Comparative Pathogenomics of Aeromonas veronii from Pigs in South Africa: Dominance of the Novel ST657 Clone

 ,
,  ,
,  ,
,  , ,
, ,
Abstract
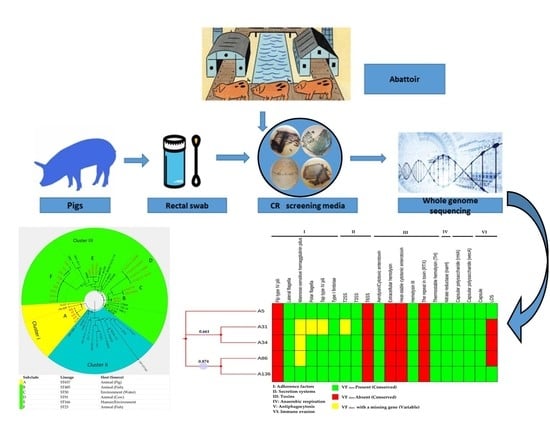
1. Introduction
2. Materials and Methods
2.1. Ethical Approval
2.2. Sampling
2.3. Isolation and Identification
2.3.1. Screening of Carbapenemase-Producing Isolates
2.3.2. Confirmation of Isolates and Determination of Antibiotic Susceptibility Profiles
2.4. DNA Purification, Genome Sequencing and Pre-Processing of Sequence Data
2.5. Genome Visualization and Annotation
2.6. Whole Genome Sequencing-Based Confirmation and Molecular Typing of Aeromonas Veronii
2.7. Antibiotic Resistance Genes, Efflux Genes, and Mobile Elements Identification
2.8. Assessment of Pathogenic Potential and Prediction of Putative Virulence Factors
2.9. Global Phylogenomic Relationship and Metadata Analysis
2.10. Data Availability
3. Results
3.1. Prevalence and Phenotypic Resistance Profiles of A. Veronii Isolates
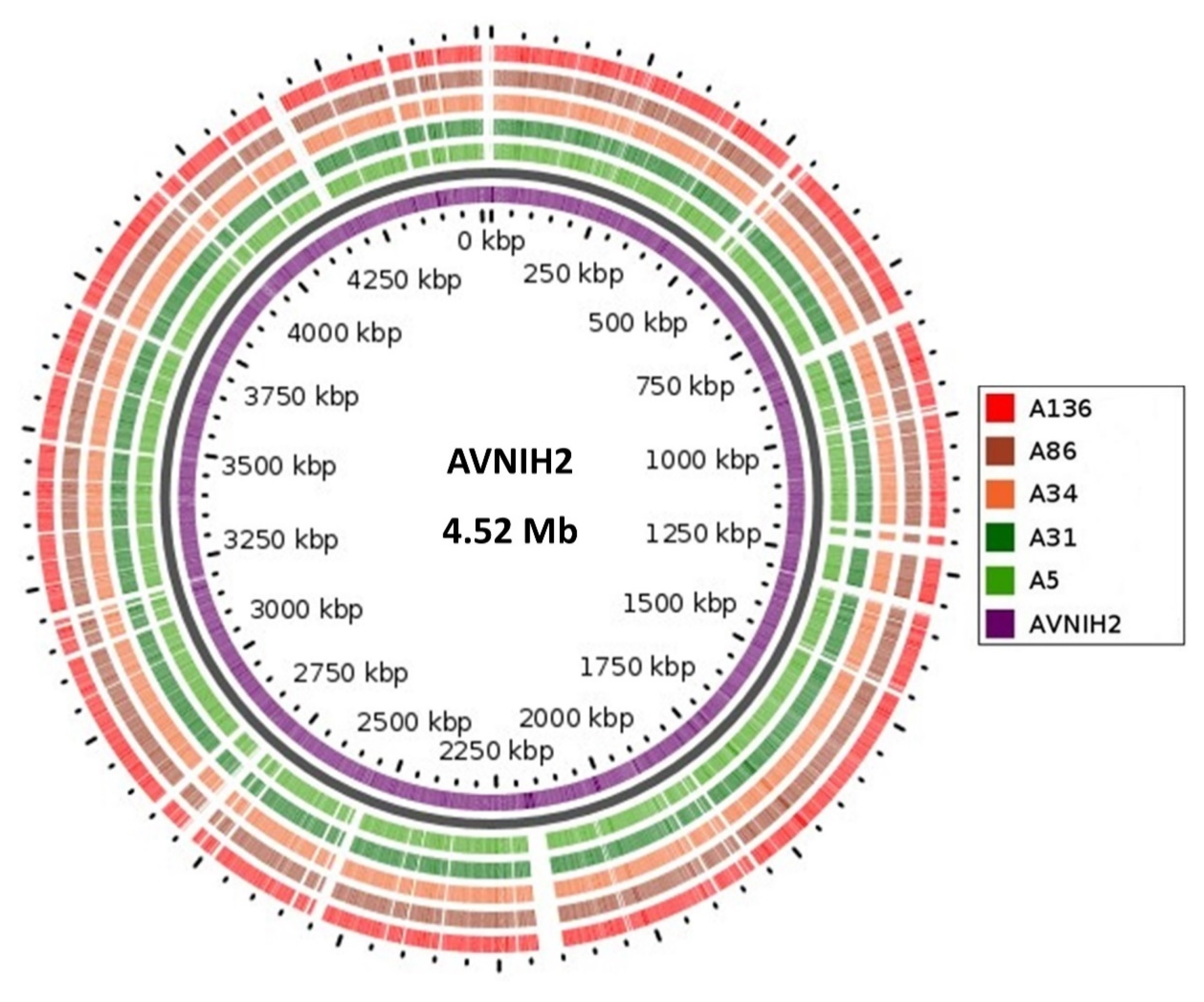
3.2. Genome Confirmation, Statistics, Annotation, and Visualization
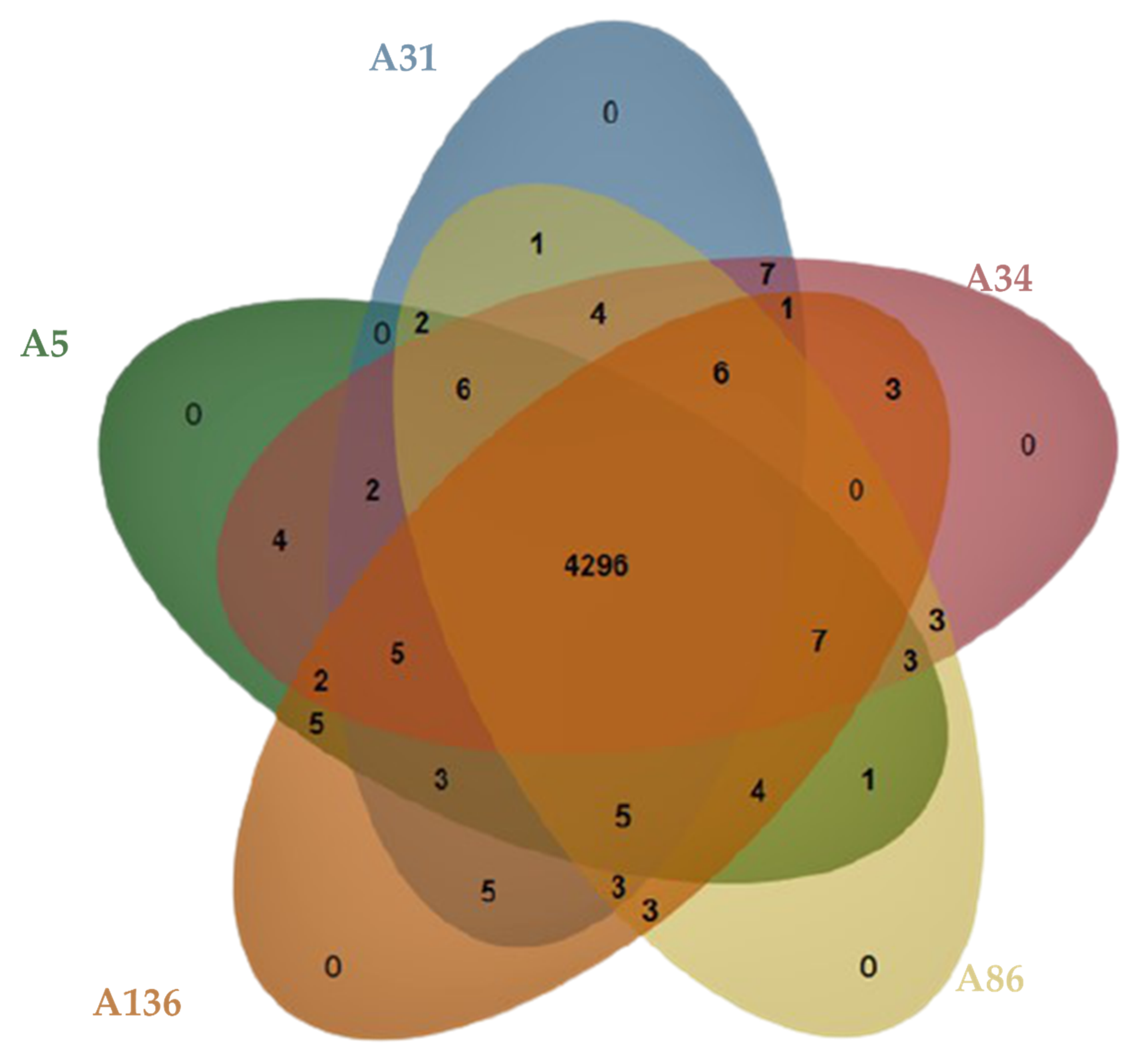
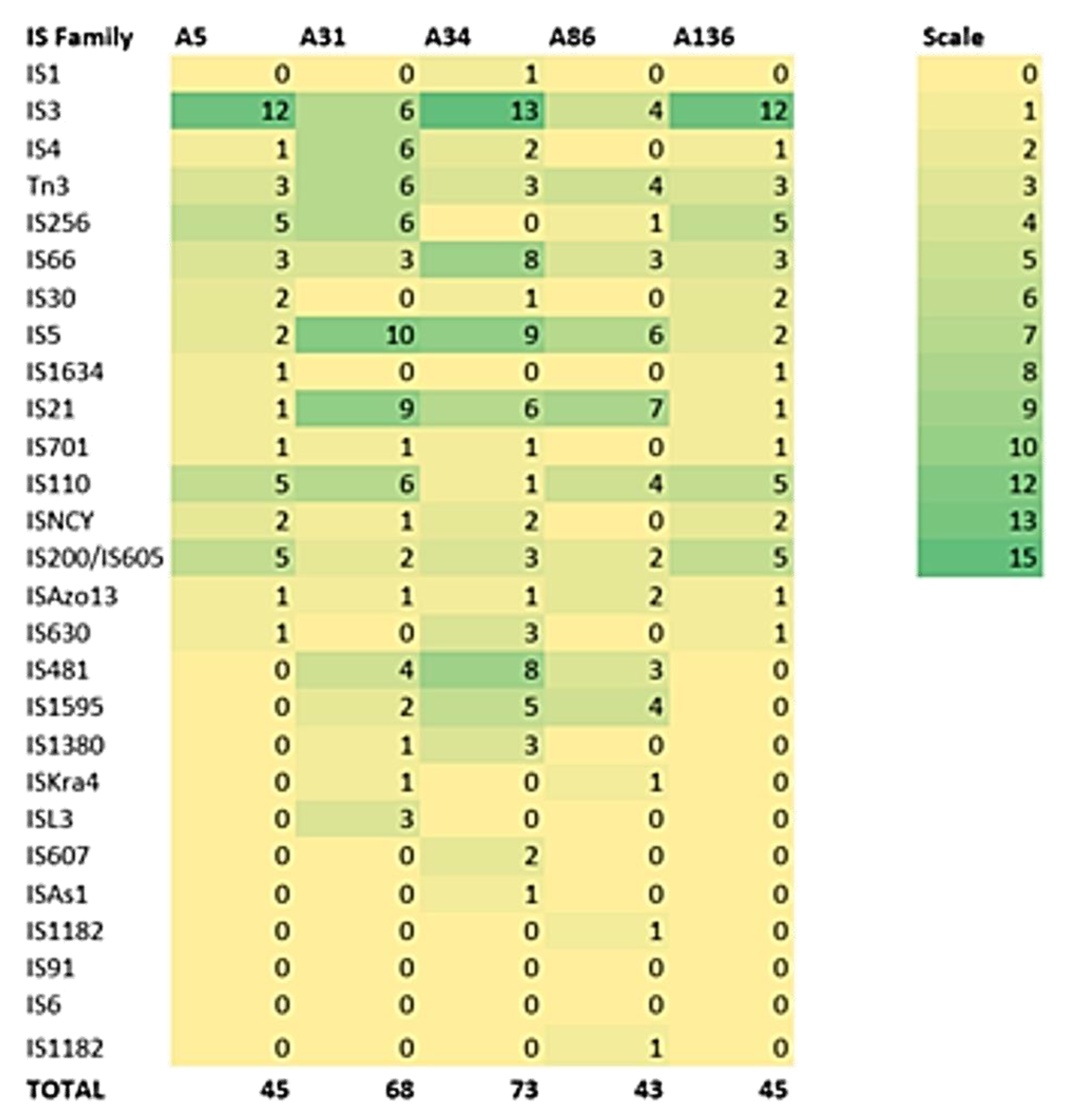
3.3. Defence Systems (CRISPRCas Cluster and Restriction-Modification (R-M) System), Antibiotic Resistome, Mobilome and Genetic Environment Analysis
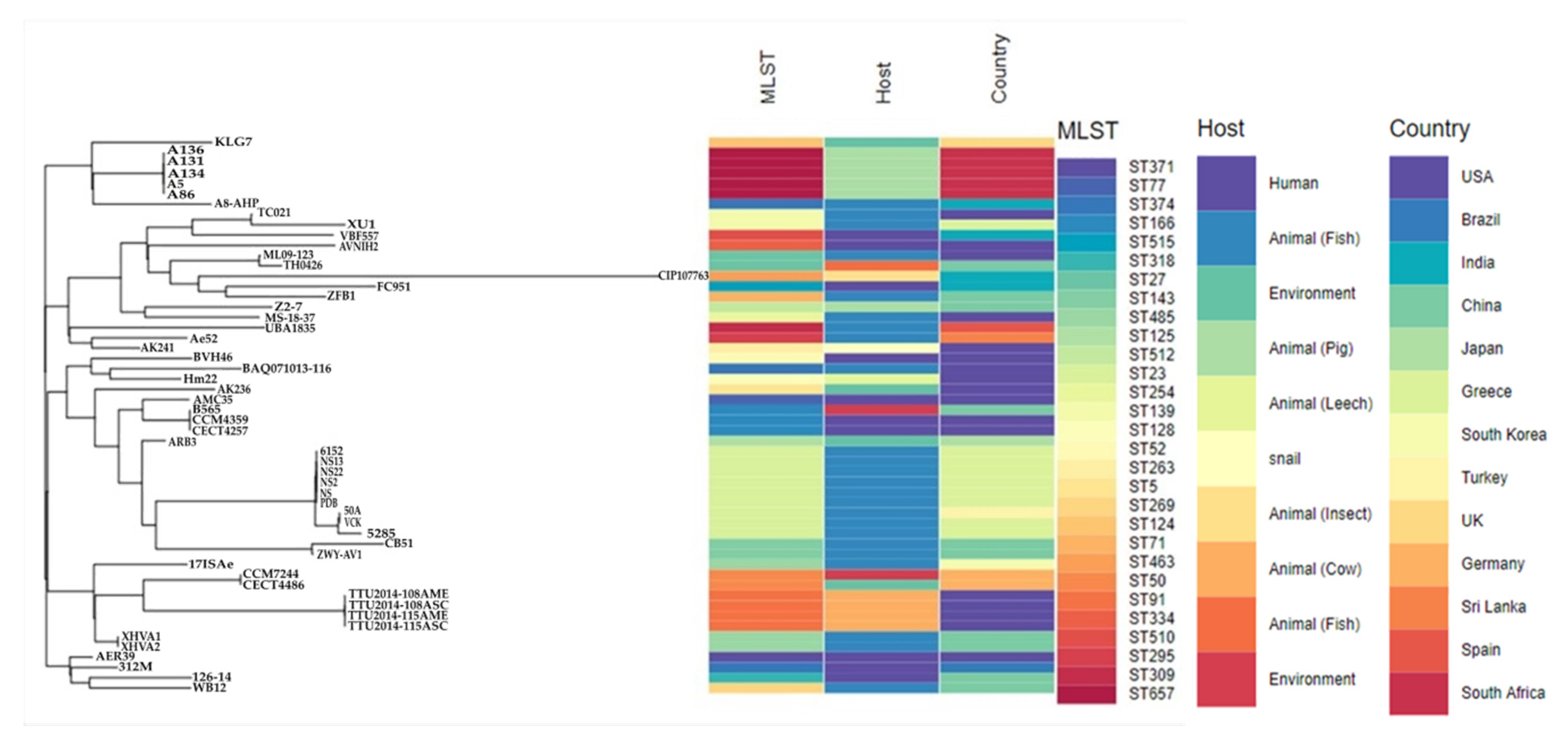
3.4. Molecular Typing, Global Epidemiological and Phylogenomic Analysis
