The Missing Tailed Phages: Prediction of Small Capsid Candidates



Abstract
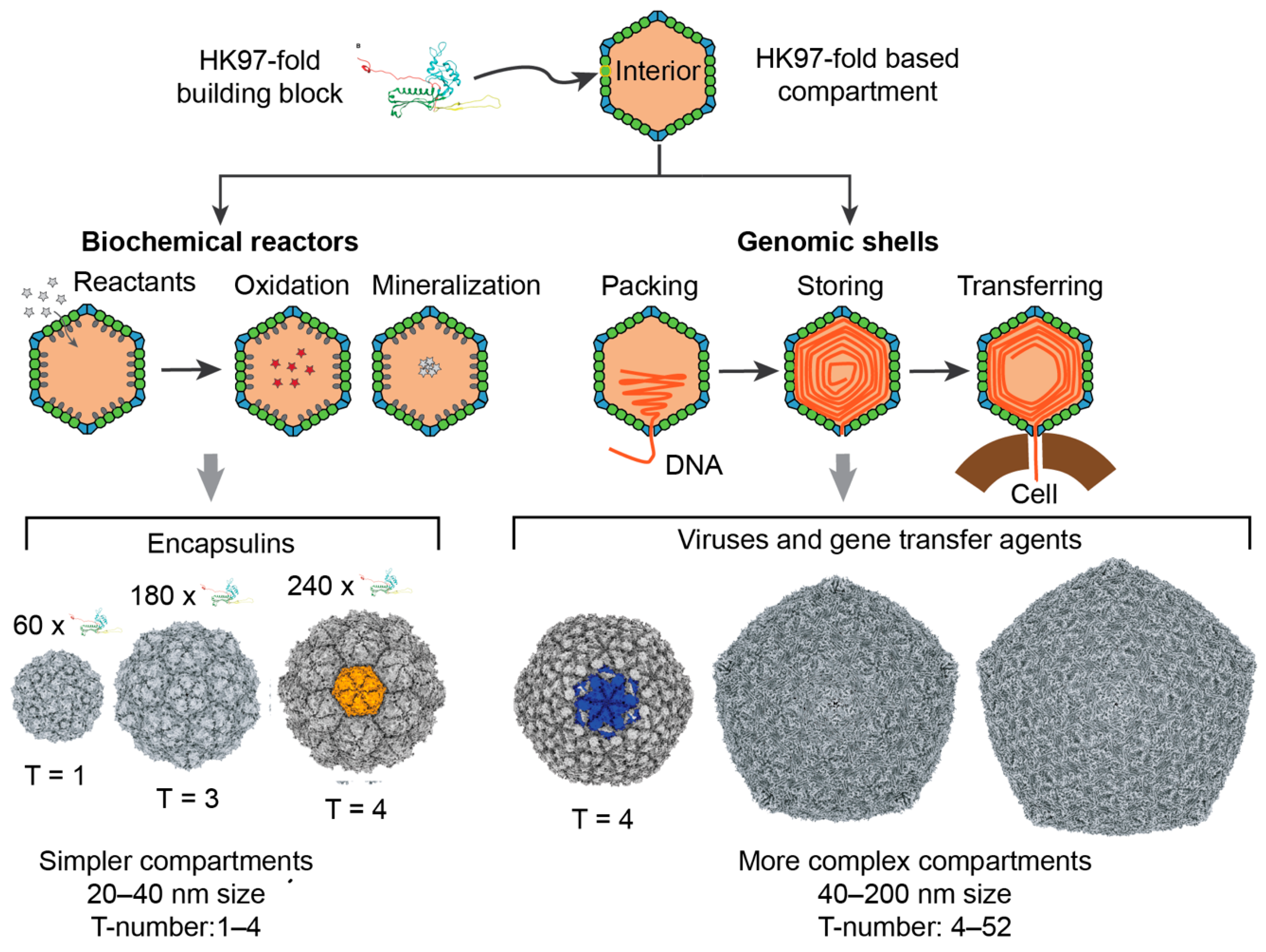
1. Introduction
2. Materials and Methods
3. Results
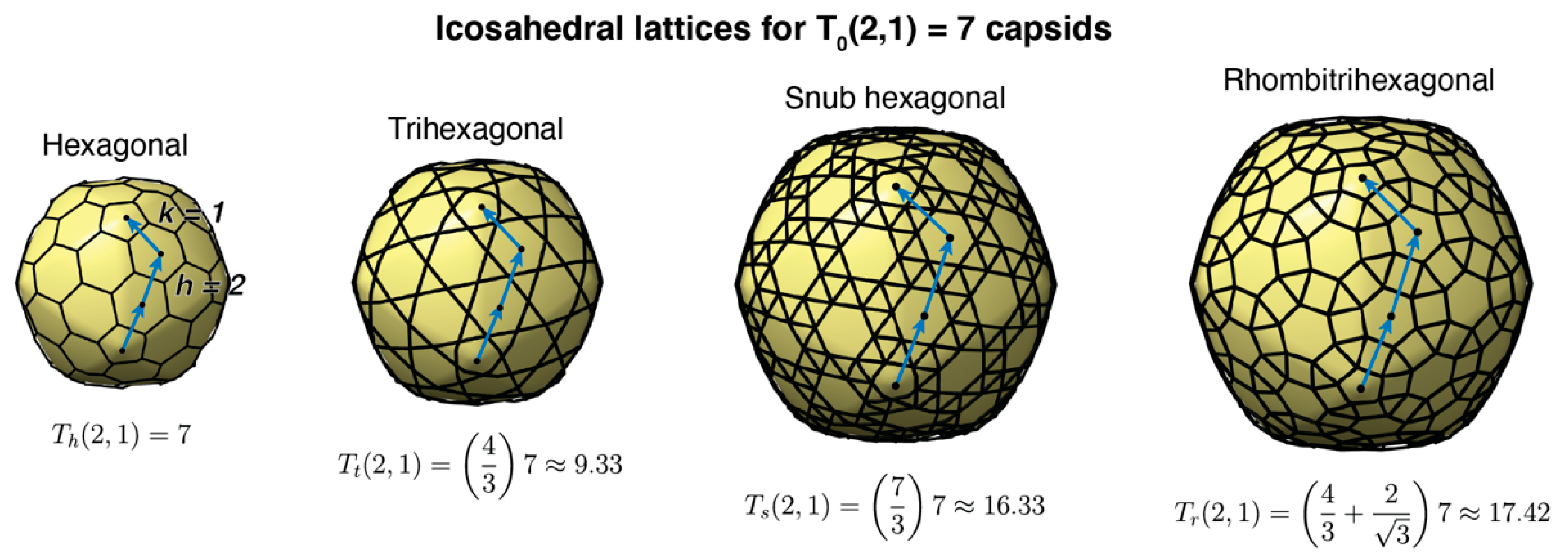
3.1. Structural Properties of Characterized Tailed Phage Capsids
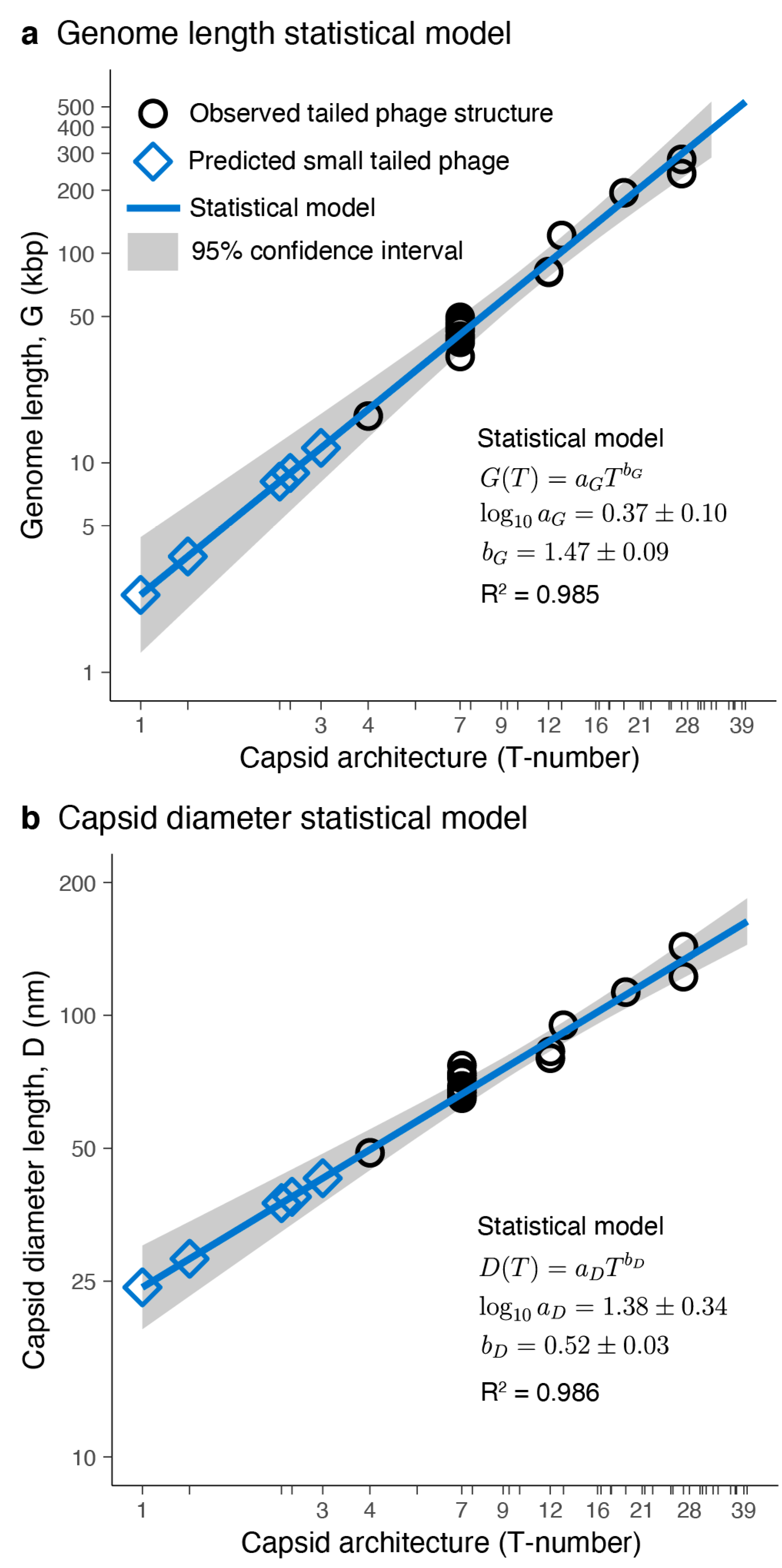
3.2. Tailed Phage Capsids: Statistical Models and Predictions
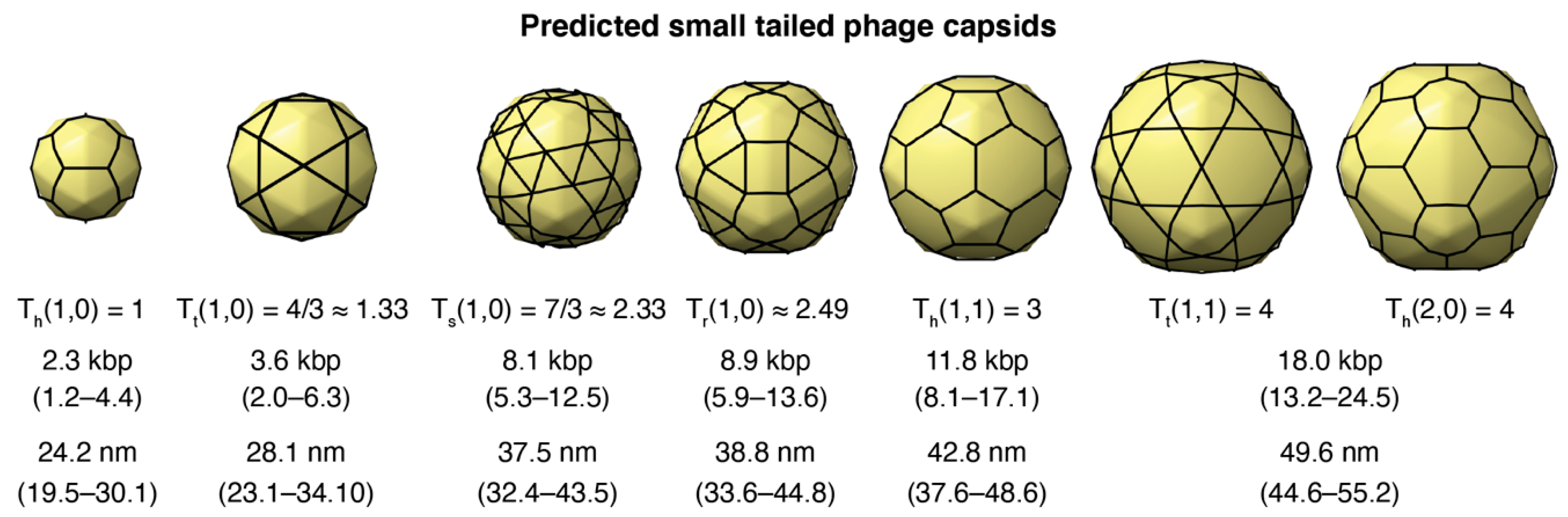
3.3. Putative Small-Tailed Phage Candidates
3.3.1. Predictions from Isolated Tailed Phages
3.3.2. Predictions from Metagenome-Assembled Tailed Phages
3.3.3. Small-Tailed Phage Capsid Candidates
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cobián Güemes, A.G.; Youle, M.; Cantú, V.A.; Felts, B.; Nulton, J.; Rohwer, F. Viruses as winners in the game of life. Annu. Rev. Virol. 2016, 3, 197–214. [Google Scholar] [CrossRef] [PubMed]
- Wommack, K.E.; Colwell, R.R. Virioplankton: Viruses in aquatic ecosystems. Microbiol. Mol. Biol. Rev. 2000, 64, 69–114. [Google Scholar] [CrossRef] [PubMed]
- Danovaro, R.; Corinaldesi, C.; Dell’Anno, A.; Fuhrman, J.A.; Middelburg, J.J.; Noble, R.T.; Suttle, C.A. Marine viruses and global climate change. FEMS Microbiol. Rev. 2011, 35, 993–1034. [Google Scholar] [CrossRef] [PubMed]
- Knowles, B.; Silveira, C.B.; Bailey, B.A.; Barott, K.; Cantu, V.A.; Cobián-Güemes, A.G.; Coutinho, F.H.; Dinsdale, E.A.; Felts, B.; Furby, K.A.; et al. Lytic to temperate switching of viral communities. Nature 2016, 531, 466–470. [Google Scholar] [CrossRef]
- Luque, A.; Silveira, C. Quantification of lysogeny caused by phage coinfections in microbial communities from biophysical principles. mSystems 2020, 5, e00353-20. [Google Scholar] [CrossRef]
- Silveira, C.B.; Coutinho, F.H.; Cavalcanti, G.S.; Benler, S.; Doane, M.P.; Dinsdale, E.A.; Edwards, R.A.; Francini-Filho, R.B.; Thompson, C.C.; Luque, A.; et al. Genomic and ecological attributes of marine bacteriophages encoding bacterial virulence genes. BMC Genomics 2020, 21, 126. [Google Scholar] [CrossRef]
- Paez-Espino, D.; Eloe-Fadrosh, E.A.; Pavlopoulos, G.A.; Thomas, A.D.; Huntemann, M.; Mikhailova, N.; Rubin, E.; Ivanova, N.N.; Kyrpides, N.C. Uncovering Earth’s virome. Nature 2016, 5636, 425–430. [Google Scholar] [CrossRef]
- Hua, J.; Huet, A.; Lopez, C.A.; Toropova, K.; Pope, W.H.; Duda, R.L.; Hendrix, R.W.; Conway, J.F. Capsids and genomes of jumbo-sized bacteriophages reveal the evolutionary reach of the HK97 fold. MBio 2017, 8. [Google Scholar] [CrossRef]
- Briani, F.; Dehò, G.; Forti, F.; Ghisotti, D. The plasmid status of satellite bacteriophage P4. Plasmid 2001, 45, 1–17. [Google Scholar] [CrossRef]
