bech Hansen, E.; De Vuyst, L.; Laulund, S.; Lähteenmäki, L.; Lindgren, S.; Mollet, B.; Salminen, S.; von Wright, A. The lactic acid bacteria, the food chain, and their regulation. Trends Food Sci. Technol. 2004, 15, 498–505. [Google Scholar] [CrossRef]
- Leroy, F.; De Vuyst, L. Lactic acid bacteria as functional starter cultures for the food fermentation industry. Trends Food Sci. Technol. 2004, 15, 67–78. [Google Scholar] [CrossRef]
- Song, A.A.; In, L.L.A.; Lim, S.H.E.; Rahim, R.A. A review on Lactococcus lactis: From food to factory. Microb. Cell Fact. 2017, 16, 55. [Google Scholar] [CrossRef] [PubMed]
- Kimoto, H.; Kurisaki, J.; Tsuji, N.M.; Ohmomo, S.; Okamoto, T. Lactococci as probiotic strains: Adhesion to human enterocyte-like Caco-2 cells and tolerance to low pH and bile. Lett. Appl. Microbiol. 1999, 29, 313–316. [Google Scholar] [CrossRef] [PubMed]
- Kimoto, H.; Mizumachi, K.; Okamoto, T.; Kurisaki, J. New Lactococcus Strain with Immunomodulatory Activity: Enhancement of Th1-Type Immune Response. Microbiol. Immunol. 2004, 48, 75–82. [Google Scholar] [CrossRef]
- Kimoto-Nira, H.; Mizumachi, K.; Nomura, M.; Kobayashi, M.; Fujita, Y.; Okamoto, T.; Suzuki, I.; Tsuji, N.M.; Kurisaki, J.; Ohmomo, S. Lactococcus sp. as potential probiotic lactic acid bacteria. Jpn. Agric. Res. Q. JARQ 2007, 41, 181–189. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, Y.; Yao, J.; Li, W. Inhibition ability of probiotic, Lactococcus lactis, against A. hydrophila and study of its immunostimulatory effect in tilapia (Oreochromis niloticus). Int. J. Eng. Sci. Technol. 2010, 2. [Google Scholar] [CrossRef]
- Cavanagh, D.; Fitzgerald, G.F.; McAuliffe, O. From field to fermentation: The origins of Lactococcus lactis and its domestication to the dairy environment. Food Microbiol. 2015, 47, 45–61. [Google Scholar] [CrossRef]
- Dussault, D.; Vu, K.D.; Lacroix, M. Enhancement of Nisin Production by Lactococcus lactis subsp. Lactis. Probiotics Antimicrob. Proteins 2016, 8, 170–175. [Google Scholar] [CrossRef]
- Ganesan, B.; Weimer, B.C. Biotechnology of flavor formation in fermented dairy products. In Biotechnology in Flavor Production, 2nd ed.; Daphna Havkin-Frenkel, N.D., Ed.; Wiley Press: Singapore, 2015; pp. 133–158. [Google Scholar]
- Ganesan, B.; Weimer, B.C. Amino acid metabolism in relationship to cheese flavor development. Improv. Flavour Cheese 2007, 70–101. [Google Scholar] [CrossRef]
- Mills, S.; O’Sullivan, O.; Hill, C.; Fitzgerald, G.; Ross, R.P. The changing face of dairy starter culture research: From genomics to economics. Int. J. Dairy Technol. 2010, 63, 149–170. [Google Scholar] [CrossRef]
- Klijn, N.; Weerkamp, A.H.; De Vos, W. Genetic marking of Lactococcus lactis shows its survival in the human gastrointestinal tract. Appl. Environ. Microbiol. 1995, 61, 2771–2774. [Google Scholar] [CrossRef] [PubMed]
- Meyrand, M.; Guillot, A.; Goin, M.; Furlan, S.; Armalyte, J.; Kulakauskas, S.; Cortes-Perez, N.G.; Thomas, G.; Chat, S.; Pechoux, C. Surface proteome analysis of a natural isolate of Lactococcus lactis reveals the presence of pili able to bind human intestinal epithelial cells. Mol. Cell. Proteom. 2013, 12, 3935–3947. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, B.; Weimer, B.C.; Pinzon, J.; Dao Kong, N.; Rompato, G.; Brothersen, C.; McMahon, D.J. Probiotic bacteria survive in Cheddar cheese and modify populations of other lactic acid bacteria. J. Appl. Microbiol. 2014, 116, 1642–1656. [Google Scholar] [CrossRef]
