The Microbiota Profile in Inflamed and Non-Inflamed Ileal Pouch–Anal Anastomosis

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Trial Design
2.2. Participants
2.3. Sample Preparation
2.4. Data Analysis
2.5. Ethics
3. Results
3.1. Patient Population
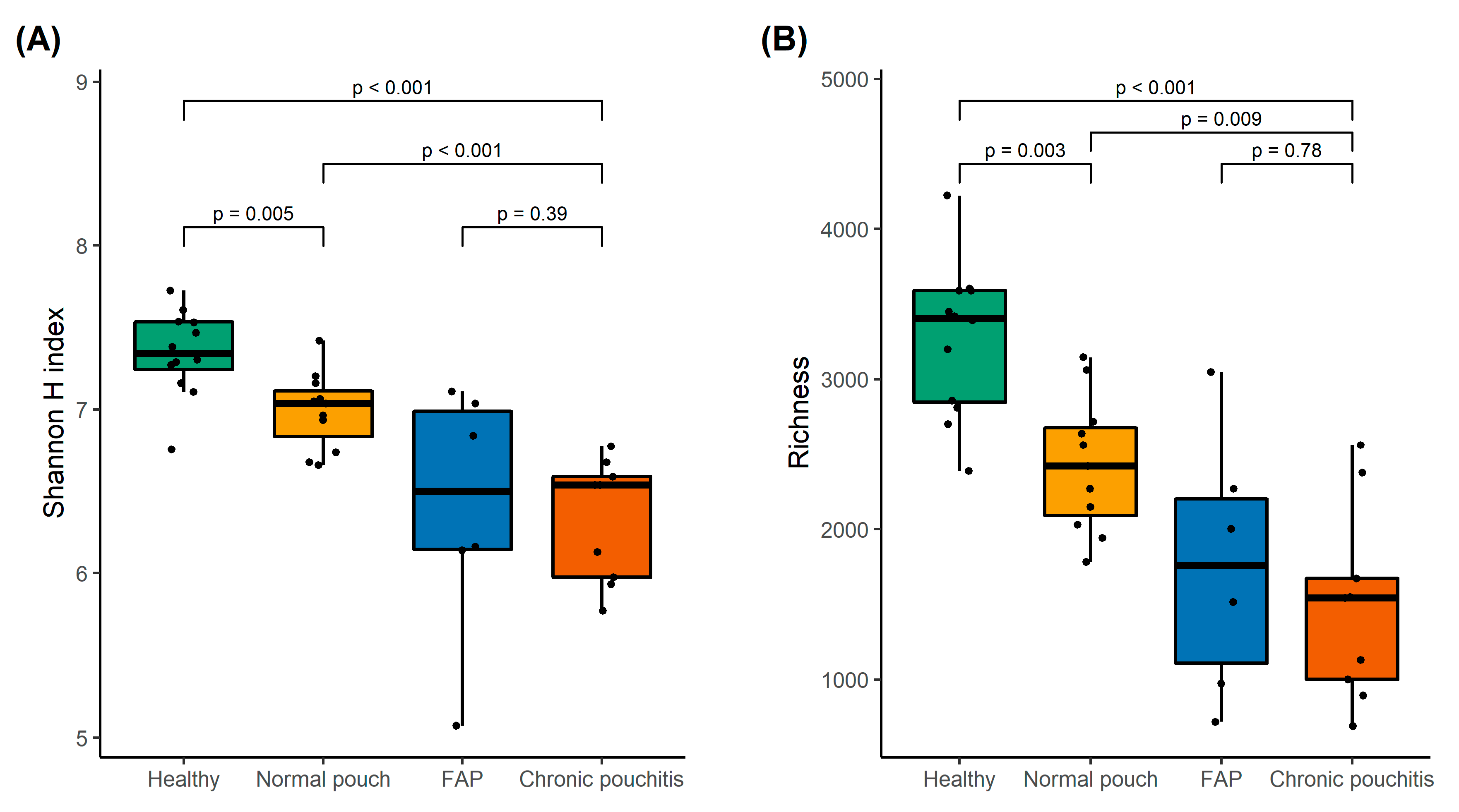
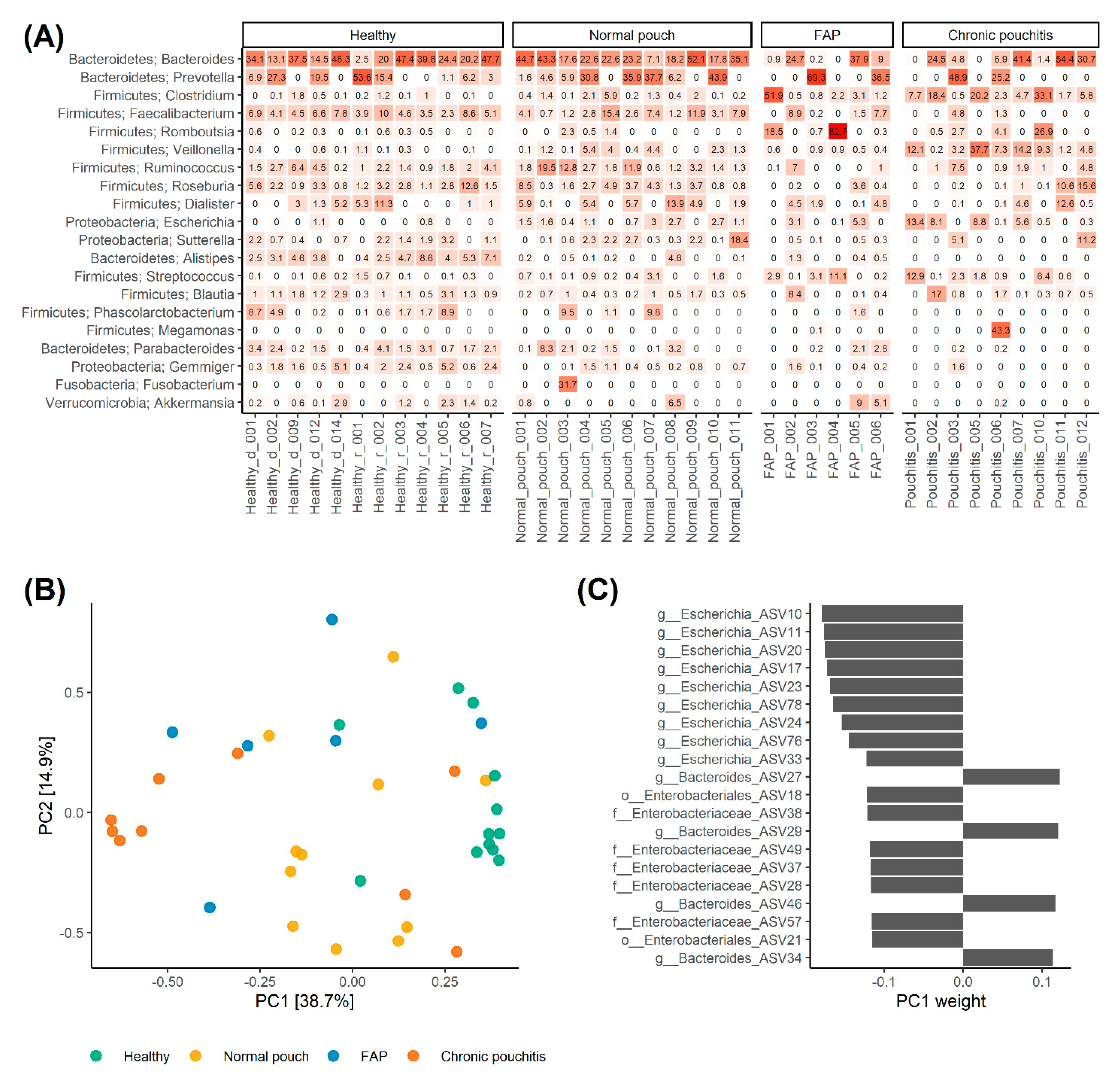
3.2. Analysis of Gut Microbiota
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hata, K.; Ishihara, S.; Nozawa, H.; Kawai, K.; Kiyomatsu, T.; Tanaka, T.; Kishikawa, J.; Anzai, H.; Watanabe, T. Pouchitis after ileal pouch-anal anastomosis in ulcerative colitis: Diagnosis, management, risk factors, and incidence. Dig. Endosc. 2017, 29, 26–34. [Google Scholar] [CrossRef]
- Fazio, V.; Kiran, R.; Remzi, F.; Coffey, J.; Heneghan, H.; Kirat, H.; Manilich, E.; Shen, B.; Martin, S. Ileal pouch anal anastomosis: Analysis of outcome and quality of life in 3707 patients. Ann. Surg. 2013, 257, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W. Pouchitis following ileal pouch-anal anastomosis: Definition, pathogenesis, and treatment. Gastroenterology 1994, 107, 1856–1860. [Google Scholar] [CrossRef]
- Shen, B. Acute and chronic pouchitis--pathogenesis, diagnosis and treatment. Nat. Rev. Gastroenterol. Hepatol. 2012, 17, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Segal, J.; Ding, N.; Worley, G.; Mclaughlin, S.; Preston, S.; Faiz, O.; Clark, S.; Hart, A. Systematic review with meta-analysis: The management of chronic refractory pouchitis with an