- Suhanovsky, M.M.; Teschke, C.M. Nature’s favorite building block: Deciphering folding and capsid assembly of proteins with the HK97-fold. Virology 2015, 479–480, 487–497. [Google Scholar] [CrossRef]
- Ackermann, H.W. 5500 Phages examined in the electron microscope. Arch. Virol. 2007, 152, 227–243. [Google Scholar] [CrossRef] [PubMed]
- Luque, A.; Reguera, D. The structure of elongated viral capsids. Biophys. J. 2010, 98, 2993–3003. [Google Scholar] [CrossRef]
- Luque, A.; Zandi, R.; Reguera, D. Optimal architectures of elongated viruses. Proc. Natl. Acad. Sci. USA 2010, 107, 5323–5328. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Koonin, E. V Multiple origins of viral capsid proteins from cellular ancestors. Proc. Natl. Acad. Sci. USA 2017, 114, E2401–E2410. [Google Scholar] [CrossRef]
- Ho, P.T.; Montiel-Garcia, D.J.; Wong, J.J.; Carrillo-Tripp, M.; Brooks III, C.L.; Johnson, J.E.; Reddy, V.S. VIPERdb: A Tool for Virus Research. Annu. Rev. Virol. 2018, 5, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Sutter, M.; Boehringer, D.; Gutmann, S.; Günther, S.; Prangishvili, D.; Loessner, M.J.; Stetter, K.O.; Weber-Ban, E.; Ban, N. Structural basis of enzyme encapsulation into a bacterial nanocompartment. Nat. Struct. Mol. Biol. 2008, 15, 939–947. [Google Scholar] [CrossRef] [PubMed]
- Giessen, T.W.; Orlando, B.J.; Verdegaal, A.A.; Chambers, M.G.; Gardener, J.; Bell, D.C.; Birrane, G.; Liao, M.; Silver, P.A. Large protein organelles form a new iron sequestration system with high storage capacity. Elife 2019, 8, e46070. [Google Scholar] [CrossRef] [PubMed]
- Caspar, D.L.; Klug, A. Physical principles in the construction of regular viruses. Cold Spring Harb. Symp. Quant. Biol. 1962, 27, 1–24. [Google Scholar] [CrossRef]
- Twarock, R.; Luque, A. Structural puzzles in virology solved with an overarching icosahedral design principle. Nat. Commun. 2019, 10, 4414. [Google Scholar] [CrossRef]
- Podgorski, J.; Calabrese, J.; Alexandrescu, L.; Jacobs-Sera, D.; Pope, W.; Hatfull, G.; White, S. Structures of three actinobacteriophage capsids: Roles of symmetry and accessory proteins. Viruses 2020, 12, 294. [Google Scholar] [CrossRef]
- Tao, Y.; Olson, N.H.; Xu, W.; Anderson, D.L.; Rossmann, M.G.; Baker, T.S. Assembly of a tailed bacterial virus and its genome release studied in three dimensions. Cell 1998, 95, 431–437. [Google Scholar] [CrossRef]
- Choi, K.H.; Morais, M.C.; Anderson, D.L.; Rossmann, M.G. Determinants of bacteriophage phi29 head morphology. Structure 2006, 14, 1723–1727. [Google Scholar] [CrossRef] [PubMed]