- Ganesan, B.; Bart, C.W. Amino acid catabolism. In Cheese: Chemistry, Physics & Microbiology, 4th ed.; Fox, P.F., McSweeney, P.L.H., Everett, D.W., Cotter, P., Eds.; Elsevier: Oxford, UK, 2014; Volume 1. [Google Scholar]
- Ganesan, B.; Stuart, M.R.; Weimer, B.C. Carbohydrate starvation causes a metabolically active but nonculturable state in Lactococcus lactis. Appl. Environ. Microbiol. 2007, 73, 2498–2512. [Google Scholar] [CrossRef]
- Ganesan, B.; Dobrowolski, P.; Weimer, B.C. Identification of the leucine-to-2-methylbutyric acid catabolic pathway of Lactococcus lactis. Appl. Environ. Microbiol. 2006, 72, 4264–4273. [Google Scholar] [CrossRef]
- Salama, M.S.; Musafija-Jeknic, T.; Sandine, W.E.; Giovannoni, S.J. An Ecological Study of Lactic Acid Bacteria: Isolation of New Strains of Lactococcus Including Lactococcus lactis subspecies cremoris. J. Dairy Sci. 1995, 78, 1004–1017. [Google Scholar] [CrossRef]
- Lamont, J.R.; Wilkins, O.; Bywater-Ekegärd, M.; Smith, D.L. From yogurt to yield: Potential applications of lactic acid bacteria in plant production. Soil Biol. Biochem. 2017, 111, 1–9. [Google Scholar] [CrossRef]
- Nomura, M.; Kobayashi, M.; Narita, T.; Kimoto-Nira, H.; Okamoto, T. Phenotypic and molecular characterization of Lactococcus lactis from milk and plants. J. Appl. Microbiol. 2006, 101, 396–405. [Google Scholar] [CrossRef]
- Kelly, W.J.; Ward, L.J.H.; Leahy, S.C. Chromosomal Diversity in Lactococcus lactis and the Origin of Dairy Starter Cultures. Genome Biol. Evol. 2010, 2, 729–744. [Google Scholar] [CrossRef]
- Yu, A.O.; Leveau, J.H.J.; Marco, M.L. Abundance, diversity and plant-specific adaptations of plant-associated lactic acid bacteria. Environ. Microbiol. Rep. 2020, 12, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Siezen, R.J.; Starrenburg, M.J.; Boekhorst, J.; Renckens, B.; Molenaar, D.; van Hylckama Vlieg, J.E. Genome-scale genotype-phenotype matching of two Lactococcus lactis isolates from plants identifies mechanisms of adaptation to the plant niche. Appl. Environ. Microbiol. 2008, 74, 424–436. [Google Scholar] [CrossRef] [PubMed]
- Minervini, F.; Celano, G.; Lattanzi, A.; Tedone, L.; De Mastro, G.; Gobbetti, M.; De Angelis, M. Lactic Acid Bacteria in Durum Wheat Flour Are Endophytic Components of the Plant during Its Entire Life Cycle. Appl. Environ. Microbiol. 2015, 81, 6736–6748. [Google Scholar] [CrossRef] [PubMed]
- Siezen, R.J.; Bayjanov, J.; Renckens, B.; Wels, M.; van Hijum, S.A.; Molenaar, D.; van Hylckama Vlieg, J.E. Complete genome sequence of Lactococcus lactis subsp. lactis KF147, a plant-associated lactic acid bacterium. J. Bacteriol. 2010, 192, 2649–2650. [Google Scholar] [CrossRef] [PubMed]
- Higdon, S.M.; Pozzo, T.; Kong, N.; Huang, B.; Yang, M.L.; Jeannotte, R.; Brown, C.T.; Bennett, A.B.; Weimer, B.C. Genomic characterization of a diazotrophic microbiota associated with maize aerial root mucilage. arXiv 2020, arXiv:bio/064337. [Google Scholar] [CrossRef]
- Dos Santos, P.C.; Fang, Z.; Mason, S.W.; Setubal, J.C.; Dixon, R. Distribution of nitrogen fixation and nitrogenase-like sequences amongst microbial genomes. BMC Genom. 2012, 13, 162. [Google Scholar] [CrossRef] [PubMed]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Brynildsrud, O.; Bohlin, J.; Scheffer, L.; Eldholm, V. Rapid scoring of genes in microbial pan-genome-wide association studies with Scoary. Genome Biol. 2016, 17, 238. [Google Scholar] [CrossRef]