evidence-based treatment algorithm. Aliment. Pharmacol. Ther. 2017, 45, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.; Zhang, B.; Holubar, S.D.; Pardi, D.S.; Singh, S. Treatment and prevention of pouchitis after ileal pouch-anal anastomosis for chronic ulcerative colitis. Cochrane Database Syst. Rev. 2019, 5, CD001176. [Google Scholar] [CrossRef]
- Shen, B.; Achkar, J.; Lashner, B.; Ormsby, A.; Remzi, F.; Bevins, C.; Brzezinski, A.; Petras, R.; Fazio, V. Endoscopic and histologic evaluation together with symptom assessment are required to diagnose pouchitis. Gastroenterology 2001, 121, 261–267. [Google Scholar] [CrossRef]
- Mimura, T.; Rizzello, F.; Helwig, U.; Poggioli, G.; Schreiber, S.; Talbot, I.; Nicholls, R.; Gionchetti, P.; Campieri, M.; Kamm, M. Once daily high dose probiotic therapy (VSL#3) for maintaining remission in recurrent or refractory pouchitis. Gut 2004, 53, 108–114. [Google Scholar] [CrossRef]
- Stallmach, A.; Lange, K.; Buening, J.; Sina, C.; Vital, M.; Pieper, D.H. Fecal Microbiota Transfer in Patients With Chronic Antibiotic-Refractory Pouchitis. Am. J. Gastroenterol. 2016, 111, 441–443. [Google Scholar] [CrossRef]
- Marchesi, J.; Adams, D.; Fava, F.; Hermes, G.; Hirschfield, G.; Hold, G.; Quraishi, M.; Kinross, J.; Smidt, H.; Tuohy, K.; et al. The gut microbiota and host health: A new clinical frontier. Gut 2016, 65, 330–339. [Google Scholar] [CrossRef]
