Role of Infections in the Pathogenesis of Rheumatoid Arthritis: Focus on Mycobacteria

 , , , ,
, , , , 
Abstract
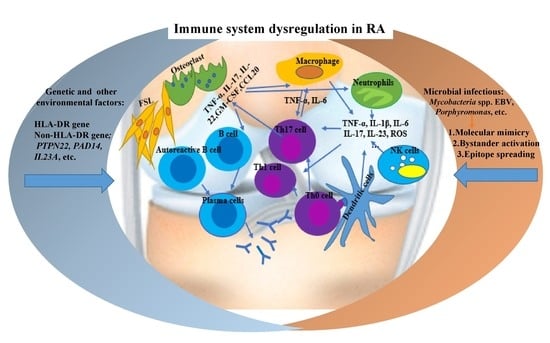
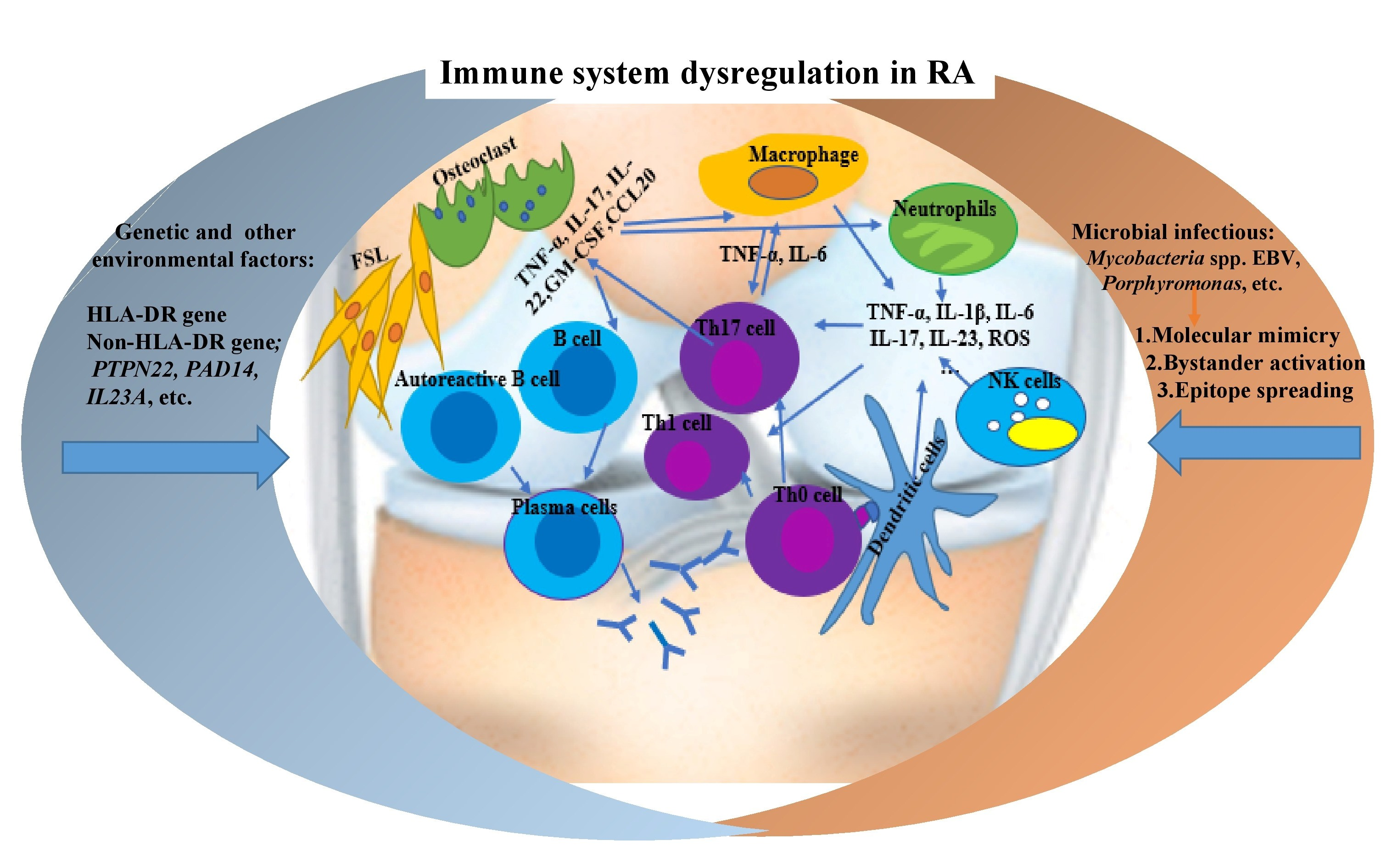
1. Introduction
2. RA Immunopathogenesis
3. The Role of Mycobacterial Infections in Rheumatoid Arthritis
4. Other Infections Associated with RA
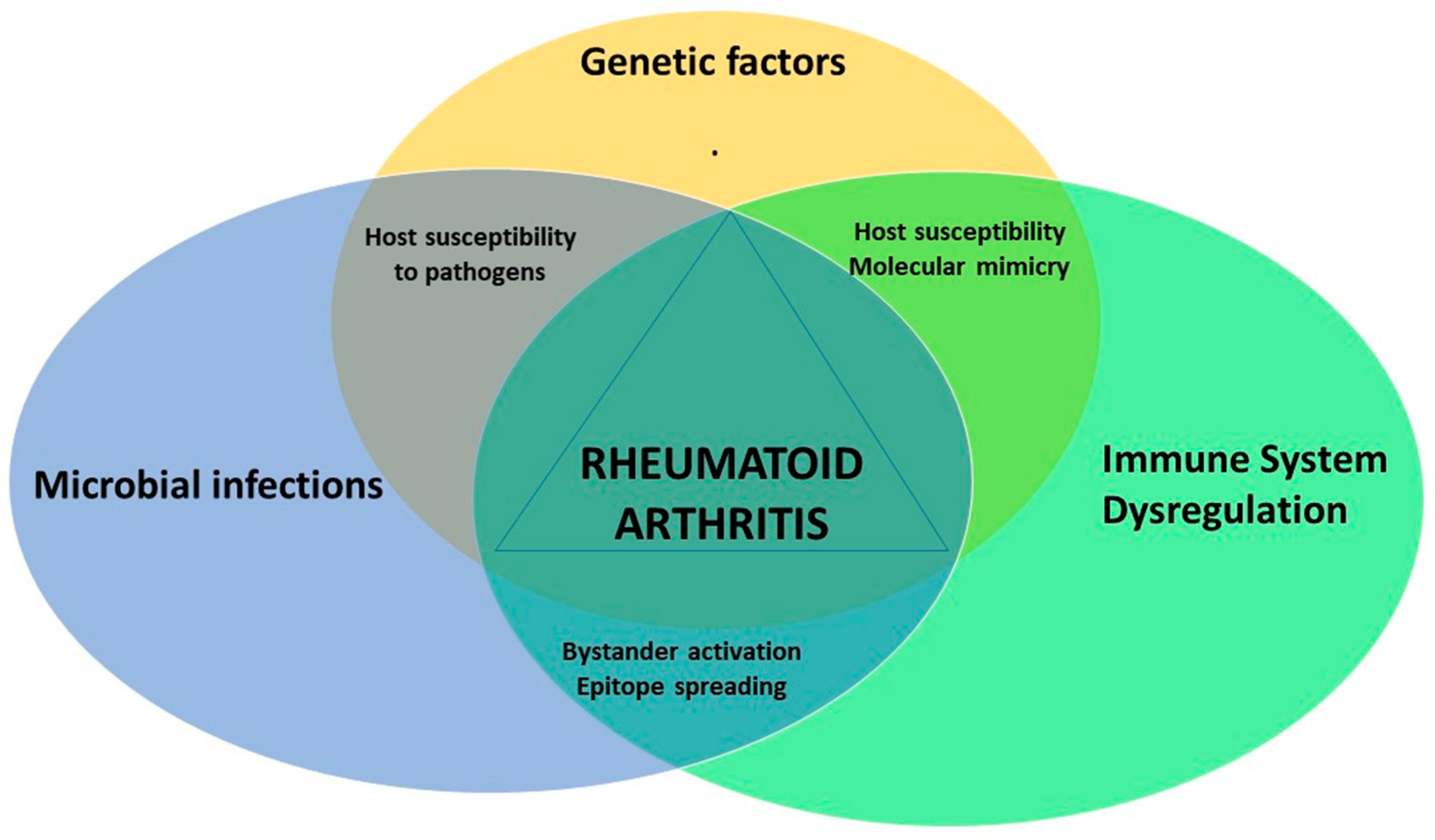
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Simon, T.A.; Kawabata, H.; Ray, N.; Baheti, A.; Suissa, S.; Esdaile, J.M. Prevalence of co-existing autoimmune disease in rheumatoid arthritis: A cross-sectional study. Adv. Ther. 2017, 34, 2481–2490. [Google Scholar] [CrossRef]
- Alivernini, S.; Tolusso, B.; Petricca, L.; Ferraccioli, G.; Gremese, E. Chapter 46—Rheumatoid arthritis. In Mosaic of Autoimmunity; Elsevier: Amsterdam, The Netherlands, 2019; pp. 501–526. [Google Scholar]
- de Brito Rocha, S.; Baldo, D.C.; Andrade, L.E.C. Clinical and pathophysiologic relevance of autoantibodies in rheumatoid arthritis. Adv. Rheumatol. 2019, 59, 2. [Google Scholar] [CrossRef]
- Huizinga, T.W.; Pincus, T. Rheumatoid arthritis. Ann. Intern. Med. 2010, 153, ITC1-1. [Google Scholar] [CrossRef]
- Grassi, W.; De Angelis, R.; Lamanna, G.; Cervini, C. The clinical featuRes. of rheumatoid arthritis. Eur. J. Radiol. 1998, 27, S18–S24. [Google Scholar] [CrossRef]
- van Delft, M.A.M.; Huizinga, T.W.J. An overview of autoantibodies in rheumatoid arthritis. J. Autoimmun. 2020, 110, 102392. [Google Scholar] [CrossRef]
- Ma, X.; Xu, S. TNF inhibitor therapy for rheumatoid arthritis. Biomed. Rep. 2013, 1, 177–184. [Google Scholar] [CrossRef]
- Croia, C.; Bursi, R.; Sutera, D.; Petrelli, F.; Alunno, A.; Puxeddu, I. One year in review 2019: Pathogenesis of rheumatoid arthritis. Clin. Exp. Rheumatol. 2019, 37, 347–357. [Google Scholar]
- Hussein, H.M.; Rahal, E.A. The role of viral infections in the development of autoimmune diseases. Crit Rev. Microbiol. 2019, 45, 394–412. [Google Scholar] [CrossRef]
- Atkin, S.L.; Welbury, R.R.; Stanfield, E.; Beavis, D.; Iwais, B.; Dick, W.C. Clinical and laboratory studies of inflammatory polyarthritis in patients with leprosy in Papua New Guinea. Ann. Rheum. Dis. 1987, 46, 688–690. [Google Scholar] [CrossRef]
- Rook, G.A. Rheumatoid arthritis, mycobacterial antigens and agalactosyl IgG. Scand. J. Immunol. 1988, 28, 487–493. [Google Scholar] [CrossRef]
- Shoenfeld, Y.; Isenberg, D.A. Mycobacteria and autoimmunity. Immunol. Today 1988, 9, 178–182. [Google Scholar] [CrossRef]
- Liao, T.L.; Lin, C.H.; Shen, G.H.; Chang, C.L.; Lin, C.F.; Chen, D.Y. Risk for mycobacterial disease among patients with rheumatoid arthritis, Taiwan, 2001–2011. Emerg. Infect. Dis. 2015, 21, 1387–1395. [Google Scholar] [CrossRef]
- Listing, J.; Gerhold, K.; Zink, A. The risk of infections associated with rheumatoid arthritis, with its comorbidity and treatment. Rheumatology 2012, 52, 53–61. [Google Scholar] [CrossRef]
- Mehta, B.; Pedro, S.; Ozen, G.; Kalil, A.; Wolfe, F.; Mikuls, T.; Michaud, K. Serious infection risk in rheumatoid arthritis compared with non-inflammatory rheumatic and musculoskeletal diseases: A US national cohort study. RMD Open 2019, 5, e000935. [Google Scholar] [CrossRef]
- McInnes, I.B.; Schett, G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat. Rev. Immunol. 2007, 7, 429–442. [Google Scholar] [CrossRef]
- Brennan, F.M.; McInnes, I.B. Evidence that cytokines play a role in rheumatoid arthritis. J. Clin. Investig. 2008, 118, 3537–3545. [Google Scholar] [CrossRef]
- Coutant, F.; Miossec, P. Evolving concepts of the pathogenesis of rheumatoid arthritis with focus on the early and late stages. Curr. Opin. Rheumatol. 2020, 32, 57–63. [Google Scholar] [CrossRef]
- Karami, J.; Aslani, S.; Jamshidi, A.; Garshasbi, M.; Mahmoudi, M. Genetic implications in the pathogenesis of rheumatoid arthritis; an updated review. Gene 2019, 702, 8–16. [Google Scholar] [CrossRef]
- Lee, J.C.; Espéli, M.; Anderson, C.A.; Linterman, M.A.; Pocock, J.M.; Williams, N.J.; Roberts, R.; Viatte, S.; Fu, B.; Peshu, N.; et al. Human SNP links differential outcomes in inflammatory and infectious disease to a FOXO3-regulated pathway. Cell 2013, 155, 57–69. [Google Scholar] [CrossRef]
- Gregersen, P.K.; Silver, J.; Winchester, R.J. The shared epitope hypothesis. an approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987, 30, 1205–1213. [Google Scholar] [CrossRef]
- van der Helm-van Mil, A.H.M.; Verpoort, K.N.; le Cessie, S.; Huizinga, T.W.J.; de Vries, R.R.P.; Toes, R.E.M. The HLA–DRB1 shared epitope alleles differ in the interaction with smoking and predisposition to antibodies to cyclic citrullinated peptide. Arthritis Rheum. 2007, 56, 425–432. [Google Scholar] [CrossRef]
- Albani, S.; Keystone, E.C.; Nelson, J.L.; Ollier, W.E.; La Cava, A.; Montemayor, A.C.; Weber, D.A.; Montecucco, C.; Martini, A.; Carson, D.A. Positive selection in autoimmunity: Abnormal immune responses to a bacterial dnaJ antigenic determinant in patients with early rheumatoid arthritis. Nat. Med. 1995, 1, 448–452. [Google Scholar] [CrossRef]
- Bax, M.; van Heemst, J.; Huizinga, T.W.J.; Toes, R.E.M. Genetics of rheumatoid arthritis: What have we learned? Immunogenetics 2011, 63, 459–466. [Google Scholar] [CrossRef]
- Cha, S.; Choi, C.-B.; Han, T.-U.; Kang, C.P.; Kang, C.; Bae, S.-C. Association of Anti–Cyclic citrullinated peptide antibody levels with PADI4 haplotypes in early rheumatoid arthritis and with shared epitope alleles in very late rheumatoid arthritis. Arthritis Rheum. 2007, 56, 1454–1463. [Google Scholar] [CrossRef]
- Faragó, B.; Magyari, L.; Sáfrány, E.; Csöngei, V.; Járomi, L.; Horvatovich, K.; Sipeky, C.; Maász, A.; Radics, J.; Gyetvai, Á.; et al. Functional variants of interleukin-23 receptor gene confer risk for rheumatoid arthritis but not for systemic sclerosis. Ann. Rheum. Dis. 2008, 67, 248–250. [Google Scholar] [CrossRef]
- Farago, B.; Talian, G.C.; Komlosi, K.; Nagy, G.; Berki, T.; Gyetvai, A.; Szekanecz, Z.; Nyarady, Z.; Kiss, C.G.; Nemeth, P.; et al. Protein tyrosine phosphatase gene C1858T allele confers risk for rheumatoid arthritis in Hungarian subjects. Rheumatol. Int. 2009, 29, 793–796. [Google Scholar] [CrossRef]
- Alivernini, S.; Tolusso, B.; Petricca, L.; Ferraccioli, G.; Gremese, E. Chapter 16—Rheumatoid arthritis. In Mosaic of Autoimmunity; Elsevier: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Klareskog, L.; Catrina, A.I.; Paget, S. Rheumatoid arthritis. Lancet 2009, 373, 659–672. [Google Scholar] [CrossRef]
- McInnes, I.B.; Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef]