- Bandoy, D.D.R.; Weimer, B.C. Biological Machine Learning Combined with Campylobacter Population Genomics Reveals Virulence Gene Allelic Variants Cause Disease. Microorganisms 2020, 8, 549. [Google Scholar] [CrossRef]
- Goldstein, B.A.; Polley, E.C.; Briggs, F.B.S. Random forests for genetic association studies. Stat. Appl. Genet. Mol. Biol. 2011, 10, 32. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Wheeler, N.E.; Gardner, P.P.; Barquist, L. Machine learning identifies signatures of host adaptation in the bacterial pathogen Salmonella enterica. PLoS Genet. 2018, 14, e1007333. [Google Scholar] [CrossRef] [PubMed]
- Moradigaravand, D.; Palm, M.; Farewell, A.; Mustonen, V.; Warringer, J.; Parts, L. Prediction of antibiotic resistance in Escherichia coli from large-scale pan-genome data. PLoS Comput. Biol. 2018, 14, e1006258. [Google Scholar] [CrossRef] [PubMed]
- Obolski, U.; Gori, A.; Lourenço, J.; Thompson, C.; Thompson, R.; French, N.; Heyderman, R.S.; Gupta, S. Identifying genes associated with invasive disease in S. pneumoniae by applying a machine learning approach to whole genome sequence typing data. Sci. Rep. 2019, 9, 4049. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.; Slesarev, A.; Wolf, Y.; Sorokin, A.; Mirkin, B.; Koonin, E.; Pavlov, A.; Pavlova, N.; Karamychev, V.; Polouchine, N.; et al. Comparative genomics of the lactic acid bacteria. Proc. Natl. Acad. Sci. USA 2006, 103, 15611. [Google Scholar] [CrossRef]
- Chou, L.-S.; Weimer, B.C.; Cutler, R. Relationship of arginine and lactose utilization by Lactococcus lactis ssp. lactis ML3. Int. Dairy J. 2001, 11, 253–258. [Google Scholar] [CrossRef]
- Ganesan, B.; Weimer, B.C. Role of aminotransferase IlvE in production of branched-chain fatty acids by Lactococcus lactis subsp. lactis. Appl. Environ. Microbiol. 2004, 70, 638–641. [Google Scholar] [CrossRef]
- Weimer, B.; Xie, Y.; Chou, L.; Cutler, A. Gene Expression Arrays in Food. In Microbial Processes and Products. Methods in Biotechnology; Barredo, J.L., Ed.; Humana Press: Totowa, NJ, USA, 2005; Volume 18, pp. 333–343. [Google Scholar] [CrossRef]
- Xie, Y.; Chou, L.-S.; Cutler, A.; Weimer, B. DNA macroarray profiling of Lactococcus lactis subsp. lactis IL1403 gene expression during environmental stresses. Appl. Environ. Microbiol. 2004, 70, 6738–6747. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.T.; Irber, L. sourmash: A library for MinHash sketching of DNA. J. Open Source Softw. 2016, 1, 27. [Google Scholar] [CrossRef]
- Pierce, N.T.; Irber, L.; Reiter, T.; Brooks, P.; Brown, C.T. Large-scale sequence comparisons with sourmash. F1000Research 2019, 8, 1006. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Hadfield, J.; Croucher, N.J.; Goater, R.J.; Abudahab, K.; Aanensen, D.M.; Harris, S.R. Phandango: An interactive viewer for bacterial population genomics. Bioinformatics 2018, 34, 292–293. [Google Scholar] [CrossRef]
- Wickham, H.; François, R.; Henry, L.; Müller, K. dplyr: A Grammar of Data Manipulation. R Package version 0.8.5. R Project. Available online: https://CRAN.R-project.org/package=dplyr (accessed on 7 March 2020).