3.5. Pathogenic Potential and Putative Virulence Factors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Batra, P.; Mathur, P.; Misra, M.C. Aeromonas spp.: An Emerging Nosocomial Pathogen. J. Lab. Phys. 2016, 8, 1. [Google Scholar]
- Martin-Carnahan, A.; Joseph, S.W. Aeromonadales ord.nov. In Bergey’s Manual® of Systematic Bacteriology; Springer US: Boston, MA, USA, 2005; pp. 556–587. [Google Scholar]
- Fernández-Bravo, A.; Figueras, M.J. An Update on the Genus Aeromonas: Taxonomy, Epidemiology, and Pathogenicity. Microorganisms 2020, 8, 129. [Google Scholar] [CrossRef]
- Igbinosa, I.H.; Igumbor, E.U.; Aghdasi, F.; Tom, M.; Okoh, A.I. Emerging Aeromonas Species Infections and Their Significance in Public Health. Sci. World J. 2012, 2012, 1–13. [Google Scholar]
- Janda, J.M.; Abbott, S.L. The Genus Aeromonas: Taxonomy, Pathogenicity, and Infection. Clin. Microbiol. Rev. 2010, 23, 35–73. [Google Scholar] [CrossRef]
- Martino, M.E.; Fasolato, L.; Montemurro, F.; Rosteghin, M.; Manfrin, A.; Patarnello, T.; Novelli, E.; Cardazzo, B. Determination of Microbial Diversity of Aeromonas Strains on the Basis of Multilocus Sequence Typing, Phenotype, and Presence of Putative Virulence Genes. Appl. Environ. Microbiol. 2011, 77, 4986–5000. [Google Scholar] [CrossRef]
- Puah, S.M.; Khor, W.C.; Kee, B.P.; Tan, J.A.M.A.; Puthucheary, S.D.; Chua, K.H. Development of a species-specific PCR-RFLP targeting rpoD gene fragment for discrimination of Aeromonas species. J. Med. Microbiol. 2018, 67, 1271–1278. [Google Scholar] [CrossRef]