- Farrugia, M.; Baron, B. The role of TNF-α in rheumatoid arthritis: A focus on regulatory T cells. J. Clin. Transl. Res. 2016, 2, 84–90. [Google Scholar] [CrossRef]
- Calabresi, E.; Petrelli, F.; Bonifacio, A.F.; Puxeddu, I.; Alunno, A. One year in review 2018: Pathogenesis of rheumatoid arthritis. Clin. Exp. Rheumatol. 2018, 36, 175–184. [Google Scholar]
- Huang, Q.Q.; Pope, R.M. The role of toll-like receptors in rheumatoid arthritis. Curr. Rheumatol. Rep. 2009, 11, 357–364. [Google Scholar] [CrossRef]
- Elshabrawy, H.A.; Essani, A.E.; Szekanecz, Z.; Fox, D.A.; Shahrara, S. TLRs, future potential therapeutic targets for RA. AutoImmun. Rev. 2017, 16, 103–113. [Google Scholar] [CrossRef]
- Ospelt, C.; Brentano, F.; Rengel, Y.; Stanczyk, J.; Kolling, C.; Tak, P.P.; Gay, R.E.; Gay, S.; Kyburz, D. Overexpression of toll-like receptors 3 and 4 in synovial tissue from patients with early rheumatoid arthritis: Toll-like receptor expression in early and longstanding arthritis. Arthritis Rheum. 2008, 58, 3684–3692. [Google Scholar] [CrossRef]
- Huang, Q.; Ma, Y.; Adebayo, A.; Pope, R.M. Increased macrophage activation mediated through toll-like receptors in rheumatoid arthritis. Arthritis Rheum. 2007, 56, 2192–2201. [Google Scholar] [CrossRef]
- Kowalski, M.L.; Wolska, A.; Grzegorczyk, J.; Hilt, J.; Jarzebska, M.; Drobniewski, M.; Synder, M.; Kurowski, M. Increased responsiveness to toll-like receptor 4 stimulation in peripheral blood mononuclear cells from patients with recent onset rheumatoid arthritis. Mediat. Inflamm. 2008, 2008, 132732. [Google Scholar] [CrossRef]
- Sacre, S.M.; Lo, A.; Gregory, B.; Simmonds, R.E.; Williams, L.; Feldmann, M.; Brennan, F.M.; Foxwell, B.M. Inhibitors of TLR8 reduce TNF production from human rheumatoid synovial membrane cultures. J. Immunol. 2008, 181, 8002–8009. [Google Scholar] [CrossRef]
- Doorenspleet, M.E.; Klarenbeek, P.L.; de Hair, M.J.; van Schaik, B.D.; Esveldt, R.E.; van Kampen, A.H.; Gerlag, D.M.; Musters, A.; Baas, F.; Tak, P.P.; et al. Rheumatoid arthritis synovial tissue harbours dominant B-cell and plasma-cell clones associated with autoreactivity. Ann. Rheum. Dis. 2014, 73, 756–762. [Google Scholar] [CrossRef]
- Lubberts, E. The IL-23-IL-17 axis in inflammatory arthritis. Nat. Rev. Rheumatol. 2015, 11, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Gaffen, S.L.; Jain, R.; Garg, A.V.; Cua, D.J. The IL-23-IL-17 immune axis: From mechanisms to therapeutic testing. Nat. Rev. Immunol. 2014, 14, 585–600. [Google Scholar] [CrossRef]
- Paulissen, S.M.; van Hamburg, J.P.; Dankers, W.; Lubberts, E. The role and modulation of CCR6+ Th17 cell populations in rheumatoid arthritis. Cytokine 2015, 74, 43–53. [Google Scholar] [CrossRef]
- Andersson, A.K.; Li, C.; Brennan, F.M. Recent developments in the immunobiology of rheumatoid arthritis. Arthritis Res. Ther. 2008, 10, 204. [Google Scholar] [CrossRef] [PubMed]
- Anaya, J.M.; Shoenfeld, Y.; Rojas-Villarraga, A.; Levy, R.A.; Cervera, R. (Eds.) Autoimmunity: From Bench to Bedside; El Rosario University Press© 2020 Universidad del Rosario: Bogota, Colombia, 2013. [Google Scholar]
- Abbas, A.K.L.A.; Pillai, S. Cellular and Molecularimmunology, 7th ed.; Elsevier: Philadelphia, PA, USA, 2012. [Google Scholar]
- Demoruelle, M.K.; Deane, K.D.; Holers, V.M. When and where does inflammation begin in rheumatoid arthritis? Curr. Opin. Rheumatol. 2014, 26, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Deane, K.D.; Demoruelle, M.K.; Kelmenson, L.B.; Kuhn, K.A.; Norris, J.M.; Holers, V.M. Genetic and environmental risk factors for rheumatoid arthritis. Best Pract. Res. Clin. Rheumatol. 2017, 31, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Klareskog, L.; Stolt, P.; Lundberg, K.; Källberg, H.; Bengtsson, C.; Grunewald, J.; Rönnelid, J.; Harris, H.E.; Ulfgren, A.K.; Rantapää-Dahlqvist, S.; et al. A new model for an etiology of rheumatoid arthritis: Smoking may trigger HLA-DR (shared epitope)-restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum. 2006, 54, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Rosenstein, E.D.; Greenwald, R.A.; Kushner, L.J.; Weissmann, G. Hypothesis: The humoral immune response to oral bacteria provides a stimulus for the development of rheumatoid arthritis. Inflammation 2004, 28, 311–318. [Google Scholar] [CrossRef]
- Malmström, V.; Catrina, A.I.; Klareskog, L. The immunopathogenesis of seropositive rheumatoid arthritis: From triggering to targeting. Nat. Rev. Immunol. 2017, 17, 60–75. [Google Scholar] [CrossRef]
- Gizinski, A.M.; Mascolo, M.; Loucks, J.L.; Kervitsky, A.; Meehan, R.T.; Brown, K.K.; Holers, V.M.; Deane, K.D. Rheumatoid arthritis (RA)-specific autoantibodies in patients with interstitial lung disease and absence of clinically apparent articular RA. Clin. Rheumatol. 2009, 28, 611–613. [Google Scholar] [CrossRef]
- Oldstone, M.B. Molecular mimicry and autoimmune disease. Cell 1987, 50, 819–820. [Google Scholar] [CrossRef]
- Venigalla, S.S.K.; Premakumar, S.; Janakiraman, V. A possible role for autoimmunity through molecular mimicry in alphavirus mediated arthritis. Sci. Rep. 2020, 10, 938. [Google Scholar] [CrossRef]
- Root-Bernstein, R.; Fairweather, D. Complexities in the relationship between infection and autoimmunity. Curr. Allergy Asthma Rep. 2014, 14, 407. [Google Scholar] [CrossRef]
- Thaper, D.; Prabha, V. Molecular mimicry: An explanation for autoimmune diseases and infertility. Scand. J. Immunol. 2018, 88, e12697. [Google Scholar] [CrossRef] [PubMed]
- Arleevskaya, M.I.; Kravtsova, O.A.; Lemerle, J.; Renaudineau, Y.; Tsibulkin, A.P. How rheumatoid arthritis can result from provocation of the immune system by microorganisms and viruses. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef]
- Ercolini, A.M.; Miller, S.D. The role of infections in autoimmune disease. Clin. Exp. Immunol. 2009, 155, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Fujinami, R.S.; von Herrath, M.G.; Christen, U.; Whitton, J.L. Molecular mimicry, bystander activation, or viral persistence: Infections and autoimmune disease. Clin. Microbiol. Rev. 2006, 19, 80–94. [Google Scholar] [CrossRef] [PubMed]
- Bo, M.; Niegowska, M.; Erre, G.L.; Piras, M.; Longu, M.G.; Manchia, P.; Manca, M.; Passiu, G.; Sechi, L.A. Rheumatoid arthritis patient antibodies highly recognize IL-2 in the immune response pathway involving IRF5 and EBV antigens. Sci. Rep. 2018, 8, 1789. [Google Scholar] [CrossRef]
- Bo, M.; Niegowska, M.; Eames, H.L.; Almuttaqi, H.; Arru, G.; Erre, G.L.; Passiu, G.; Khoyratty, T.E.; van Grinsven, E.; Udalova, I.A.; et al. Antibody response to homologous epitopes of Epstein-Barr virus, Mycobacterium avium subsp. paratuberculosis and IRF5 in patients with different connective tissue diseases and in mouse model of antigen-induced arthritis. J. Transl. Autoimmun. 2020, 3, 100048. [Google Scholar] [CrossRef]