- Liaw, A.; Wiener, M. Classification and regression by randomForest. R News 2002, 2, 18–22. [Google Scholar]
- Kuhn, M.; Wing, J.; Weston, S.; Williams, A.; Keefer, C.; Engelhardt, A.; Cooper, T.; Mayer, Z.; Kenkel, B. Caret: Classification and Regression Training. R Package Version 6.0–84. R Project. Available online: https://cran.r-project.org/web/packages/caret (accessed on 27 April 2019).
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef] [PubMed]
- Bushnell, B. BBTools Software Package. Available online: http://sourceforge.net/projects/bbmap (accessed on 1 March 2020).
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Vernikos, G.; Medini, D.; Riley, D.R.; Tettelin, H. Ten years of pan-genome analyses. Curr. Opin. Microbiol. 2015, 23, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Miethke, M.; Klotz, O.; Linne, U.; May, J.J.; Beckering, C.L.; Marahiel, M.A. Ferri-bacillibactin uptake and hydrolysis in Bacillus subtilis. Mol. Microbiol. 2006, 61, 1413–1427. [Google Scholar] [CrossRef]
- Hu, Y.; Ribbe, M.W. Maturation of nitrogenase cofactor—The role of a class E radical SAM methyltransferase NifB. Curr. Opin. Chem. Biol. 2016, 31, 188–194. [Google Scholar] [CrossRef]
- Schirner, K.; Eun, Y.-J.; Dion, M.; Luo, Y.; Helmann, J.D.; Garner, E.C.; Walker, S. Lipid-linked cell wall precursors regulate membrane association of bacterial actin MreB. Nat. Chem. Biol. 2015, 11, 38–45. [Google Scholar] [CrossRef]
- John, M.; Rohrig, H.; Schmidt, J.; Wieneke, U.; Schell, J. Rhizobium NodB protein involved in nodulation signal synthesis is a chitooligosaccharide deacetylase. Proc. Natl. Acad. Sci. USA 1993, 90, 625–629. [Google Scholar] [CrossRef]
- Severi, E.; Hood, D.W.; Thomas, G.H. Sialic acid utilization by bacterial pathogens. Microbiology 2007, 153, 2817–2822. [Google Scholar] [CrossRef]
- Amicucci, M.J.; Galermo, A.G.; Guerrero, A.; Treves, G.; Nandita, E.; Kailemia, M.J.; Higdon, S.M.; Pozzo, T.; Labavitch, J.M.; Bennett, A.B.; et al. Strategy for Structural Elucidation of Polysaccharides: Elucidation of a Maize Mucilage that Harbors Diazotrophic Bacteria. Anal. Chem. 2019, 91, 7254–7265. [Google Scholar] [CrossRef]
- Berk, H.; Thauer, R.K. Function of coenzyme F420-dependent NADP reductase in methanogenic archaea containing an NADP-dependent alcohol dehydrogenase. Arch. Microbiol. 1997, 168, 396–402. [Google Scholar] [CrossRef]
- Van Hylckama Vlieg, J.E.T.; Rademaker, J.L.W.; Bachmann, H.; Molenaar, D.; Kelly, W.J.; Siezen, R.J. Natural diversity and adaptive responses of Lactococcus lactis. Curr. Opin. Biotechnol. 2006, 17, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Wels, M.; Siezen, R.; Van Hijum, S.; Kelly, W.; Bachmann, H. Comparative genome analysis of Lactococcus lactis indicates niche adaptation and resolves genotype/phenotype disparity. Front. Microbiol. 2019, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, J.H.; Seabolt, E.; Kunitomi, M.; Agarwal, A.; Beck, K.; Krishnareddy, H.; Weimer, B.C. Exploiting functional context in biology: Reconsidering classification of bacterial life. In Proceedings of the IEEE 34th International Conference on Data Engineering Workshops, ICDEW 2018, Paris, France, 16–20 April 2018; pp. 17–20. [Google Scholar]