- Wu, C.-J.; Chen, P.-L.; Hsueh, P.-R.; Chang, M.-C.; Tsai, P.-J.; Shih, H.-I.; Wang, H.-C.; Chou, P.-H.; Ko, W.-C. Clinical Implications of Species Identification in Monomicrobial Aeromonas Bacteremia. PLoS ONE 2015, 10, e0117821. [Google Scholar] [CrossRef]
- Liao, K.-C.; Yen, P.-T.; Liu, C. Necrotizing Fasciitis Caused by Inconspicuous Infection of Aeromonas hydrophila in an Immunocompromised Host. J. Surg. Case Rep. 2010, 2010, 2. [Google Scholar] [CrossRef] [PubMed]
- Gowda, T.K.G.M.; Reddy, V.R.A.P.; Devleesschauwer, B.; Zade, N.N.; Chaudhari, S.P.; Khan, W.A.; Shinde, S.V.; Patil, A.R. Isolation and Seroprevalence of Aeromonas spp. Among Common Food Animals Slaughtered in Nagpur, Central India. Foodborne Pathog. Dis. 2015, 12, 626–630. [Google Scholar] [CrossRef] [PubMed]
- Cabié, A.; Hope-Rapp, E.; Beaucaire, G.; Hochedez, P.; Nicolas, M.; Olive, C. Bacteremia Caused by Aeromonas hydrophila Complex in the Caribbean Islands of Martinique and Guadeloupe. Am. J. Trop. Med. Hyg. 2010, 83, 1123–1127. [Google Scholar]
- Li, T.; Raza, S.H.A.; Yang, B.; Sun, Y.; Wang, G.; Sun, W.; Qian, A.; Wang, C.; Kang, Y.; Shan, X. Aeromonas veronii Infection in Commercial Freshwater Fish: A Potential Threat to Public Health. Animals 2020, 10, 608. [Google Scholar] [CrossRef]
- Figueras, M.J.; Beaz-Hidalgo, R. Aeromonas infections in humans. In Aeromonas; Caister Academic Press: Norfolk, UK, 2015; pp. 65–108. [Google Scholar]