- Gagneux, S. Ecology and evolution of Mycobacterium tuberculosis. Nat. Rev. Microbiol. 2018, 16, 202–213. [Google Scholar] [CrossRef]
- Cronan, M.R.; Beerman, R.W.; Rosenberg, A.F.; Saelens, J.W.; Johnson, M.G.; Oehlers, S.H.; Sisk, D.M.; Jurcic Smith, K.L.; Medvitz, N.A.; Miller, S.E.; et al. Macrophage epithelial reprogramming underlies mycobacterial granuloma formation and promotes infection. Immunity 2016, 45, 861–876. [Google Scholar] [CrossRef]
- Birnbaum, G.; Kotilinek, L.; Albrecht, L. Spinal fluid lymphocytes from a subgroup of multiple sclerosis patients respond to mycobacterial antigens. Ann. Neurol. 1993, 34, 18–24. [Google Scholar] [CrossRef]
- Mor, F.; Cohen, I.R. T cells in the lesion of experimental autoimmune encephalomyelitis. Enrichment for reactivities to myelin basic protein and to heat shock proteins. J. Clin. Investig. 1992, 90, 2447–2455. [Google Scholar] [CrossRef]
- Res, P.C.; Schaar, C.G.; Breedveld, F.C.; van Eden, W.; van Embden, J.D.; Cohen, I.R.; de Vries, R.R. Synovial fluid T cell reactivity against 65 kD heat shock protein of mycobacteria in early chronic arthritis. Lancet 1988, 2, 478–480. [Google Scholar] [CrossRef]
- Salvetti, M.; Ristori, G.; Buttinelli, C.; Fiori, P.; Falcone, M.; Britton, W.; Adams, E.; Paone, G.; Grasso, M.G.; Pozzilli, C. The immune response to mycobacterial 70-kDa heat shock proteins frequently involves autoreactive T cells and is quantitatively disregulated in multiple sclerosis. J. NeuroImmunol. 1996, 65, 143–153. [Google Scholar] [CrossRef]
- Poncet, A. De la polyarthrite tuberculeuse deformante oupseudorheumatisme chronique tuberculeux. Congr. Fr. Chir. 1897, 1, 732–739. [Google Scholar]
- Torisu, M.; Miyahara, T.; Shinohara, N.; Ohsato, K.; Sonozaki, H. A new side effect of BCG immunotherapy —BCG-induced arthritis in man. Cancer Immunol. Immunother. 1978, 5, 77–83. [Google Scholar] [CrossRef]
- Kempsell, K.E.; Cox, C.J.; McColm, A.A.; Bagshaw, J.A.; Reece, R.; Veale, D.J.; Emery, P.; Isaacs, J.D.; Gaston, J.S.; Crowe, J.S. Detection of Mycobacterium tuberculosis group organisms in human and mouse joInt. tissue by reverse transcriptase PCR: Prevalence in diseased synovial tissue suggests lack of specific association with rheumatoid arthritis. Infect. Immun. 2001, 69, 1821–1831. [Google Scholar] [CrossRef] [PubMed]
- van der Heijden, I.M.; Wilbrink, B.; Schouls, L.M.; van Embden, J.D.; Breedveld, F.C.; Tak, P.P. Detection of mycobacteria in joInt. samples from patients with arthritis using a genus-specific polymerase chain reaction and sequence analysis. Rheumatology 1999, 38, 547–553. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wu, C.H.; Jeng, K.C.; Lan, J.L. Mycobacterium tuberculosis antigen, interleukin 2 and interleukin 2 inhibitor in patients with rheumatoid arthritis. Immunol. Invest. 1995, 24, 957–964. [Google Scholar] [CrossRef]
- Erre, G.L.; Cossu, D.; Masala, S.; Mameli, G.; Cadoni, M.L.; Serdino, S.; Longu, M.G.; Passiu, G.; Sechi, L.A. Mycobacterium tuberculosis lipoarabinomannan antibodies are associated to rheumatoid arthritis in Sardinian patients. Clin. Rheumatol. 2014, 33, 1725–1729. [Google Scholar] [CrossRef]
- Bahr, G.M.; Rook, G.A.; al-Saffar, M.; Van Embden, J.; Stanford, J.L.; Behbehani, K. Antibody levels to mycobacteria in relation to HLA type: Evidence for non-HLA-linked high levels of antibody to the 65 kD heat shock protein of M. bovis in rheumatoid arthritis. Clin. Exp. Immunol. 1988, 74, 211–215. [Google Scholar]
- Tsoulfa, G.; Rook, G.A.; Bahr, G.M.; Sattar, M.A.; Behbehani, K.; Young, D.B.; Mehlert, A.; Van-Embden, J.D.; Hay, F.C.; Isenberg, D.A.; et al. Elevated IgG antibody levels to the mycobacterial 65-kDa heat shock protein are characteristic of patients with rheumatoid arthritis. Scand. J. Immunol. 1989, 30, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Bo, M.; Erre, G.L.; Bach, H.; Slavin, Y.N.; Manchia, P.A.; Passiu, G.; Sechi, L.A. PtpA and PknG proteins secreted by Mycobacterium avium subsp. paratuberculosis are recognized by sera from patients with rheumatoid arthritis: A case-control study. J. Inflamm. Res. 2019, 12, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Gaston, J.S.; Life, P.F.; Bailey, L.C.; Bacon, P.A. In vitro responses to a 65-kilodalton mycobacterial protein by synovial T cells from inflammatory arthritis patients. J. Immunol. 1989, 143, 2494–2500. [Google Scholar] [PubMed]
- Holoshitz, J.; Koning, F.; Coligan, J.E.; De Bruyn, J.; Strober, S. Isolation of CD4- CD8- mycobacteria-reactive T lymphocyte clones from rheumatoid arthritis synovial fluid. Nature 1989, 339, 226–229. [Google Scholar] [CrossRef]
- Kanagawa, H.; Niki, Y.; Kobayashi, T.; Sato, Y.; Katsuyama, E.; Fujie, A.; Hao, W.; Miyamoto, K.; Tando, T.; Watanabe, R.; et al. Mycobacterium tuberculosis promotes arthritis development through Toll-like receptor 2. J. Bone Min. Metab. 2015, 33, 135–141. [Google Scholar] [CrossRef]
- Brand, D.D.; Latham, K.A.; Rosloniec, E.F. Collagen-induced arthritis. Nat. Protoc. 2007, 2, 1269–1275. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; He, W.; Du, X.; Yang, J.; Wen, Q.; Zhong, X.P.; Ma, L. IL-17 Production of neutrophils enhances antibacteria ability but promotes arthritis development during mycobacterium tuberculosis infection. EBioMedicine 2017, 23, 88–99. [Google Scholar] [CrossRef]
- Billiau, A.; Matthys, P. Modes of action of Freund’s adjuvants in experimental models of autoimmune diseases. J. Leukoc. Biol. 2001, 70, 849–860. [Google Scholar]
- Celis, L.; Vandevyver, C.; Geusens, P.; Dequeker, J.; Raus, J.; Zhang, J. Clonal expansion of mycobacterial heat-shock protein-reactive T lymphocytes in the synovial fluid and blood of rheumatoid arthritis patients. Arthritis Rheum. 1997, 40, 510–519. [Google Scholar] [CrossRef]
- Kogure, A.; Miyata, M.; Nishimaki, T.; Kasukawa, R. Proliferative response of synovial fluid mononuclear cells of patients with rheumatoid arthritis to mycobacterial 65 kDa heat shock protein and its association with HLA-DR+.gamma delta+ T cells. J. Rheumatol. 1994, 21, 1403–1408. [Google Scholar]
- Wucherpfennig, K.W.; Strominger, J.L. Molecular mimicry in T cell-mediated autoimmunity: Viral peptides activate human T cell clones specific for myelin basic protein. Cell 1995, 80, 695–705. [Google Scholar] [CrossRef]
- Tsuchiya, N.; Williams, R.C., Jr. Molecular mimicry--hypothesis or reality? West. J. Med. 1992, 157, 133–138. [Google Scholar] [PubMed]