- Ni, J.; Wu, G.; Albenberg, L.; Tomov, V. Gut microbiota and IBD: Causation or correlation? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Suau, A.; Bonnet, R.; Sutren, M.; Godon, J.; Gibson, G.; Collins, M.; Doré, J. Direct analysis of genes encoding 16S rRNA from complex communities reveals many novel molecular species within the human gut. Appl. Environ. Microbiol. 1999, 65, 4799–4807. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.; Raes, J.; Bork, P. The Human Gut Microbiome: From Association to Modulation. Cell 2018, 172, 1198–1215. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Lashner, B. Diagnosis and treatment of pouchitis. Gastroenterol. Hepatol. 2008, 4, 355–361. [Google Scholar]
- Kousgaard, S.J.; Michaelsen, T.Y.; Nielsen, H.L.; Kirk, K.F.; Brandt, J.; Albertsen, M.; Thorlacius-Ussing, O. Clinical results and microbiota changes after faecal microbiota transplantation for chronic pouchitis: A pilot study. Scand. J. Gastroenterol. 2020, 55, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Parada, A.E.; Needham, D.M.; Fuhrman, J.A. Every base matters: Assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 2016, 18, 1403–1414. [Google Scholar] [CrossRef]
- Apprill, A.; Mcnally, S.; Parsons, R.; Weber, L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol. 2015, 75, 129–137. [Google Scholar] [CrossRef]
- Edgar, R. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Edgar, R. SINTAX: A simple non-Bayesian taxonomy classifier for 16S and ITS sequences. bioRxiv 2016. Available online: https://www.biorxiv.org/content/10.1101/074161v1 (accessed on 20 September 2020).