- Praveen, P.K.; Debnath, C.; Shekhar, S.; Dalai, N.; Ganguly, S. Incidence of Aeromonas spp. infection in fish and chicken meat and its related public health hazards: A review. Vet. World 2016, 9, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Xu, C.; Sun, Q.; Schwarz, S.; Ou, Y.; Yang, L.; Huang, Z.; Eichhorn, I.; Walsh, T.R.; Wang, Y.; et al. Prevalence and Genetic Analysis of mcr-3-Positive Aeromonas Species from Humans, Retail Meat, and Environmental Water Samples. Antimicrob. Agents Chemother. 2018, 62, 1. [Google Scholar] [CrossRef] [PubMed]
- Svenungsson, B.; Lagergren, A.; Ekwall, E.; Evengard, B.; Hedlund, K.O.; Karnell, A.; Lofdahl, S.; Svensson, L.; Weintraub, A. Enteropathogens in Adult Patients with Diarrhea and Healthy Control Subjects: A 1-Year Prospective Study in a Swedish Clinic for Infectious Diseases. Clin. Infect. Dis. 2000, 30, 770–778. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.J.; Ansaruzzaman, M.; Talukder, K.A.; Chopra, A.K.; Kuhn, I.; Rahman, M.; Faruque, A.S.G.; Islam, M.S.; Sack, R.B.; Mollby, R. Prevalence of Enterotoxin Genes in Aeromonas spp. Isolated from Children with Diarrhea, Healthy Controls, and the Environment. J. Clin. Microbiol. 2000, 38, 3785–3790. [Google Scholar] [CrossRef]
- Tomás, J.M. The Main Aeromonas Pathogenic Factors. ISRN Microbiol. 2012, 2012, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Beaz-Hidalgo, R.; Figueras, M.J. Aeromonas spp. whole genomes and virulence factors implicated in fish disease. J. Fish Dis. 2013, 36, 371–388. [Google Scholar] [CrossRef]