- Esaguy, N.; Aguas, A.P.; van Embden, J.D.; Silva, M.T. Mycobacteria and human autoimmune disease: Direct evidence of cross-reactivity between human lactoferrin and the 65-kilodalton protein of tubercle and leprosy bacilli. Infect. Immun. 1991, 59, 1117–1125. [Google Scholar] [CrossRef] [PubMed]
- Bizzaro, N.; Mazzanti, G.; Tonutti, E.; Villalta, D.; Tozzoli, R. Diagnostic accuracy of the anti-citrulline antibody assay for rheumatoid arthritis. Clin. Chem. 2001, 47, 1089–1093. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.K.; Shim, T.S.; Sheen, D.H.; Na, D.J.; Min, S.S.; Shim, S.C. Anti-cyclic citrulline peptide antibody in non-tuberculous mycobacteria sera: A negative association. Clin. Rheumatol. 2010, 29, 335–336. [Google Scholar] [CrossRef]
- Elkayam, O.; Segal, R.; Bendayan, D.; van Uitert, R.; Onnekink, C.; Pruijn, G.J. The anti-cyclic citrullinated peptide response in tuberculosis patients is not citrulline-dependent and sensitive to treatment. Arthritis Res. Ther. 2010, 12, R12. [Google Scholar] [CrossRef]
- Silva, A.F.D.; Matos, A.N.; Lima, Á.M.S.; Lima, E.F.; Gaspar, A.P.; Braga, J.A.F.; Carvalho, E.M. Valor diagnóstico do anticorpo antipeptídeo citrulinado cíclico na artrite reumatóide. Revista Brasileira de Reumatologia 2006, 46, 174–180. [Google Scholar] [CrossRef]
- Aguas, A.; Esaguy, N.; Sunkel, C.E.; Silva, M.T. Cross-reactivity and sequence homology between the 65-kilodalton mycobacterial heat shock protein and human lactoferrin, transferrin, and DR beta subsets of major histocompatibility complex class II molecules. Infect. Immun. 1990, 58, 1461–1470. [Google Scholar] [CrossRef]
- van Eden, W.; Holoshitz, J.; Nevo, Z.; Frenkel, A.; Klajman, A.; Cohen, I.R. Arthritis induced by a T-lymphocyte clone that responds to Mycobacterium tuberculosis and to cartilage proteoglycans. Proc. Natl. Acad. Sci. USA 1985, 82, 5117–5120. [Google Scholar] [CrossRef]
- van Eden, W.; Hogervorst, E.J.; van der Zee, R.; van Embden, J.D.; Hensen, E.J.; Cohen, I.R. The mycobacterial 65 kD heat-shock protein and autoimmune arthritis. Rheumatol. Int. 1989, 9, 187–191. [Google Scholar] [CrossRef]
- Dow, C.T.M. paratuberculosis Heat Shock Protein 65 and Human Diseases: Bridging Infection and Autoimmunity. Autoimmune Dis. 2012, 2012, 150824. [Google Scholar] [CrossRef]
- Valdez, M.M.; Clark, J.I.; Wu, G.J.; Muchowski, P.J. Functional similarities between the small heat shock proteins Mycobacterium tuberculosis HSP 16.3 and human alphaB-crystallin. Eur. J. BioChem. 2002, 269, 1806–1813. [Google Scholar] [CrossRef] [PubMed]
- Dubaniewicz, A.; Trzonkowski, P.; Dubaniewicz-Wybieralska, M.; Dubaniewicz, A.; Singh, M.; Myśliwski, A. Comparative analysis of mycobacterial heat shock proteins-induced apoptosis of peripheral blood mononuclear cells in sarcoidosis and tuberculosis. J. Clin. Immunol. 2006, 26, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Hill, H.M.; Kirshbaum, J.D. Military tuberculosis developing during prolonged cortisone therapy of systemic lupus erythematosus. Ann. Intern. Med. 1956, 44, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, F.M.; Szyper-Kravitz, M.; Klumb, E.M.; Lannes, G.; Ribeiro, F.R.; Albuquerque, E.M.; Shoenfeld, Y. Can lupus flaRes. be associated with tuberculosis infection? Clin. Rev. Allergy Immunol. 2010, 38, 163–168. [Google Scholar] [CrossRef]
- van Eden, W.; Thole, J.E.; van der Zee, R.; Noordzij, A.; van Embden, J.D.; Hensen, E.J.; Cohen, I.R. Cloning of the mycobacterial epitope recognized by T lymphocytes in adjuvant arthritis. Nature 1988, 331, 171–173. [Google Scholar] [CrossRef]
- Zügel, U.; Kaufmann, S.H. Role of heat shock proteins in protection from and pathogenesis of infectious diseases. Clin. Microbiol. Rev. 1999, 12, 19–39. [Google Scholar] [CrossRef]
- Dubaniewicz, A.; Dubaniewicz-Wybieralska, M.; Sternau, A.; Zwolska, Z.; Izycka-Swieszewska, E.; Augustynowicz-Kopec, E.; Skokowski, J.; Singh, M.; Zimnoch, L. Mycobacterium tuberculosis complex and mycobacterial heat shock proteins in lymph node tissue from patients with pulmonary sarcoidosis. J. Clin. Microbiol. 2006, 44, 3448–3451. [Google Scholar] [CrossRef]
- van Eden, W.; van der Zee, R.; Prakken, B. Heat-shock proteins induce T-cell regulation of chronic inflammation. Nat. Rev. Immunol. 2005, 5, 318–330. [Google Scholar] [CrossRef]
- Shoda, H.; Hanata, N.; Sumitomo, S.; Okamura, T.; Fujio, K.; Yamamoto, K. Immune responses to Mycobacterial heat shock protein 70 accompany self-reactivity to human BiP in rheumatoid arthritis. Sci. Rep. 2016, 6, 22486. [Google Scholar] [CrossRef]
- Chodisetti, S.B.; Rai, P.K.; Gowthaman, U.; Pahari, S.; Agrewala, J.N. Potential T cell epitopes of Mycobacterium tuberculosis that can instigate molecular mimicry against host: Implications in autoimmune pathogenesis. BMC Immunol. 2012, 13, 13. [Google Scholar] [CrossRef]
- Gutlapalli, V.R.; Sykam, A.; Nayarisseri, A.; Suneetha, S.; Suneetha, L.M. Insights from the predicted epitope similarity between Mycobacterium tuberculosis virulent factors and its human homologs. Bioinformation 2015, 11, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, A.S.; Al-Attiyah, R.; Hanif, S.N.; Shaban, F.A. Efficient testing of large pools of Mycobacterium tuberculosis RD1 peptides and identification of major antigens and immunodominant peptides recognized by human Th1 cells. Clin. Vaccine Immunol. 2008, 15, 916–924. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mustafa, A.S. In silico binding predictions for identification of HLA-DR-promiscuous regions and epitopes of Mycobacterium tuberculosis protein MPT64 (Rv1980c) and their recognition by human Th1 cells. Med. Princ. Pract. 2010, 19, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Gowthaman, U.; Agrewala, J.N. In silico methods for predicting T-cell epitopes: Dr Jekyll or Mr Hyde? Expert Rev. Proteom. 2009, 6, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Ates, O.; Dalyan, L.; Müsellim, B.; Hatemi, G.; Türker, H.; Ongen, G.; Hamuryudan, V.; Topal-Sarikaya, A. NRAMP1 (SLC11A1) gene polymorphisms that correlate with autoimmune versus infectious disease susceptibility in tuberculosis and rheumatoid arthritis. Int. J. Immunogenet. 2009, 36, 15–19. [Google Scholar] [CrossRef]
- Sechi, L.A.; Gazouli, M.; Sieswerda, L.E.; Molicotti, P.; Ahmed, N.; Ikonomopoulos, J.; Scanu, A.M.; Paccagnini, D.; Zanetti, S. Relationship between Crohn’s disease, infection with Mycobacterium avium subspecies paratuberculosis and SLC11A1 gene polymorphisms in Sardinian patients. World J. Gastroenterol. 2006, 12, 7161–7164. [Google Scholar] [CrossRef]