- Callahan, B.; McMurdie, P.; Holmes, S. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 2017, 11, 2639–2643. [Google Scholar] [CrossRef]
- Edgar, R.; Flyvbjerg, H. Error filtering, pair assembly and error correction for next-generation sequencing reads. Bioinformatics 2015, 31, 3476–3482. [Google Scholar] [CrossRef]
- DeSantis, T.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef]
- Edgar, R. UNOISE2: Improved error-correction for Illumina 16S and ITS amplicon sequencing. bioRxiv 2016. Available online: https://www.biorxiv.org/content/biorxiv/early/2016 (accessed on 20 September 2020).
- Andersen, K.; Kirkegaard, R.; Karst, S.; Albertsen, M. ampvis2: An R package to analyse and visualise 16S rRNA amplicon data. bioRxiv 2018. Available online: https://www.biorxiv.org/content/10.1101/299537v1.a (accessed on 20 September 2020).
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.D.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse 2019. J. Open Source Sofeware 2019, 4, 1686. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’hara, R.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. R Package Version 2.4-3 2016, Vienna: R Foundation for Statistical Computing. Available online: https://www.researchgate.net/publication/282247686_Vegan_Community_Ecology_Package_R_package_version_20-2 (accessed on 20 September 2020).
- Hervé, M. RVAideMemoire: Testing and Plotting Procedures for Biostatistics. R Package Version 0.9-78. 2020. Available online: https://cran.r-project.org/web/packages/RVAideMemoire/RVAideMemoire.pdf (accessed on 20 September 2020).
- Holm, S. A Simple Sequentially Rejective Multiple Test Procedure. Scand. J. Stat. 1979, 6, 65–70. [Google Scholar]
- Landy, J.; Walker, A.W.; Li, J.V.; Al-Hassi, H.O.; Ronde, E.; English, N.R.; Mann, E.R.; Bernardo, D.; McLaughlin, S.D.; Parkhill, J.; et al. Variable alterations of the microbiota, without metabolic or immunological change, following faecal microbiota transplantation in patients with chronic pouchitis. Sci. Rep. 2015, 5, 12955. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.J.; Lim, J.H.; Kang, D.; Lim, S.; Park, S.J.; Kim, J.H. Is stool frequency associated with the richness and community composition of gut microbiota? Intest. Res. 2019, 17, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Petersen, A.M.; Mirsepasi-Lauridsen, H.C.; Vester-Andersen, M.K.; Sørensen, N.; Krogfelt, K.A.; Bendtsen, F. High abundance of proteobacteria in ileo-anal pouch anastomosis and increased abundance of fusobacteria associated with increased pouch inflammation. Antibiotics 2020, 9, 237. [Google Scholar] [CrossRef]
- Pawełka, D.; Bednarz, W.; Krawczyk, Z.; Rzeszutko, M.; Olewiński, R.; Czopnik, P. Ileal pouch morphology and microbiology in ulcerative colitis patients. Adv. Clin. Exp. Med. 2015, 24, 267–274. [Google Scholar] [CrossRef]
- Tannock, G.W.; Lawley, B.; Munro, K.; Lay, C.; Taylor, C.; Daynes, C.; Baladjay, L.; Mcleod, R.; Thompson-Fawcett, M. Comprehensive analysis of the bacterial content of stool from patients with chronic pouchitis, normal pouches, or familial adenomatous polyposis pouches. Inflamm. Bowel Dis. 2012, 18, 925–934. [Google Scholar] [CrossRef]