- Aksyuk, A.; Bowman, V.D.; Kaufmann, B.; Fields, C.; Klose, T.; Holdaway, H.; Fischetti, V.; Rossmann, M.G. Structural investigations of a Podoviridae streptococcus phage C1, implications for the mechanism of viral entry. Proc. Natl. Acad. Sci. USA 2012, 109, 14001–14006. [Google Scholar] [CrossRef] [PubMed]
- Zandi, R.; Reguera, D.; Bruinsma, R.F.; Gelbart, W.M.; Rudnick, J. Origin of icosahedral symmetry in viruses. Proc. Natl. Acad. Sci. USA 2004, 101, 15556–15560. [Google Scholar] [CrossRef] [PubMed]
- Luque, A.; Reguera, D.; Morozov, A.; Rudnick, J.; Bruinsma, R. Physics of shell assembly: Line tension, hole implosion, and closure catastrophe. J. Chem. Phys. 2012, 136, 184507. [Google Scholar] [CrossRef] [PubMed]
- Hagan, M.F. Modeling viral capsid assembly. Adv. Chem. Phys. 2014, 155, 1. [Google Scholar] [CrossRef]
- Aznar, M.; Reguera, D. Physical ingredients controlling stability and structural selection of empty viral capsids. J. Phys. Chem. B 2016, 120, 6147–6159. [Google Scholar] [CrossRef]
- Rice, G.; Tang, L.; Stedman, K.; Roberto, F.; Spuhler, J.; Gillitzer, E.; Johnson, J.E.; Douglas, T.; Young, M. The structure of a thermophilic archaeal virus shows a double-stranded DNA viral capsid type that spans all domains of life. Proc. Natl. Acad. Sci. USA 2004, 101, 7716–7720. [Google Scholar] [CrossRef]
- Koonin, E.V.; Dolja, V.V.; Krupovic, M.; Varsani, A.; Wolf, Y.I.; Yutin, N.; Zerbini, F.M.; Kuhn, J.H. Global organization and proposed megataxonomy of the virus world. Microbiol. Mol. Biol. Rev. 2020, 84. [Google Scholar] [CrossRef]
- Krupovic, M.; Dolja, V.V.; Koonin, E.V. The LUCA and its complex virome. Nat. Rev. Microbiol. 2020, 18, 661–670. [Google Scholar] [CrossRef]
- Doore, S.M.; Fane, B.A. The microviridae: Diversity, assembly, and experimental evolution. Virology 2016, 491, 45–55. [Google Scholar] [CrossRef]
- Creasy, A.; Rosario, K.; Leigh, B.A.; Dishaw, L.J.; Breitbart, M. Unprecedented diversity of ssDNA phages from the family Microviridae detected within the gut of a protochordate model organism (Ciona robusta). Viruses 2018, 10, 404. [Google Scholar] [CrossRef] [PubMed]
- Mavrich, T.N.; Hatfull, G.F. Evolution of Superinfection Immunity in Cluster A Mycobacteriophages. Am. Soc. Microbiol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, T.; Karambelkar, S.; Bondy-Denomy, J.; Wiedenheft, B. Structures and Strategies of Anti-CRISPR-Mediated Immune Suppression. Annu. Rev. Microbiol. 2020, 74, E5122–E5128. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M.; Thompson, L.R.; Suttle, C.A.; Sullivan, M.B. Exploring the vast diversity of marine viruses. Oceanography 2007, 20, 135–139. [Google Scholar] [CrossRef]