- Tekedar, H.C.; Kumru, S.; Blom, J.; Perkins, A.D.; Griffin, M.J.; Abdelhamed, H.; Karsi, A.; Lawrence, M.L. Comparative genomics of Aeromonas veronii: Identification of a pathotype impacting aquaculture globally. PLoS ONE 2019, 14, e0221018. [Google Scholar] [CrossRef]
- Albarral, V.; Sanglas, A.; Palau, M.; Miñana-Galbis, D.; Fusté, M.C. Potential pathogenicity of Aeromonas hydrophila complex strains isolated from clinical, food, and environmental sources. Can. J. Microbiol. 2016, 62, 296–306. [Google Scholar] [CrossRef]
- Papadimitriou-Olivgeris, M.; Bartzavali, C.; Christofidou, M.; Bereksi, N.; Hey, J.; Zambardi, G.; Spiliopoulou, I. Performance of chromID® CARBA medium for carbapenemases-producing enterobacteriaceae detection during rectal screening. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 35–40. [Google Scholar] [CrossRef]
- Simner, P.J.; Martin, I.; Opene, B.; Tamma, P.D.; Carroll, K.C.; Milstone, A.M. Evaluation of Multiple Methods for Detection of Gastrointestinal Colonization of Carbapenem-Resistant Organisms from Rectal Swabs. J. Clin. Microbiol. 2016, 54, 1664–1667. [Google Scholar] [CrossRef] [PubMed]
- Clinical and Laboratory Standards Institute Performance Standards for Antimicrobial Susceptibility Testing: 27th Edition Informational Supplement M100-S27; CLSI: Wayne, PA, USA, 2017.