- Mumolo, M.G.; Bertani, L.; Ceccarelli, L.; Laino, G.; Fluri, G.D.; Albano, E.; Tapete, G.; Costa, F. From bench to bedside: Fecal calprotectin in inflammatory bowel diseases clinical setting. World J. Gastroenterol. 2018, 24, 3681–3694. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, R.; Rani, A.; Metwally, A.; McGee, H.S.; Perkins, D.L. Analysis of the microbiome: Advantages of whole genome shotgun versus 16S amplicon sequencing. Biochem. Biophys. Res. Commun. 2016, 469, 967–977. [Google Scholar] [CrossRef] [PubMed]
- Dueholm, M.S.; Andersen, K.S.; Petriglieri, F.; McIlroy, S.J.; Nierychlo, M.; Petersen, J.F.; Kristensen, J.M.; Yashiro, E.; Karst, S.M.; Albertsen, M.; et al. Comprehensive ecosystem-specific 16S rRNA gene databases with automated taxonomy assignment (AutoTax) provide species-level resolution in microbial ecology. bioRxiv 2019, 672873. [Google Scholar] [CrossRef]
- Johnson, J.S.; Spakowicz, D.J.; Hong, B.Y.; Petersen, L.M.; Demkowicz, P.; Chen, L.; Leopold, S.R.; Hanson, B.M.; Agresta, H.O.; Gerstein, M.; et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat. Commun. 2019, 10, 5029. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Group | Normal Pouch (n = 11) | Chronic Pouchitis (n = 9) | FAP (n = 6) | HCs (n = 12) |
|---|---|---|---|---|
| Age mean (SD) | 47.1 (11.0) | 52.9 (13.7) | 54.8 (16.3) | 42.3 (13.9) |
| Male n (%) | 7 (64) | 3 (33) | 1 (17) | 8 (69) |
| Years since surgery mean (range) | 12.9 (5–21) | 17.6 (8–28) | 14.2 (3–30) | - |
| Daily bowel movements mean (range) | 5.5 (3–8) | 11.2 (5–20) | 5.5 (1–8) | 1.2 (1–2) |
| cPDAI score mean (range) | 0.6 (0–1) | 3.7 (3–5) | 1.0 (1–1) | - |
| Continues antibiotics n (%) | 0 (0) | 3 (33) | 0 (0) | 0 (0) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kousgaard, S.J.; Michaelsen, T.Y.; Nielsen, H.L.; Kirk, K.F.; Albertsen, M.; Thorlacius-Ussing, O. The Microbiota Profile in Inflamed and Non-Inflamed Ileal Pouch–Anal Anastomosis. Microorganisms 2020, 8, 1611. https://doi.org/10.3390/microorganisms8101611
Kousgaard SJ, Michaelsen TY, Nielsen HL, Kirk KF, Albertsen M, Thorlacius-Ussing O. The Microbiota Profile in Inflamed and Non-Inflamed Ileal Pouch–Anal Anastomosis. Microorganisms. 2020; 8(10):1611. https://doi.org/10.3390/microorganisms8101611
Chicago/Turabian StyleKousgaard, Sabrina Just, Thomas Yssing Michaelsen, Hans Linde Nielsen, Karina Frahm Kirk, Mads Albertsen, and Ole Thorlacius-Ussing. 2020. "The Microbiota Profile in Inflamed and Non-Inflamed Ileal Pouch–Anal Anastomosis" Microorganisms 8, no. 10: 1611. https://doi.org/10.3390/microorganisms8101611
APA StyleKousgaard, S. J., Michaelsen, T. Y., Nielsen, H. L., Kirk, K. F., Albertsen, M., & Thorlacius-Ussing, O. (2020). The Microbiota Profile in Inflamed and Non-Inflamed Ileal Pouch–Anal Anastomosis. Microorganisms, 8(10), 1611. https://doi.org/10.3390/microorganisms8101611