- Paccagnini, D.; Sieswerda, L.; Rosu, V.; Masala, S.; Pacifico, A.; Gazouli, M.; Ikonomopoulos, J.; Ahmed, N.; Zanetti, S.; Sechi, L.A. Linking chronic infection and autoimmune diseases: Mycobacterium avium subspecies paratuberculosis, SLC11A1 polymorphisms and type-1 diabetes mellitus. PLoS ONE 2009, 4, e7109. [Google Scholar] [CrossRef]
- Sharp, R.C.; Beg, S.A.; Naser, S.A. Polymorphisms in protein tyrosine phosphatase non-receptor type 2 and 22 (PTPN2/22) are linked to Hyper-Proliferative T-Cells and susceptibility to mycobacteria in rheumatoid arthritis. Front. Cell. Infect. Microbiol. 2018, 8, 11. [Google Scholar] [CrossRef]
- Wyllie, S.; Seu, P.; Goss, J.A. The natural resistance-associated macrophage protein 1 Slc11a1 (formerly Nramp1) and iron metabolism in macrophages. Microbes Infect. 2002, 4, 351–359. [Google Scholar] [CrossRef]
- Hackam, D.J.; Rotstein, O.D.; Zhang, W.; Gruenheid, S.; Gros, P.; Grinstein, S. Host resistance to intracellular infection: Mutation of natural resistance-associated macrophage protein 1 (Nramp1) impairs phagosomal acidification. J. Exp. Med. 1998, 188, 351–364. [Google Scholar] [CrossRef]
- Yang, Y.S.; Kim, S.J.; Kim, J.W.; Koh, E.M. NRAMP1 gene polymorphisms in patients with rheumatoid arthritis in Koreans. J. Korean Med. Sci. 2000, 15, 83–87. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kotze, M.J.; de Villiers, J.N.; Rooney, R.N.; Grobbelaar, J.J.; Mansvelt, E.P.; Bouwens, C.S.; Carr, J.; Stander, I.; du Plessis, L. Analysis of the NRAMP1 gene implicated in iron transport: Association with multiple sclerosis and age effects. Blood Cells Mol. Dis. 2001, 27, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Gazouli, M.; Sechi, L.; Paccagnini, D.; Sotgiu, S.; Arru, G.; Nasioulas, G.; Vassilopoulos, D. NRAMP1 polymorphism and viral factors in Sardinian multiple sclerosis patients. Can. J. Neurol. Sci. 2008, 35, 491–494. [Google Scholar] [CrossRef]
- Kotlowski, R.; Bernstein, C.N.; Silverberg, M.S.; Krause, D.O. Population-based case-control study of alpha 1-antitrypsin and SLC11A1 in Crohn’s disease and ulcerative colitis. Inflamm. Bowel Dis. 2008, 14, 1112–1117. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Satoh, J.; Kojima, Y.; Negoro, K.; Hirai, M.; Hinokio, Y.; Kinouchi, Y.; Suzuki, S.; Matsuura, N.; Shimosegawa, T.; et al. Promoter polymorphism of SLC11A1 (formerly NRAMP1) confers susceptibility to autoimmune type 1 diabetes mellitus in Japanese. Tissue Antigens 2004, 63, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Qasem, A.; Ramesh, S.; Naser, S.A. Genetic polymorphisms in tumour necrosis factor receptors (TNFRSF1A/1B) illustrate differential treatment response to TNFα inhibitors in patients with Crohn’s disease. BMJ Open Gastroenterol. 2019, 6, e000246. [Google Scholar] [CrossRef]
- Matsui, T.; Ohsumi, K.; Ozawa, N.; Shimada, K.; Sumitomo, S.; Shimane, K.; Kawakami, M.; Nakayama, H.; Sugii, S.; Ozawa, Y.; et al. CD64 on neutrophils is a sensitive and specific marker for detection of infection in patients with rheumatoid arthritis. J. Rheumatol. 2006, 33, 2416–2424. [Google Scholar]
- Caporali, R.; Caprioli, M.; Bobbio-Pallavicini, F.; Montecucco, C. DMARDS and infections in rheumatoid arthritis. AutoImmun. Rev. 2008, 8, 139–143. [Google Scholar] [CrossRef]
- Solovic, I.; Sester, M.; Gomez-Reino, J.J.; Rieder, H.L.; Ehlers, S.; Milburn, H.J.; Kampmann, B.; Hellmich, B.; Groves, R.; Schreiber, S.; et al. The risk of tuberculosis related to tumour necrosis factor antagonist therapies: A TBNET consensus statement. Eur. Respir. J. 2010, 36, 1185–1206. [Google Scholar] [CrossRef] [PubMed]
- Askling, J.; Fored, C.M.; Brandt, L.; Baecklund, E.; Bertilsson, L.; Cöster, L.; Geborek, P.; Jacobsson, L.T.; Lindblad, S.; Lysholm, J.; et al. Risk and case characteristics of tuberculosis in rheumatoid arthritis associated with tumor necrosis factor antagonists in Sweden. Arthritis Rheum. 2005, 52, 1986–1992. [Google Scholar] [CrossRef]
- Ingraham, N.E.; Schneider, B.; Alpern, J.D. Prosthetic JoInt. Infection due to Mycobacterium avium-intracellulare in a Patient with Rheumatoid Arthritis: A Case Report and Review of the Literature. Case Rep. Infect. Dis. 2017, 2017, 8682354. [Google Scholar] [CrossRef] [PubMed]
- Iwata, K.; Oka, S.; Tsuno, H.; Furukawa, H.; Shimada, K.; Hashimoto, A.; Komiya, A.; Tsuchiya, N.; Katayama, M.; Tohma, S. Biomarker for nontuberculous mycobacterial pulmonary disease in patients with rheumatoid arthritis: Anti-glycopeptidolipid core antigen immunoglobulin A antibodies. Mod. Rheumatol. 2018, 28, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Schubert, N.; Schill, T.; Plüß, M.; Korsten, P. Flare or foe?—Mycobacterium marinum infection mimicking rheumatoid arthritis tenosynovitis: Case report and literature review. BMC Rheumatol. 2020, 4, 11. [Google Scholar] [CrossRef]
- Chen, H.W.; Lai, C.C.; Tan, C.K. Arthritis caused by Mycobacterium terrae in a patient with rheumatoid arthritis. Int. J. Infect Dis. 2009, 13, e145–e147. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.; Toma, W.; Schlesinger, N. Mycobacterium marinum arthritis mimicking rheumatoid arthritis. J. Rheumatol. 2006, 33, 817–819. [Google Scholar]
- DeMerieux, P.; Keystone, E.C.; Hutcheon, M.; Laskin, C. Polyarthritis due to Mycobacterium kansasii in a patient with rheumatoid arthritis. Ann. Rheum. Dis. 1980, 39, 90–94. [Google Scholar] [CrossRef]
- Dos Santos Sobrín, R.; Pérez Gómez, N.; Vilas, A.S.; Suárez, M.P.; Pampín, E.P.; Antúnez López, J.R.; Mera Varela, A. Infection by Mycobacterium chelonae at the site of administration of sarilumab for rheumatoid arthritis. Rheumatology 2020, 59, 265. [Google Scholar] [CrossRef]
- Dutertre, M.; Delobel, P.; Marchou, B.; Boyer, J.F.; Mougari, F.; Martin-Blondel, G. Olecranon bursitis secondary to Mycobacterium europaeum infection in a patient receiving immunosuppressive drugs for rheumatoid arthritis. Med. Et Mal. Infect. 2019, 49, 358–359. [Google Scholar] [CrossRef]
- Benedek, T.G. The history of bacteriologic concepts of rheumatic fever and rheumatoid arthritis. Semin Arthritis Rheum. 2006, 36, 109–123. [Google Scholar] [CrossRef]
- Martinez-Martinez, R.E.; Abud-Mendoza, C.; Patiño-Marin, N.; Rizo-Rodríguez, J.C.; Little, J.W.; Loyola-Rodríguez, J.P. Detection of periodontal bacterial DNA in serum and synovial fluid in refractory rheumatoid arthritis patients. J. Clin. Periodontol. 2009, 36, 1004–1010. [Google Scholar] [CrossRef]
- Totaro, M.C.; Cattani, P.; Ria, F.; Tolusso, B.; Gremese, E.; Fedele, A.L.; D’Onghia, S.; Marchetti, S.; Sante, G.D.; Canestri, S.; et al. Porphyromonas gingivalis and the pathogenesis of rheumatoid arthritis: Analysis of various compartments including the synovial tissue. Arthritis Res. Ther. 2013, 15, R66. [Google Scholar] [CrossRef] [PubMed]