- Kaufman, J.H.; Elkins, C.A.; Davis, M.; Weis, A.M.; Huang, B.C.; Mammel, M.K.; Patel, I.R.; Beck, K.L.; Edlund, S.; Chambliss, D.; et al. Insular Microbiogeography: Three Pathogens as Exemplars. In Microbial Ecology: Current Advances from Genomics, Metagenomics and Other Omics; Marco, D.E., Ed.; Caister Academic Press: Poole, UK, 2019; pp. 45–64. [Google Scholar]
- Bayjanov, J.R.; Starrenburg, M.J.C.; van der Sijde, M.R.; Siezen, R.J.; van Hijum, S.A.F.T. Genotype-phenotype matching analysis of 38 Lactococcus lactisstrains using random forest methods. BMC Microbiol. 2013, 13, 68. [Google Scholar] [CrossRef] [PubMed]
- Van Deynze, A.; Zamora, P.; Delaux, P.M.; Heitmann, C.; Jayaraman, D.; Rajasekar, S.; Graham, D.; Maeda, J.; Gibson, D.; Schwartz, K.D.; et al. Nitrogen fixation in a landrace of maize is supported by a mucilage-associated diazotrophic microbiota. PLoS Biol. 2018, 16, e2006352. [Google Scholar] [CrossRef]
- Lapujade, P.; Cocaign-Bousquet, M.; Loubiere, P. Glutamate Biosynthesis in Lactococcus lactis subsp. lactis NCDO 2118. Appl. Environ. Microbiol. 1998, 64, 2485–2489. [Google Scholar] [CrossRef]
- Gaudu, P.; Lamberet, G.; Poncet, S.; Gruss, A. CcpA regulation of aerobic and respiration growth in Lactococcus lactis. Mol. Microbiol. 2003, 50, 183–192. [Google Scholar] [CrossRef]
- Cretenet, M.; Le Gall, G.; Wegmann, U.; Even, S.; Shearman, C.; Stentz, R.; Jeanson, S. Early adaptation to oxygen is key to the industrially important traits of Lactococcus lactis ssp. cremoris during milk fermentation. BMC Genom. 2014, 15, 1054. [Google Scholar] [CrossRef]
- Duwat, P.; Sourice, S.; Cesselin, B.; Lamberet, G.; Vido, K.; Gaudu, P.; Le Loir, Y.; Violet, F.; Loubiere, P.; Gruss, A. Respiration capacity of the fermenting bacterium Lactococcus lactis and its positive effects on growth and survival. J. Bacteriol. 2001, 183, 4509–4516. [Google Scholar] [CrossRef]
- Rezaïki, L.; Cesselin, B.; Yamamoto, Y.; Vido, K.; Van West, E.; Gaudu, P.; Gruss, A. Respiration metabolism reduces oxidative and acid stress to improve long-term survival of Lactococcus lactis. Mol. Microbiol. 2004, 53, 1331–1342. [Google Scholar] [CrossRef]
- Pedersen, M.B.; Gaudu, P.; Lechardeur, D.; Petit, M.-A.; Gruss, A. Aerobic respiration metabolism in lactic acid bacteria and uses in biotechnology. Annu. Rev. Food Sci. Technol. 2012, 3, 37–58. [Google Scholar] [CrossRef]