- Edwards, K.F.; Steward, G.F.; Schvarcz, C.A. Making sense of virus size and the tradeoffs shaping viral fitness. Ecol. Lett. 2020. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Q.; Murata, K.; Baker, M.L.; Sullivan, M.B.; Fu, C.; Dougherty, M.T.; Schmid, M.F.; Osburne, M.S.; Chisholm, S.W.; et al. Structural changes in a marine podovirus associated with release of its genome into Prochlorococcus. Nat. Struct. Mol. Biol. 2010, 17, 830–836. [Google Scholar] [CrossRef]
- Bebeacua, C.; Lai, L.; Vegge, C.S.; Brøndsted, L.; van Heel, M.; Veesler, D.; Cambillau, C. Visualizing a complete Siphoviridae member by single-particle electron microscopy: The structure of lactococcal phage TP901-1. J. Virol. 2013, 87, 1061–1068. [Google Scholar] [CrossRef]
- Parent, K.N.; Tang, J.; Cardone, G.; Gilcrease, E.B.; Janssen, M.E.; Olson, N.H.; Casjens, S.R.; Baker, T.S. Three-dimensional reconstructions of the bacteriophage CUS-3 virion reveal a conserved coat protein I-domain but a distinct tailspike receptor-binding domain. Virology 2014, 464, 55–66. [Google Scholar] [CrossRef]
- Spilman, M.S.; Dearborn, A.D.; Chang, J.R.; Damle, P.K.; Christie, G.E.; Dokland, T. A conformational switch involved in maturation of Staphylococcus aureus bacteriophage 80α capsids. J. Mol. Biol. 2011, 405, 863–876. [Google Scholar] [CrossRef]
- Effantin, G.; Figueroa-Bossi, N.; Schoehn, G.; Bossi, L.; Conway, J.F. The tripartite capsid gene of Salmonella phage Gifsy-2 yields a capsid assembly pathway engaging features from HK97 and λ. Virology 2010, 402, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Shen, P.S.; Domek, M.J.; Sanz-García, E.; Makaju, A.; Taylor, R.M.; Hoggan, R.; Culumber, M.D.; Oberg, C.J.; Breakwell, D.P.; Prince, J.T. Sequence and structural characterization of great salt lake bacteriophage CW02, a member of the T7-like supergroup. J. Virol. 2012, 86, 7907–7917. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Hodes, A.; Hui, W.H.; Gingery, M.; Miller, J.F.; Zhou, Z.H. Three-dimensional structure of tropism-switching Bordetella bacteriophage. Proc. Natl. Acad. Sci. USA 2010, 107, 4347–4352. [Google Scholar] [CrossRef] [PubMed]
- White, H.E.; Sherman, M.B.; Brasilès, S.; Jacquet, E.; Seavers, P.; Tavares, P.; Orlova, E. V Capsid Structure and Its Stability at the Late Stages of Bacteriophage SPP1 Assembly. J. Virol. 2012, 86, 6768–6777. [Google Scholar] [CrossRef]