- Petkau, A.; Stuart-Edwards, M.; Stothard, P.; Van Domselaar, G. Interactive microbial genome visualization with GView. Bioinformatics 2010, 26, 3125–3126. [Google Scholar] [CrossRef] [PubMed]
- Tatusova, T.; Dicuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef] [PubMed]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef]
- Xu, L.; Dong, Z.; Fang, L.; Luo, Y.; Wei, Z.; Guo, H.; Zhang, G.; Gu, Y.Q.; Coleman-Derr, D.; Xia, Q.; et al. OrthoVenn2: A web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 2019, 47, W52–W58. [Google Scholar] [CrossRef]
- Neher, R.A.; Bedford, T. Real-Time Analysis and Visualization of Pathogen Sequence Data. J. Clin. Microbiol. 2018, 56, 1–15. [Google Scholar] [CrossRef]
- Larsen, M.V.; Cosentino, S.; Rasmussen, S.; Friis, C.; Hasman, H.; Marvig, R.L.; Jelsbak, L.; Sicheritz-Pontén, T.; Ussery, D.W.; Aarestrup, F.M.; et al. Multilocus Sequence Typing of Total-Genome-Sequenced Bacteria. J. Clin. Microbiol. 2012, 50, 1355–1361. [Google Scholar] [CrossRef]
- Feil, E.J.; Li, B.C.; Aanensen, D.M.; Hanage, W.P.; Spratt, B.G. eBURST: Inferring Patterns of Evolutionary Descent among Clusters of Related Bacterial Genotypes from Multilocus Sequence Typing Data. J. Bacteriol. 2004, 186, 1518–1530. [Google Scholar] [CrossRef]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef]
- Gupta, S.K.; Padmanabhan, B.R.; Diene, S.M.; Lopez-Rojas, R.; Kempf, M.; Landraud, L.; Rolain, J.M. ARG-annot, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob. Agents Chemother. 2014, 58, 212–220. [Google Scholar] [CrossRef]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A Fast Phage Search Tool. Nucleic Acids Res. 2011, 39, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Yu, Z.; Xu, Z. Study the features of 57 confirmed CRISPR Loci in 38 strains of Staphylococcus aureus. Front. Microbiol. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Grissa, I.; Vergnaud, G.; Pourcel, C. CRISPRFinder: A web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007, 35, W52–W57. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Cosentino, S.; Voldby Larsen, M.; Møller Aarestrup, F.; Lund, O. PathogenFinder--distinguishing friend from foe using bacterial whole genome sequence data. PLoS ONE 2013, 8, e77302. [Google Scholar] [CrossRef]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef]
- Codjoe, F.; Donkor, E. Carbapenem Resistance: A Review. Med. Sci. 2017, 6, 1. [Google Scholar] [CrossRef]
- Dias, C.; Serra, C.R.; Simões, L.C.; Simões, M.; Martinez-Murcia, A.; Saavedra, M.J. Extended-spectrum β-lactamase and carbapenemase-producing Aeromonas species in wild animals from Portugal. Vet. Rec. 2014, 174, 532. [Google Scholar] [CrossRef]
- Skwor, T.; Stringer, S.; Haggerty, J.; Johnson, J.; Duhr, S.; Johnson, M.; Seckinger, M.; Stemme, M. Prevalence of Potentially Pathogenic Antibiotic-Resistant Aeromonas spp. in Treated Urban Wastewater Effluents versus Recipient Riverine Populations: A 3-Year Comparative Study. Appl. Environ. Microbiol. 2019, 86, 1–15. [Google Scholar] [CrossRef]
- Rosso, F.; Cedano, J.A.; Parra-Lara, L.G.; Sanz, A.M.; Toala, A.; Velez, J.F.; Hormaza, M.P.; Moncada, P.A.; Correa, A. Emerging carbapenem-resistant Aeromonas spp. infections in Cali, Colombia. Braz. J. Infect. Dis. 2019, 23, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Uechi, K.; Tada, T.; Sawachi, Y.; Hishinuma, T.; Takaesu, R.; Nakama, M.; Nakasone, I.; Kirikae, T.; Fujita, J. A carbapenem-resistant clinical isolate of Aeromonas hydrophila in Japan harbouring an acquired gene encoding GES-24 β-lactamase. J. Med. Microbiol. 2018, 67, 1535–1537. [Google Scholar] [CrossRef] [PubMed]