- Kawahito, Y.; Ichinose, S.; Sano, H.; Tsubouchi, Y.; Kohno, M.; Yoshikawa, T.; Tokunaga, D.; Hojo, T.; Harasawa, R.; Nakano, T.; et al. Mycoplasma fermentans glycolipid-antigen as a pathogen of rheumatoid arthritis. BioChem. Biophys. Res. Commun. 2008, 369, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Schaeverbeke, T.; Renaudin, H.; Clerc, M.; Lequen, L.; Vernhes, J.P.; De Barbeyrac, B.; Bannwarth, B.; Bébéar, C.; Dehais, J. Systematic detection of mycoplasmas by culture and polymerase chain reaction (PCR) proceduRes. in 209 synovial fluid samples. Rheumatology 1997, 36, 310–314. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hoffman, R.W.; O’Sullivan, F.X.; Schafermeyer, K.R.; Moore, T.L.; Roussell, D.; Watson-McKown, R.; Kim, M.F.; Wise, K.S. Mycoplasma infection and rheumatoid arthritis: Analysis of their relationship using immunoblotting and an ultrasensitive polymerase chain reaction detection method. Arthritis Rheum. 1997, 40, 1219–1228. [Google Scholar] [CrossRef]
- Wilkinson, N.Z.; Kingsley, G.H.; Jones, H.W.; Sieper, J.; Braun, J.; Ward, M.E. The detection of DNA from a range of bacterial species in the joints of patients with a variety of arthritides using a nested, broad-range polymerase chain reaction. Rheumatology 1999, 38, 260–266. [Google Scholar] [CrossRef]
- van der Heijden, I.M.; Wilbrink, B.; Tchetverikov, I.; Schrijver, I.A.; Schouls, L.M.; Hazenberg, M.P.; Breedveld, F.C.; Tak, P.P. Presence of bacterial DNA and bacterial peptidoglycans in joints of patients with rheumatoid arthritis and other arthritides. Arthritis Rheum. 2000, 43, 593–598. [Google Scholar] [CrossRef]
- Saal, J.G.; Steidle, M.; Einsele, H.; Müller, C.A.; Fritz, P.; Zacher, J. Persistence of B19 parvovirus in synovial membranes of patients with rheumatoid arthritis. Rheumatol. Int. 1992, 12, 147–151. [Google Scholar] [CrossRef]
- Jobanputra, P.; Davidson, F.; Graham, S.; O’Neill, H.; Simmonds, P.; Yap, P.L. High frequency of parvovirus B19 in patients tested for rheumatoid factor. BMJ 1995, 311, 1542. [Google Scholar] [CrossRef]
- Takeda, T.; Mizugaki, Y.; Matsubara, L.; Imai, S.; Koike, T.; Takada, K. Lytic Epstein-Barr virus infection in the synovial tissue of patients with rheumatoid arthritis. Arthritis Rheum. 2000, 43, 1218–1225. [Google Scholar] [CrossRef]
- Mehraein, Y.; Lennerz, C.; Ehlhardt, S.; Remberger, K.; Ojak, A.; Zang, K.D. Latent Epstein-Barr virus (EBV) infection and cytomegalovirus (CMV) infection in synovial tissue of autoimmune chronic arthritis determined by RNA- and DNA-in situ hybridization. Mod. Pathol. 2004, 17, 781–789. [Google Scholar] [CrossRef]
- Sorgato, C.C.; Lins, E.S.M.; Leão, J.C.; Vasconcelos, L.R.; Melo, T.R.; Duarte, A.L.; Gueiros, L.A. EBV and CMV Viral Load in Rheumatoid Arthritis and Their Role Associated Sjögren’s Syndrome. J. Oral. Pathol. Med. 2020. [Google Scholar] [CrossRef]
- Kuusela, E.; Kouri, V.P.; Olkkonen, J.; Koivuniemi, R.; Äyräväinen, L.; Rajamäki, K.; Valleala, H.; Nordström, D.; Leirisalo-Repo, M.; Ainola, M.; et al. Serum Epstein-Barr virus DNA, detected by droplet digital PCR, correlates with disease activity in patients with rheumatoid arthritis. Clin. Exp. Rheumatol. 2018, 36, 778–784. [Google Scholar]
- Erre, G.L.; Mameli, G.; Cossu, D.; Muzzeddu, B.; Piras, C.; Paccagnini, D.; Passiu, G.; Sechi, L.A. Increased Epstein-Barr Virus DNA Load and Antibodies Against EBNA1 and EA in Sardinian Patients with Rheumatoid Arthritis. Viral. Immunol. 2015, 28, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Mikuls, T.R.; Payne, J.B.; Reinhardt, R.A.; Thiele, G.M.; Maziarz, E.; Cannella, A.C.; Holers, V.M.; Kuhn, K.A.; O’Dell, J.R. Antibody responses to Porphyromonas gingivalis (P. gingivalis) in subjects with rheumatoid arthritis and periodontitis. Int. Immunopharmacol. 2009, 9, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Ogrendik, M.; Kokino, S.; Ozdemir, F.; Bird, P.S.; Hamlet, S. Serum antibodies to oral anaerobic bacteria in patients with rheumatoid arthritis. MedGenMed 2005, 7, 2. [Google Scholar] [PubMed]
- Quirke, A.M.; Lugli, E.B.; Wegner, N.; Hamilton, B.C.; Charles, P.; Chowdhury, M.; Ytterberg, A.J.; Zubarev, R.A.; Potempa, J.; Culshaw, S.; et al. Heightened immune response to autocitrullinated Porphyromonas gingivalis peptidylarginine deiminase: A potential mechanism for breaching immunologic tolerance in rheumatoid arthritis. Ann. Rheum. Dis. 2014, 73, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Klatt, T.; Ouyang, Q.; Flad, T.; Koetter, I.; Bühring, H.J.; Kalbacher, H.; Pawelec, G.; Müller, C.A. Expansion of peripheral CD8+ CD28- T cells in response to Epstein-Barr virus in patients with rheumatoid arthritis. J. Rheumatol. 2005, 32, 239–251. [Google Scholar] [PubMed]
- Rickinson, A.B.; Moss, D.J. Human cytotoxic T lymphocyte responses to Epstein-Barr virus infection. Annu. Rev. Immunol. 1997, 15, 405–431. [Google Scholar] [CrossRef]
- Scotet, E.; David-Ameline, J.; Peyrat, M.A.; Moreau-Aubry, A.; Pinczon, D.; Lim, A.; Even, J.; Semana, G.; Berthelot, J.M.; Breathnach, R.; et al. T cell response to Epstein-Barr virus transactivators in chronic rheumatoid arthritis. J. Exp. Med. 1996, 184, 1791–1800. [Google Scholar] [CrossRef]
- Lünemann, J.D.; Frey, O.; Eidner, T.; Baier, M.; Roberts, S.; Sashihara, J.; Volkmer, R.; Cohen, J.I.; Hein, G.; Kamradt, T.; et al. Increased frequency of EBV-specific effector memory CD8+ T cells correlates with higher viral load in rheumatoid arthritis. J. Immunol. 2008, 181, 991–1000. [Google Scholar] [CrossRef]
- Trier, N.H.; Holm, B.E.; Heiden, J.; Slot, O.; Locht, H.; Lindegaard, H.; Svendsen, A.; Nielsen, C.T.; Jacobsen, S.; Theander, E.; et al. Antibodies to a strain-specific citrullinated Epstein-Barr virus peptide diagnoses rheumatoid arthritis. Sci. Rep. 2018, 8, 3684. [Google Scholar] [CrossRef]
- Sternbæk, L.; Draborg, A.H.; Østerlund, M.T.; Iversen, L.V.; Troelsen, L.; Theander, E.; Nielsen, C.T.; Jacobsen, S.; Houen, G. Increased antibody levels to stage-specific Epstein-Barr virus antigens in systemic autoimmune diseases reveal a common pathology. Scand. J. Clin. Lab. Investig. 2019, 79, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Motokawa, S.; Hasunuma, T.; Tajima, K.; Krieg, A.M.; Ito, S.; Iwasaki, K.; Nishioka, K. High prevalence of arthropathy in HTLV-I carriers on a Japanese island. Ann. Rheum. Dis. 1996, 55, 193–195. [Google Scholar] [CrossRef] [PubMed]