- Grose, J.H.; Belnap, D.M.; Jensen, J.D.; Mathis, A.D.; Prince, J.T.; Merrill, B.D.; Burnett, S.H.; Breakwell, D.P. The Genomes, Proteomes, and Structures of Three Novel Phages That Infect the Bacillus cereus Group and Carry Putative Virulence Factors. J. Virol. 2014, 88, 11846–11860. [Google Scholar] [CrossRef]
- Lander, G.C.; Baudoux, A.; Azam, F.; Potter, C.S.; Carragher, B.; Johnson, J.E. Article Capsomer Dynamics and Stabilization in the T = 12 Marine Bacteriophage SIO-2 and Its Procapsid Studied by CryoEM. Struct. Des. 2012, 20, 498–503. [Google Scholar] [CrossRef][Green Version]
- Vernhes, E.; Renouard, M.; Gilquin, B.; Cuniasse, P.; Durand, D.; England, P.; Hoos, S.; Huet, A.; Conway, J.F.; Glukhov, A. High affinity anchoring of the decoration protein pb10 onto the bacteriophage T5 capsid. Sci. Rep. 2017, 7, 41662. [Google Scholar] [CrossRef]
- Lander, G.C.; Tang, L.; Casjens, S.R.; Gilcrease, E.B.; Prevelige, P.; Poliakov, A.; Potter, C.S.; Carragher, B.; Johnson, J.E. The structure of an infectious P22 virion shows the signal for headful DNA packaging. Science 2006, 312, 1791–1795. [Google Scholar] [CrossRef]
- Stroupe, M.E.; Brewer, T.E.; Sousa, D.R.; Jones, K.M. The structure of Sinorhizobium meliloti phage ΦM12, which has a novel T= 19l triangulation number and is the founder of a new group of T4-superfamily phages. Virology 2014, 450, 205–212. [Google Scholar] [CrossRef]
- Effantin, G.; Hamasaki, R.; Kawasaki, T.; Bacia, M.; Moriscot, C.; Weissenhorn, W.; Yamada, T.; Schoehn, G. Cryo-electron microscopy three-dimensional structure of the jumbo phage ΦRSL1 infecting the phytopathogen Ralstonia solanacearum. Structure 2013, 21, 298–305. [Google Scholar] [CrossRef]
- Fokine, A.; Kostyuchenko, V.; Efimov, A.V.; Kurochkina, L.P.; Sykilinda, N.N.; Robben, J.; Volckaert, G.; Hoenger, A.; Chipman, P.R.; Battisti, A.J.; et al. A three-dimensional cryo-electron microscopy structure of the bacteriophage φKZ head. J. Mol. Biol. 2005, 352, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Pietilä, M.; Laurinmäki, P.; Russell, D.A.; Ching-Chung, K.; Jacobs-Sera, D.; Hendrix, R.W.; Bamford, D.H.; Butcher, S.J. Structure of the archaeal head-tailed virus HSTV-1 completes the HK97 fold story. Proc. Natl. Acad. Sci. USA 2013, 110, 10604–10609. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Liu, Z.; Fang, P.-A.; Zhang, Q.; Wright, E.T.; Wu, W.; Zhang, C.; Vago, F.; Ren, Y.; Jakana, J. Capsid expansion mechanism of bacteriophage T7 revealed by multistate atomic models derived from cryo-EM reconstructions. Proc. Natl. Acad. Sci. USA 2014, 111, E4606–E4614. [Google Scholar] [CrossRef] [PubMed]