- Martín Talavera, B.M.; Benassi, F.O.; von Specht, M.H.; Quiroga, M.I.; García, M.A.; Pucciarelli, A.B.; Zubreski, E.; Laczeski, M.E.; Gutkind, G. Susceptibilities to carbapenems and presence of cphA gene on food-borne Aeromonas. Braz. Arch. Biol. Technol. 2006, 49, 677–682. [Google Scholar] [CrossRef]
- Evangelista-Barreto, N.S.; Carvalho, F.C.T.D.; Vieira, R.H.S.; dos Reis, C.M.; Macrae, A.; Rodrigues, D.D.P. Characterization of Aeromonas species isolated from an estuarine environment. Braz. J. Microbiol. 2010, 41, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Amoako, D.G.; Somboro, A.M.; Abia, A.L.K.; Allam, M.; Ismail, A.; Bester, L.A.; Essack, S.Y. Genome Mining and Comparative Pathogenomic Analysis of An Endemic Methicillin-Resistant Staphylococcus Aureus (MRSA) Clone, ST612-CC8-t1257-SCCmec_IVd(2B), Isolated in South Africa. Pathogens 2019, 8, 166. [Google Scholar] [CrossRef] [PubMed]
- Bakr Shabbir, M.A.; Hao, H.; Shabbir, M.Z.; Hussain, H.I.; Iqbal, Z.; Ahmed, S.; Sattar, A.; Iqbal, M.; Li, J.; Yuan, Z. Survival and evolution of CRISPR-Cas system in prokaryotes and its applications. Front. Immunol. 2016, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Horvath, P.; Barrangou, R. CRISPR/Cas, the Immune System of Bacteria and Archaea. Science 2010, 327, 167–170. [Google Scholar] [CrossRef]
- Burstein, D.; Sun, C.L.; Brown, C.T.; Sharon, I.; Anantharaman, K.; Probst, A.J.; Thomas, B.C.; Banfield, J.F. Major bacterial lineages are essentially devoid of CRISPR-Cas viral defence systems. Nat. Commun. 2016, 7, 10613. [Google Scholar] [CrossRef]
- Osman, K.; Aly, M.; Kheader, A.; Mabrok, K. Molecular detection of the Aeromonas virulence aerolysin gene in retail meats from different animal sources in Egypt. World J. Microbiol. Biotechnol. 2012, 28, 1863–1870. [Google Scholar] [CrossRef]
- Köck, R.; Daniels-Haardt, I.; Becker, K.; Mellmann, A.; Friedrich, A.W.; Mevius, D.; Schwarz, S.; Jurke, A. Carbapenem-resistant Enterobacteriaceae in wildlife, food-producing, and companion animals: A systematic review. Clin. Microbiol. Infect. 2018, 24, 1241–1250. [Google Scholar] [CrossRef]
- Bonardi, S.; Pitino, R. Carbapenemase-producing bacteria in food-producing animals, wildlife and environment: A challenge for human health. Ital. J. Food Saf. 2019, 8, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Ghatak, S.; Blom, J.; Das, S.; Sanjukta, R.; Puro, K.; Mawlong, M.; Shakuntala, I.; Sen, A.; Goesmann, A.; Kumar, A.; et al. Pan-genome analysis of Aeromonas hydrophila, Aeromonas veronii and Aeromonas caviae indicates phylogenomic diversity and greater pathogenic potential for Aeromonas hydrophila. Antonie Van Leeuwenhoek 2016, 109, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Ghenghesh, K.S.; El-Mohammady, H.; Levin, S.Y.; Zorgani, A.; Tawil, K. Antimicrobial resistance profile of Aeromonas species isolated from Libya. Libyan J. Med. 2013, 8, 21320. [Google Scholar] [CrossRef] [PubMed]
- Vandecraen, J.; Chandler, M.; Aertsen, A.; Van Houdt, R. The impact of insertion sequences on bacterial genome plasticity and adaptability. Crit. Rev. Microbiol. 2017, 43, 709–730. [Google Scholar] [CrossRef] [PubMed]
- Touchon, M.; Rocha, E.P.C. Causes of Insertion Sequences Abundance in Prokaryotic Genomes. Mol. Biol. Evol. 2007, 24, 969–981. [Google Scholar] [CrossRef] [PubMed]
- Siguier, P.; Gourbeyre, E.; Chandler, M. Bacterial insertion sequences: Their genomic impact and diversity. FEMS Microbiol. Rev. 2014, 38, 865–891. [Google Scholar] [CrossRef]