- Koebmann, B.; Blank, L.M.; Solem, C.; Petranovic, D.; Nielsen, L.K.; Jensen, P.R. Increased biomass yield of Lactococcus lactis during energetically limited growth and respiratory conditions. Biotechnol. Appl. Biochem. 2008, 50, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Blank, L.M.; Koebmann, B.J.; Michelsen, O.; Nielsen, L.K.; Jensen, P.R. Hemin Reconstitutes Proton Extrusion in an H+-ATPase-Negative Mutant of Lactococcus lactis. J. Bacteriol. 2001, 183, 6707–6709. [Google Scholar] [CrossRef] [PubMed]
- Raymond, J.; Siefert, J.L.; Staples, C.R.; Blankenship, R.E. The natural history of nitrogen fixation. Mol. Biol. Evol. 2004, 21, 541–554. [Google Scholar] [CrossRef] [PubMed]
- Siezen, R.J.; Renckens, B.; van Swam, I.; Peters, S.; van Kranenburg, R.; Kleerebezem, M.; de Vos, W.M. Complete sequences of four plasmids of Lactococcus lactis subsp. cremoris SK11 reveal extensive adaptation to the dairy environment. Appl. Environ. Microbiol. 2005, 71, 8371–8382. [Google Scholar] [CrossRef]
- Golomb, B.L.; Marco, M.L. Lactococcus lactis metabolism and gene expression during growth on plant tissues. J. Bacteriol. 2015, 197, 371–381. [Google Scholar] [CrossRef]
- Laroute, V.; Tormo, H.; Couderc, C.; Mercier-Bonin, M.; Le Bourgeois, P.; Cocaign-Bousquet, M.; Daveran-Mingot, M.-L. From genome to phenotype: An integrative approach to evaluate the biodiversity of Lactococcus lactis. Microorganisms 2017, 5, 27. [Google Scholar] [CrossRef]
- Passerini, D.; Coddeville, M.; Le Bourgeois, P.; Loubière, P.; Ritzenthaler, P.; Fontagné-Faucher, C.; Daveran-Mingot, M.-L.; Cocaign-Bousquet, M. The carbohydrate metabolism signature of Lactococcus lactis strain A12 reveals its sourdough ecosystem origin. Appl. Environ. Microbiol. 2013, 79, 5844–5852. [Google Scholar] [CrossRef]
- Siezen, R.J.; Bayjanov, J.R.; Felis, G.E.; van der Sijde, M.R.; Starrenburg, M.; Molenaar, D.; Wels, M.; van Hijum, S.A.; van Hylckama Vlieg, J.E. Genome-scale diversity and niche adaptation analysis of Lactococcus lactis by comparative genome hybridization using multi-strain arrays. Microb. Biotechnol. 2011, 4, 383–402. [Google Scholar] [CrossRef]
- Etzold, S.; Kober, O.I.; MacKenzie, D.A.; Tailford, L.E.; Gunning, A.P.; Walshaw, J.; Hemmings, A.M.; Juge, N. Structural basis for adaptation of lactobacilli to gastrointestinal mucus. Environ. Microbiol. 2014, 16, 888–903. [Google Scholar] [CrossRef]
- Ielasi, F.S.; Alioscha-Perez, M.; Donohue, D.; Claes, S.; Sahli, H.; Schols, D.; Willaert, R.G. Lectin-Glycan interaction network-based identification of host receptors of microbial pathogenic adhesins. mBio 2016, 7, e00584-16. [Google Scholar] [CrossRef]
- Chapot-Chartier, M.-P.; Kulakauskas, S. Cell wall structure and function in lactic acid bacteria. Microb. Cell Factories 2014, 13, S1–S9. [Google Scholar] [CrossRef]
- Holmer, R.; Rutten, L.; Kohlen, W.; van Velzen, R.; Geurts, R. Commonalities in symbiotic plant-microbe signalling. In Advances in Botanical Research; Elsevier: Amsterdam, The Netherlands, 2017; Volume 82, pp. 187–221. [Google Scholar]