- Parent, K.N.; Gilcrease, E.B.; Casjens, S.R.; Baker, T.S. Structural evolution of the P22-like phages: Comparison of Sf6 and P22 procapsid and virion architectures. Virology 2012, 427, 177–188. [Google Scholar] [CrossRef]
- Baker, M.L.; Hryc, C.F.; Zhang, Q.; Wu, W.; Jakana, J.; Haase-Pettingell, C.; Afonine, P.V.; Adams, P.D.; King, J.; Jiang, W.; et al. Validated near-atomic resolution structure of bacteriophage epsilon15 derived from cryo-EM and modeling. Proc. Natl. Acad. Sci. USA 2013, 110, 12301–12306. [Google Scholar] [CrossRef]
- Gipson, P.; Baker, M.L.; Raytcheva, D.; Haase-Pettingell, C.; Piret, J.; King, J.A.; Chiu, W. Protruding knob-like proteins violate local symmetries in an icosahedral marine virus. Nat. Commun. 2014, 5, 4278. [Google Scholar] [CrossRef][Green Version]
- Leiman, P.G.; Battisti, A.J.; Bowman, V.D.; Stummeyer, K.; Mühlenhoff, M.; Gerardy-Schahn, R.; Scholl, D.; Molineux, I.J. The structures of bacteriophages K1E and K1-5 explain processive degradation of polysaccharide capsules and evolution of new host specificities. J. Mol. Biol. 2007, 371, 836–849. [Google Scholar] [CrossRef]
- Lander, G.C.; Evilevitch, A.; Jeembaeva, M.; Potter, C.S.; Carragher, B.; Johnson, J.E. Bacteriophage lambda stabilization by auxiliary protein gpD: Timing, location, and mechanism of attachment determined by cryo-EM. Structure 2008, 16, 1399–1406. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Kutner, M.H.; Neter, J.; Nachtsheim, C.J.; Li, W. Applied Linear Statistical Models, 5th ed.; McGraw-Hill Education: New York, NY, USA, 2004. [Google Scholar]
- James, G.; Witten, D.; Hastie, T.; Tibshirani, R. An Introduction to Statistical Learning, 7th ed.; Springer: New York, NY, USA, 2017. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef]
- Benler, S.; Yutin, N.; Antipov, D.; Raykov, M.; Shmakov, S.A.; Gussow, A.B.; Pevzner, P.A.; Koonin, E. V Thousands of previously unknown phages discovered in whole-community human gut metagenomes. bioRxiv 2020. [Google Scholar] [CrossRef]
- Aznar, M.; Luque, A.; Reguera, D. Relevance of capsid structure in the buckling and maturation of spherical viruses. Phys. Biol. 2012, 9, 036003. [Google Scholar] [CrossRef] [PubMed]
- Dearborn, A.D.; Laurinmaki, P.; Chandramouli, P.; Rodenburg, C.M.; Wang, S.; Butcher, S.J.; Dokland, T. Structure and size determination of bacteriophage P2 and P4 procapsids: Function of size responsiveness mutations. J. Struct. Biol. 2012, 178, 215–224. [Google Scholar] [CrossRef] [PubMed]