- Pang, M.; Jiang, J.; Xie, X.; Wu, Y.; Dong, Y.; Kwok, A.H.Y.; Zhang, W.; Yao, H.; Lu, C.; Leung, F.C.; et al. Novel insights into the pathogenicity of epidemic Aeromonas hydrophila ST251 clones from comparative genomics. Sci. Rep. 2015, 5, 9833. [Google Scholar] [CrossRef]
- Cianciotto, N.P. Type II secretion: A protein secretion system for all seasons. Trends Microbiol. 2005, 13, 581–588. [Google Scholar] [CrossRef]
- Ast, V.M.; Schoenhofen, I.C.; Langen, G.R.; Stratilo, C.W.; Chamberlain, M.D.; Howard, S.P. Expression of the ExeAB complex of Aeromonas hydrophila is required for the localization and assembly of the ExeD secretion port multimer. Mol. Microbiol. 2002, 44, 217–231. [Google Scholar] [CrossRef]
- Kong, C.; Neoh, H.M.; Nathan, S. Targeting Staphylococcus aureus toxins: A potential form of anti-virulence therapy. Toxins (Basel) 2016, 8, 72. [Google Scholar] [CrossRef]
- Yu, H.B.; Zhang, Y.L.; Lau, Y.L.; Yao, F.; Vilches, S.; Merino, S.; Tomas, J.M.; Howard, S.P.; Leung, K.Y. Identification and Characterization of Putative Virulence Genes and Gene Clusters in Aeromonas hydrophila PPD134/91. Appl. Environ. Microbiol. 2005, 71, 4469–4477. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibiotics | Resistance Phenotypes 1 | ||||
|---|---|---|---|---|---|
| A5 | A31 | A34 | A86 | A136 | |
| Ampicillin (AMP) | R | R | R | R | R |
| Amoxicillin (AMX) | R | R | R | R | R |
| Amoxicillin-clavulanate (AMC) | I | S | S | S | I |
| Piperacillin-tazobactam (TZP) | S | S | S | S | R |
| Cefuroxime (CXM) | S | S | S | S | S |
| Cefotaxime (CTX) | S | S | S | S | S |
| Ceftriaxone (CRO) | S | S | S | S | S |
| Ceftazidime (CAZ) | S | S | S | S | S |
| Cefepime (FEP) | S | S | S | S | S |
| Cefoxitin (FOX) | S | S | S | S | S |
| Imipenem (IMI) | R | R | R | R | R |
| Meropenem (MER) | S | S | S | S | S |
| Isolate | In Silico Typing | Defence Systems | Resistome | Mobilome 2 | Pathogenicity 3 |
|---|---|---|---|---|---|
| ID | MLST 1 | CRISPR (Cas) | No. of Insertion Sequences | Score (Pathogenic Families) | |
| A5 | ST657 | 4 (0) | OXA-12, cphA3, | 45 | 0.607 (30) |
| A31 | ST657 | 5 (0) | OXA-12, cphA3, | 68 | 0.607 (30) |
| A34 | ST657 | 5 (0) | OXA-12, cphA3, | 73 | 0.607 (30) |
| A86 | ST657 | 4 (0) | OXA-12, cphA3, | 43 | 0.603 (29) |
| A136 | ST657 | 5 (0) | OXA-12, cphA3, | 45 | 0.607 (30) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramsamy, Y.; Mlisana, K.P.; Amoako, D.G.; Abia, A.L.K.; Allam, M.; Ismail, A.; Singh, R.; Essack, S.Y. Comparative Pathogenomics of Aeromonas veronii from Pigs in South Africa: Dominance of the Novel ST657 Clone. Microorganisms 2020, 8, 2008. https://doi.org/10.3390/microorganisms8122008
Ramsamy Y, Mlisana KP, Amoako DG, Abia ALK, Allam M, Ismail A, Singh R, Essack SY. Comparative Pathogenomics of Aeromonas veronii from Pigs in South Africa: Dominance of the Novel ST657 Clone. Microorganisms. 2020; 8(12):2008. https://doi.org/10.3390/microorganisms8122008
Chicago/Turabian StyleRamsamy, Yogandree, Koleka P. Mlisana, Daniel G. Amoako, Akebe Luther King Abia, Mushal Allam, Arshad Ismail, Ravesh Singh, and Sabiha Y. Essack. 2020. "Comparative Pathogenomics of Aeromonas veronii from Pigs in South Africa: Dominance of the Novel ST657 Clone" Microorganisms 8, no. 12: 2008. https://doi.org/10.3390/microorganisms8122008
APA StyleRamsamy, Y., Mlisana, K. P., Amoako, D. G., Abia, A. L. K., Allam, M., Ismail, A., Singh, R., & Essack, S. Y. (2020). Comparative Pathogenomics of Aeromonas veronii from Pigs in South Africa: Dominance of the Novel ST657 Clone. Microorganisms, 8(12), 2008. https://doi.org/10.3390/microorganisms8122008