- Pozzo, T.; Higdon, S.M.; Pattathil, S.; Hahn, M.G.; Bennett, A.B. Characterization of novel glycosyl hydrolases discovered by cell wall glycan directed monoclonal antibody screening and metagenome analysis of maize aerial root mucilage. PLoS ONE 2018, 13, e0204525. [Google Scholar] [CrossRef] [PubMed]
- Rubio, L.M.; Ludden, P.W. Biosynthesis of the iron-molybdenum cofactor of nitrogenase. Annu. Rev. Microbiol. 2008, 62, 93–111. [Google Scholar] [CrossRef] [PubMed]
- Rubio, L.M.; Ludden, P.W. Maturation of Nitrogenase: A Biochemical Puzzle. J. Bacteriol. 2005, 187, 405. [Google Scholar] [CrossRef] [PubMed]
- Miethke, M.; Schmidt, S.; Marahiel, M.A. The major facilitator superfamily-type transporter YmfE and the multidrug-efflux activator Mta mediate bacillibactin secretion in Bacillus subtilis. J. Bacteriol. 2008, 190, 5143–5152. [Google Scholar] [CrossRef] [PubMed]
- Bennett, A.B.; Pankievicz, V.C.S.; Ane, J.M. A Model for Nitrogen Fixation in Cereal Crops. Trends Plant Sci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Vido, K.; Le Bars, D.; Mistou, M.-Y.; Anglade, P.; Gruss, A.; Gaudu, P. Proteome analyses of heme-dependent respiration in Lactococcus lactis: Involvement of the proteolytic system. J. Bacteriol. 2004, 186, 1648–1657. [Google Scholar] [CrossRef] [PubMed]
- Gaudu, P.; Vido, K.; Cesselin, B.; Kulakauskas, S.; Tremblay, J.; Rezaïki, L.; Lamberet, G.; Sourice, S.; Duwat, P.; Gruss, A. Respiration capacity and consequences in Lactococcus lactis. Antonie Van Leeuwenhoek 2002, 82, 263–269. [Google Scholar] [CrossRef]
- Tachon, S.; Brandsma, J.B.; Yvon, M. NoxE NADH oxidase and the electron transport chain are responsible for the ability of Lactococcus lactis to decrease the redox potential of milk. Appl. Environ. Microbiol. 2010, 76, 1311–1319. [Google Scholar] [CrossRef]
- Schneegurt, M.A.; Beale, S.I. Biosynthesis of protoheme and heme a from glutamate in maize. Plant Physiol. 1986, 81, 965–971. [Google Scholar] [CrossRef]
- Bachmann, H. Regulatory and Adaptive Responses of Lactococcus Lactis In Situ; Wageningen Univeristy: Wageningen, The Netherlands, 2009; ISBN 978-90-8585-426-5. [Google Scholar]
- Riboldi, G.P.; Verli, H.; Frazzon, J. Structural studies of the Enterococcus faecalis SufU [Fe-S] cluster protein. BMC Biochem. 2009, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- Khadka, N.; Dean, D.R.; Smith, D.; Hoffman, B.M.; Raugei, S.; Seefeldt, L.C. CO2 Reduction Catalyzed by Nitrogenase: Pathways to Formate, Carbon Monoxide, and Methane. Inorg. Chem. 2016, 55, 8321–8330. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Interproscan Annotation | N Mucilage | N Dairy |
|---|---|---|---|
| araA | L-arabinose isomerase | 15 | 2 |
| btuD_9 | ABC Transporter Type 1 | 17 | 0 |
| ddrA | Rad52/22 family double-strand break repair protein | 0 | 64 |
| feuA | Iron–siderophore periplasmic ABC transporter | 16 | 0 |