- McNair, K.; Zhou, C.; Dinsdale, E.A.; Souza, B.; Edwards, R.A. PHANOTATE: A novel approach to gene identification in phage genomes. Bioinformatics 2019, 35, 4537–4542. [Google Scholar] [CrossRef]
- Dutilh, B.E.; Cassman, N.; McNair, K.; Sanchez, S.E.; Silva, G.G.Z.; Boling, L.; Barr, J.J.; Speth, D.R.; Seguritan, V.; Aziz, R.K. A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat. Commun. 2014, 5, 1–11. [Google Scholar] [CrossRef]
- Edwards, R.A.; Vega, A.A.; Norman, H.M.; Ohaeri, M.; Levi, K.; Dinsdale, E.A.; Cinek, O.; Aziz, R.K.; McNair, K.; Barr, J.J. Global phylogeography and ancient evolution of the widespread human gut virus crAssphage. Nat. Microbiol. 2019, 4, 1727–1736. [Google Scholar] [CrossRef]
- Shkoporov, A.N.; Khokhlova, E.V.; Fitzgerald, C.B.; Stockdale, S.R.; Draper, L.A.; Ross, R.P.; Hill, C. ΦCrAss001 represents the most abundant bacteriophage family in the human gut and infects Bacteroides intestinalis. Nat. Commun. 2018, 9, 1–8. [Google Scholar] [CrossRef]
- Guerin, E.; Shkoporov, A.; Stockdale, S.R.; Clooney, A.G.; Ryan, F.J.; Sutton, T.D.S.; Draper, L.A.; Gonzalez-Tortuero, E.; Ross, R.P.; Hill, C. Biology and taxonomy of crAss-like bacteriophages, the most abundant virus in the human gut. Cell Host Microbe 2018, 24, 653–664. [Google Scholar] [CrossRef]
- Yutin, N.; Makarova, K.S.; Gussow, A.B.; Krupovic, M.; Segall, A.; Edwards, R.A.; Koonin, E. V Discovery of an expansive bacteriophage family that includes the most abundant viruses from the human gut. Nat. Microbiol. 2018, 3, 38–46. [Google Scholar] [CrossRef]
- Sivanandam, V.; Mathews, D.; Garmann, R.; Erdemci-Tandogan, G.; Zandi, R.; Rao, A.L.N. Functional analysis of the N-terminal basic motif of a eukaryotic satellite RNA virus capsid protein in replication and packaging. Sci. Rep. 2016, 6, 26328. [Google Scholar] [CrossRef]
- Beren, C.; Cui, Y.; Chakravarty, A.; Yang, X.; Rao, A.L.N.; Knobler, C.M.; Zhou, Z.H.; Gelbart, W.M. Genome organization and interaction with capsid protein in a multipartite RNA virus. Proc. Natl. Acad. Sci. USA 2020, 117, 10673–10680. [Google Scholar] [CrossRef] [PubMed]
- Carrillo-Tripp, M.; Shepherd, C.M.; Borelli, I.A.; Venkataraman, S.; Lander, G.; Natarajan, P.; Johnson, J.E.; Brooks III, C.L.; Reddy, V.S. VIPERdb2: An enhanced and web API enabled relational database for structural virology. Nucleic Acids Res. 2008, 37, D436–D442. [Google Scholar] [CrossRef]
- Hulo, C.; De Castro, E.; Masson, P.; Bougueleret, L.; Bairoch, A.; Xenarios, I.; Le Mercier, P. ViralZone: A knowledge resource to understand virus diversity. Nucleic Acids Res. 2011, 39, D576–D582. [Google Scholar] [CrossRef]