- da Rocha Sobrinho, H.M.; Jarach, R.; da Silva, N.A.; Shio, M.T.; Jancar, S.; Timenetsky, J.; Oliveira, M.A.; Dorta, M.L.; Ribeiro-Dias, F. Mycoplasmal lipid-associated membrane proteins and Mycoplasma arthritidis mitogen recognition by serum antibodies from patients with rheumatoid arthritis. Rheumatol. Int. 2011, 31, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Sawitzke, A.; Joyner, D.; Knudtson, K.; Mu, H.H.; Cole, B. Anti-MAM antibodies in rheumatic disease: Evidence for a MAM-like superantigen in rheumatoid arthritis? J. Rheumatol. 2000, 27, 358–364. [Google Scholar] [PubMed]
- Tzang, B.S.; Tsai, C.C.; Tsay, G.J.; Wang, M.; Sun, Y.S.; Hsu, T.C. Anti-human parvovirus B19 nonstructural protein antibodies in patients with rheumatoid arthritis. Clin. Chim. Acta 2009, 405, 76–82. [Google Scholar] [CrossRef]
- Shi, J.; Sun, X.; Zhao, Y.; Zhao, J.; Li, Z. Prevalence and significance of antibodies to citrullinated human papilloma virus-47 E2345-362 in rheumatoid arthritis. J. AutoImmun. 2008, 31, 131–135. [Google Scholar] [CrossRef]
- Mameli, G.; Erre, G.L.; Caggiu, E.; Mura, S.; Cossu, D.; Bo, M.; Cadoni, M.L.; Piras, A.; Mundula, N.; Colombo, E.; et al. Identification of a HERV-K env surface peptide highly recognized in Rheumatoid Arthritis (RA) patients: A cross-sectional case-control study. Clin. Exp. Immunol. 2017, 189, 127–131. [Google Scholar] [CrossRef]
- Chukkapalli, S.; Rivera-Kweh, M.; Gehlot, P.; Velsko, I.; Bhattacharyya, I.; Calise, S.J.; Satoh, M.; Chan, E.K.; Holoshitz, J.; Kesavalu, L. Periodontal bacterial colonization in synovial tissues exacerbates collagen-induced arthritis in B10.RIII mice. Arthritis Res. Ther. 2016, 18, 161. [Google Scholar] [CrossRef]
- Jung, H.; Jung, S.M.; Rim, Y.A.; Park, N.; Nam, Y.; Lee, J.; Park, S.-H.; Ju, J.H. Arthritic role of Porphyromonas gingivalis in collagen-induced arthritis mice. PLoS ONE 2017, 12, e0188698. [Google Scholar] [CrossRef]
- Yamakawa, M.; Ouhara, K.; Kajiya, M.; Munenaga, S.; Kittaka, M.; Yamasaki, S.; Takeda, K.; Takeshita, K.; Mizuno, N.; Fujita, T.; et al. Porphyromonas gingivalis infection exacerbates the onset of rheumatoid arthritis in SKG mice. Clin. Exp. Immunol. 2016, 186, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Bartold, P.M.; Marino, V.; Cantley, M.; Haynes, D.R. Effect of Porphyromonas gingivalis-induced inflammation on the development of rheumatoid arthritis. J. Clin. Periodontol. 2010, 37, 405–411. [Google Scholar] [CrossRef]
- Cole, B.C.; Golightly-Rowland, L.; Ward, J.R. Arthritis of mice induced by Mycoplasma arthritidis. Humoral antibody and lymphocyte responses of CBA mice. Ann. Rheum. Dis. 1976, 35, 14–22. [Google Scholar] [CrossRef]
- Kuwana, Y.; Takei, M.; Yajima, M.; Imadome, K.-I.; Inomata, H.; Shiozaki, M.; Ikumi, N.; Nozaki, T.; Shiraiwa, H.; Kitamura, N.; et al. Epstein-Barr Virus Induces Erosive Arthritis in Humanized Mice. PLoS ONE 2011, 6, e26630. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, S.; Matsuda, G.; Imadome, K.-I. Humanized mouse models of epstein-barr virus infection and associated diseases. Pathogens 2013, 2, 153–176. [Google Scholar] [CrossRef] [PubMed]
- Röhner, E.; Detert, J.; Kolar, P.; Hocke, A.; N’Guessan, P.; Matziolis, G.; Kanitz, V.; Bernimoulin, J.P.; Kielbassa, A.; Burmester, G.R.; et al. Induced apoptosis of chondrocytes by Porphyromonas gingivalis as a possible pathway for cartilage loss in rheumatoid arthritis. Calcif. Tissue Int. 2010, 87, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Choi, I.A.; Kim, J.H.; Kim, K.H.; Lee, E.Y.; Lee, E.B.; Lee, Y.M.; Song, Y.W. Association between anti-Porphyromonas gingivalis or anti-α-enolase antibody and severity of periodontitis or rheumatoid arthritis (RA) disease activity in RA. BMC Musculoskelet. Disord. 2015, 16, 190. [Google Scholar] [CrossRef] [PubMed]
- Rojas, M.; Restrepo-Jiménez, P.; Monsalve, D.M.; Pacheco, Y.; Acosta-Ampudia, Y.; Ramírez-Santana, C.; Leung, P.S.C.; Ansari, A.A.; Gershwin, M.E.; Anaya, J.M. Molecular mimicry and autoimmunity. J. AutoImmun. 2018, 95, 100–123. [Google Scholar] [CrossRef]
- Lundberg, K.; Kinloch, A.; Fisher, B.A.; Wegner, N.; Wait, R.; Charles, P.; Mikuls, T.R.; Venables, P.J. Antibodies to citrullinated alpha-enolase peptide 1 are specific for rheumatoid arthritis and cross-react with bacterial enolase. Arthritis Rheum. 2008, 58, 3009–3019. [Google Scholar] [CrossRef] [PubMed]
- Bo, M.; Erre, G.L.; Niegowska, M.; Piras, M.; Taras, L.; Longu, M.G.; Passiu, G.; Sechi, L.A. Interferon regulatory factor 5 is a potential target of autoimmune response triggered by Epstein-barr virus and Mycobacterium avium subsp. paratuberculosis in rheumatoid arthritis: Investigating a mechanism of molecular mimicry. Clin. Exp. Rheumatol. 2018, 36, 376–381. [Google Scholar]
- Ebringer, A.; Rashid, T.; Wilson, C. Rheumatoid arthritis, Proteus, anti-CCP antibodies and Karl Popper. AutoImmun. Rev. 2010, 9, 216–223. [Google Scholar] [CrossRef]
- Ebringer, A.; Rashid, T. Rheumatoid arthritis is an autoimmune disease triggered by Proteus urinary tract infection. Clin. Dev. Immunol. 2006, 13, 41–48. [Google Scholar] [CrossRef]
- Wilson, C.; Ebringer, A.; Ahmadi, K.; Wrigglesworth, J.; Tiwana, H.; Fielder, M.; Binder, A.; Ettelaie, C.; Cunningham, P.; Joannou, C.; et al. Shared amino acid sequences between major histocompatibility complex class II glycoproteins, type XI collagen and Proteus mirabilis in rheumatoid arthritis. Ann. Rheum. Dis. 1995, 54, 216–220. [Google Scholar] [CrossRef]
- Ebringer, A.; Rashid, T. Rheumatoid arthritis is caused by a Proteus urinary tract infection. Apmis 2014, 122, 363–368. [Google Scholar] [CrossRef]
- Tiwana, H.; Wilson, C.; Alvarez, A.; Abuknesha, R.; Bansal, S.; Ebringer, A. Cross-reactivity between the rheumatoid arthritis-associated motif EQKRAA and structurally related sequences found in Proteus mirabilis. Infect. Immun. 1999, 67, 2769–2775. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Tiwana, H.; Ebringer, A. Molecular mimicry between HLA-DR alleles associated with rheumatoid arthritis and Proteus mirabilis as the Aetiological basis for autoimmunity. Microbes Infect. 2000, 2, 1489–1496. [Google Scholar] [CrossRef]
- Hou, Y.; Lin, H.; Zhu, L.; Liu, Z.; Hu, F.; Shi, J.; Yang, T.; Shi, X.; Zhu, M.; Godley, B.F.; et al. Lipopolysaccharide increases the incidence of collagen-induced arthritis in mice through induction of protease HTRA-1 expression. Arthritis Rheum. 2013, 65, 2835–2846. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, W.; Buhrmann, C.; Mobasheri, A.; Lueders, C.; Shakibaei, M. Bacterial lipopolysaccharides form procollagen-endotoxin complexes that trigger cartilage inflammation and degeneration: Implications for the development of rheumatoid arthritis. Arthritis Res. Ther. 2013, 15, R111. [Google Scholar] [CrossRef]