| group_1791 | No match | 17 | 0 |
| group_1793 | Putative Lactococcus lactis phage r1t holin | 12 | 0 |
| group_1931 | Intrinsically disordered protein | 0 | 60 |
| group_2085 | Glyoxalase/Bleomycin resistance/Dihydroxybiphenyl dioxygenase | 23 | 3 |
| group_2262 | Intrinsically disordered protein | 11 | 0 |
| group_2959 | MepB-like Protein of Unknown Function | 14 | 0 |
| group_2960 | No match | 11 | 0 |
| group_3301 | WxL domain surface cell wall-binding | 6 | 68 |
| group_3932 | Putative DNA-binding domain superfamily | 11 | 0 |
| group_5428 | Winged helix/Mga helix-turn-helix DNA binding | 17 | 1 |
| group_5436 | Membrane-bound galactosyl-transferase/GT1 | 13 | 1 |
| group_5483 | No match | 12 | 0 |
| group_5487 | Protein of unknown function DUF3892 | 12 | 0 |
| group_5497 | No match | 12 | 0 |
| group_5503 | No match | 11 | 0 |
| group_5623 | Type I DNA methyltransferase | 11 | 0 |
| group_5626 | No match | 11 | 0 |
| group_6294 | ROK (Repressor, ORF, Kinase) DNA-Binding Transcription Factor | 18 | 3 |
| group_696 | No match | 13 | 1 |
| group_7770 | Prokaryotic membrane lipoprotein lipid attachment | 15 | 1 |
| group_7798 | Extracellular arabinose binding protein | 13 | 1 |
| group_7961 | ABC Transporter Type 1 | 13 | 1 |
| group_8186 | FEMO cofactor biosynthesis protein NIFB | 13 | 1 |
| group_8190 | RGG_Cterm: transcriptional activator, Rgg/GadR/MutR family | 17 | 0 |
| group_8270 | No match | 11 | 0 |
| pspA | Histidine Phosphatase | 23 | 3 |
| scrB | Sucrose-6-phosphate hydrolase/glycosyl hydrolase family 32 | 17 | 5 |
| xylT_1 | SP: MFS transporter, sugar porter (SP) family | 15 | 2 |
| xylT_2 | SP: MFS transporter, sugar porter (SP) family | 20 | 3 |
| ybiR | Citrate transporter | 23 | 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Higdon, S.M.; Huang, B.C.; Bennett, A.B.; Weimer, B.C. Identification of Nitrogen Fixation Genes in Lactococcus Isolated from Maize Using Population Genomics and Machine Learning. Microorganisms 2020, 8, 2043. https://doi.org/10.3390/microorganisms8122043
Higdon SM, Huang BC, Bennett AB, Weimer BC. Identification of Nitrogen Fixation Genes in Lactococcus Isolated from Maize Using Population Genomics and Machine Learning. Microorganisms. 2020; 8(12):2043. https://doi.org/10.3390/microorganisms8122043
Chicago/Turabian StyleHigdon, Shawn M., Bihua C. Huang, Alan B. Bennett, and Bart C. Weimer. 2020. "Identification of Nitrogen Fixation Genes in Lactococcus Isolated from Maize Using Population Genomics and Machine Learning" Microorganisms 8, no. 12: 2043. https://doi.org/10.3390/microorganisms8122043
APA StyleHigdon, S. M., Huang, B. C., Bennett, A. B., & Weimer, B. C. (2020). Identification of Nitrogen Fixation Genes in Lactococcus Isolated from Maize Using Population Genomics and Machine Learning. Microorganisms, 8(12), 2043. https://doi.org/10.3390/microorganisms8122043