- Daufresne, M.; Lengfellner, K.; Som-mer, U. Global warming benefits the small in aquatic ecosystems, 12788-12793. Proc. Natl. Acad. Sci. USA 2009, 106, 21. [Google Scholar] [CrossRef]
- Morán, X.A.G.; López-Urrutia, Á.; Calvo-Díaz, A.; Li, W.K.W. Increasing importance of small phytoplankton in a warmer ocean. Glob. Chang. Biol. 2010, 16, 1137–1144. [Google Scholar] [CrossRef]
- Nifong, R.L.; Gillooly, J.F. Temperature effects on virion volume and genome length in dsDNA viruses. Biol. Lett. 2016, 12, 20160023. [Google Scholar] [CrossRef] [PubMed]
- Schooley, R.T.; Biswas, B.; Gill, J.J.; Hernandez-Morales, A.; Lancaster, J.; Lessor, L.; Barr, J.J.; Reed, S.L.; Rohwer, F.; Benler, S. Development and use of personalized bacteriophage-based therapeutic cocktails to treat a patient with a disseminated resistant Acinetobacter baumannii infection. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef]
- Hatfull, G.F. Actinobacteriophages: Genomics, Dynamics, and Applications. Annu. Rev. Virol. 2020, 7, 37–61. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property | Range | Property | Range |
|---|---|---|---|
| Capsids analyzed | 23 | Genome size | 17–280 kbp |
| T-number | 4–27 | Genome density | 0.34–0.62 bp/nm3 |
| Interior sphericity * | 0.25–0.75 | Interior surface | 5.08 × 103–4.32 × 104 nm2 |
| Exterior sphericity * | 0.14–0.55 | MCP interior area | 19–26 nm2 |
| Capsid diameter † | 49–143 nm | Exterior surface | 6.55 × 103–5.19 × 104 nm2 |
| Capsid thickness | 3–8 nm | MCP exterior area | 24–35 nm2 |
| Interior volume | 3.36 × 104–8.22 × 105 nm3 | MCP ratio (%) | 15–43% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luque, A.; Benler, S.; Lee, D.Y.; Brown, C.; White, S. The Missing Tailed Phages: Prediction of Small Capsid Candidates. Microorganisms 2020, 8, 1944. https://doi.org/10.3390/microorganisms8121944
Luque A, Benler S, Lee DY, Brown C, White S. The Missing Tailed Phages: Prediction of Small Capsid Candidates. Microorganisms. 2020; 8(12):1944. https://doi.org/10.3390/microorganisms8121944
Chicago/Turabian StyleLuque, Antoni, Sean Benler, Diana Y. Lee, Colin Brown, and Simon White. 2020. "The Missing Tailed Phages: Prediction of Small Capsid Candidates" Microorganisms 8, no. 12: 1944. https://doi.org/10.3390/microorganisms8121944
APA StyleLuque, A., Benler, S., Lee, D. Y., Brown, C., & White, S. (2020). The Missing Tailed Phages: Prediction of Small Capsid Candidates. Microorganisms, 8(12), 1944. https://doi.org/10.3390/microorganisms8121944