- Nakayama, M.; Niki, Y.; Kawasaki, T.; Takeda, Y.; Horiuchi, K.; Sasaki, A.; Okada, Y.; Umezawa, K.; Ikegami, H.; Toyama, Y.; et al. Enhanced susceptibility to lipopolysaccharide-induced arthritis and endotoxin shock in interleukin-32 alpha transgenic mice through induction of tumor necrosis factor alpha. Arthritis Res. Ther. 2012, 14, R120. [Google Scholar] [CrossRef]
- Yücel, G.; Zhao, Z.; El-Battrawy, I.; Lan, H.; Lang, S.; Li, X.; Buljubasic, F.; Zimmermann, W.-H.; Cyganek, L.; Utikal, J.; et al. Lipopolysaccharides induced inflammatory responses and electrophysiological dysfunctions in human-induced pluripotent stem cell derived cardiomyocytes. Sci. Rep. 2017, 7, 2935. [Google Scholar] [CrossRef]
- Frost, R.A.; Nystrom, G.J.; Lang, C.H. Lipopolysaccharide regulates proinflammatory cytokine expression in mouse myoblasts and skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 283, R698–R709. [Google Scholar] [CrossRef] [PubMed]
- Barksby, H.E.; Nile, C.J.; Jaedicke, K.M.; Taylor, J.J.; Preshaw, P.M. Differential expression of immunoregulatory genes in monocytes in response to Porphyromonas gingivalis and Escherichia coli lipopolysaccharide. Clin. Exp. Immunol. 2009, 156, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Nile, C.J.; Barksby, E.; Jitprasertwong, P.; Preshaw, P.M.; Taylor, J.J. Expression and regulation of interleukin-33 in human monocytes. Immunology 2010, 130, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Burns, E.; Bachrach, G.; Shapira, L.; Nussbaum, G. Cutting Edge: TLR2 is required for the innate response to Porphyromonas gingivalis: Activation leads to bacterial persistence and TLR2 deficiency attenuates induced alveolar bone resorption. J. Immunol. 2006, 177, 8296–8300. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Bi, L.; Yu, X.; Kawai, T.; Taubman, M.A.; Shen, B.; Han, X. Porphyromonas gingivalis exacerbates ligature-induced, RANKL-dependent alveolar bone resorption via differential regulation of Toll-like receptor 2 (TLR2) and TLR4. Infect. Immun. 2014, 82, 4127–4134. [Google Scholar] [CrossRef]
- Yang, S.; Tamai, R.; Akashi, S.; Takeuchi, O.; Akira, S.; Sugawara, S.; Takada, H. Synergistic effect of muramyldipeptide with lipopolysaccharide or lipoteichoic acid to induce inflammatory cytokines in human monocytic cells in culture. Infect. Immun. 2001, 69, 2045–2053. [Google Scholar] [CrossRef]
- Yang, S.; Takahashi, N.; Yamashita, T.; Sato, N.; Takahashi, M.; Mogi, M.; Uematsu, T.; Kobayashi, Y.; Nakamichi, Y.; Takeda, K.; et al. Muramyl Dipeptide Enhances Osteoclast Formation Induced by Lipopolysaccharide, IL-1α, and TNF-α through Nucleotide-Binding Oligomerization Domain 2-Mediated Signaling in Osteoblasts. J. Immunol. 2005, 175, 1956–1964. [Google Scholar] [CrossRef]
- Shehab, M.; Sherri, N.; Hussein, H.; Salloum, N.; Rahal, E.A. Endosomal Toll-Like Receptors Mediate Enhancement of Interleukin-17A Production Triggered by Epstein-Barr Virus DNA in Mice. J. Virol. 2019, 93, e00987-19. [Google Scholar] [CrossRef]
- Salloum, N.; Hussein, H.M.; Jammaz, R.; Jiche, S.; Uthman, I.W.; Abdelnoor, A.M.; Rahal, E.A. Epstein-Barr virus DNA modulates regulatory T-cell programming in addition to enhancing interleukin-17A production via Toll-like receptor 9. PLoS ONE 2018, 13, e0200546. [Google Scholar] [CrossRef]
- Hsiao, F.C.; Lin, M.; Tai, A.; Chen, G.; Huber, B.T. Cutting edge: Epstein-Barr virus transactivates the HERV-K18 superantigen by docking to the human complement receptor 2 (CD21) on primary B cells. J. Immunol. 2006, 177, 2056–2060. [Google Scholar] [CrossRef]
- Ford, D.K.; da Roza, D.M.; Schulzer, M.; Reid, G.D.; Denegri, J.F. Persistent synovial lymphocyte responses to cytomegalovirus antigen in some patients with rheumatoid arthritis. Arthritis Rheum. 1987, 30, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Kinloch, A.J.; Alzabin, S.; Brintnell, W.; Wilson, E.; Barra, L.; Wegner, N.; Bell, D.A.; Cairns, E.; Venables, P.J. Immunization with Porphyromonas gingivalis enolase induces autoimmunity to mammalian α-enolase and arthritis in DR4-IE-transgenic mice. Arthritis Rheum. 2011, 63, 3818–3823. [Google Scholar] [CrossRef] [PubMed]
- Wegner, N.; Wait, R.; Sroka, A.; Eick, S.; Nguyen, K.A.; Lundberg, K.; Kinloch, A.; Culshaw, S.; Potempa, J.; Venables, P.J. Peptidylarginine deiminase from Porphyromonas gingivalis citrullinates human fibrinogen and α-enolase: Implications for autoimmunity in rheumatoid arthritis. Arthritis Rheum. 2010, 62, 2662–2672. [Google Scholar] [CrossRef] [PubMed]
- Maresz, K.J.; Hellvard, A.; Sroka, A.; Adamowicz, K.; Bielecka, E.; Koziel, J.; Gawron, K.; Mizgalska, D.; Marcinska, K.A.; Benedyk, M.; et al. Porphyromonas gingivalis facilitates the development and progression of destructive arthritis through its unique bacterial peptidylarginine deiminase (PAD). PloS Pathog. 2013, 9, e1003627. [Google Scholar] [CrossRef] [PubMed]
- Courbon, G.; Rinaudo-Gaujous, M.; Blasco-Baque, V.; Auger, I.; Caire, R.; Mijola, L.; Vico, L.; Paul, S.; Marotte, H. Porphyromonas gingivalis experimentally induces periodontis and an anti-CCP2-associated arthritis in the rat. Ann. Rheum. Dis. 2019, 78, 594–599. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Presence of microbial contents in RA patients tissues and serum | Mycobacteria, P. gingivalis, EBV, Mycoplasma, Bordetalla, Haemophilus, Acinetobacter, Parvovirus, CMV, Bacterial cell wall |
| Presence of immune response to infection in RA patients tissues and serum | Mycobacteria, P. gingivalis, EBV, HTLV, Mycoplasma, Parvovirus B19, Papilloma virus, HERV |
Induction of Arthritis by Infections in Animal Models | Mycobacteria, P. gingivalis, Mycoplasma, EBV |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bo, M.; Jasemi, S.; Uras, G.; Erre, G.L.; Passiu, G.; Sechi, L.A. Role of Infections in the Pathogenesis of Rheumatoid Arthritis: Focus on Mycobacteria. Microorganisms 2020, 8, 1459. https://doi.org/10.3390/microorganisms8101459
Bo M, Jasemi S, Uras G, Erre GL, Passiu G, Sechi LA. Role of Infections in the Pathogenesis of Rheumatoid Arthritis: Focus on Mycobacteria. Microorganisms. 2020; 8(10):1459. https://doi.org/10.3390/microorganisms8101459
Chicago/Turabian StyleBo, Marco, Seyedesomaye Jasemi, Giuseppe Uras, Gian Luca Erre, Giuseppe Passiu, and Leonardo A. Sechi. 2020. "Role of Infections in the Pathogenesis of Rheumatoid Arthritis: Focus on Mycobacteria" Microorganisms 8, no. 10: 1459. https://doi.org/10.3390/microorganisms8101459
APA StyleBo, M., Jasemi, S., Uras, G., Erre, G. L., Passiu, G., & Sechi, L. A. (2020). Role of Infections in the Pathogenesis of Rheumatoid Arthritis: Focus on Mycobacteria. Microorganisms, 8(10), 1459. https://doi.org/10.3390/microorganisms8101459