Genome Improvement and Core Gene Set Refinement of Fugacium kawagutii

 , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Algal Culture and Trace Metal Treatments
2.2. RNA Extraction
2.3. Hi-C, Illumina RNA-Seq, and PacBio Sequencing
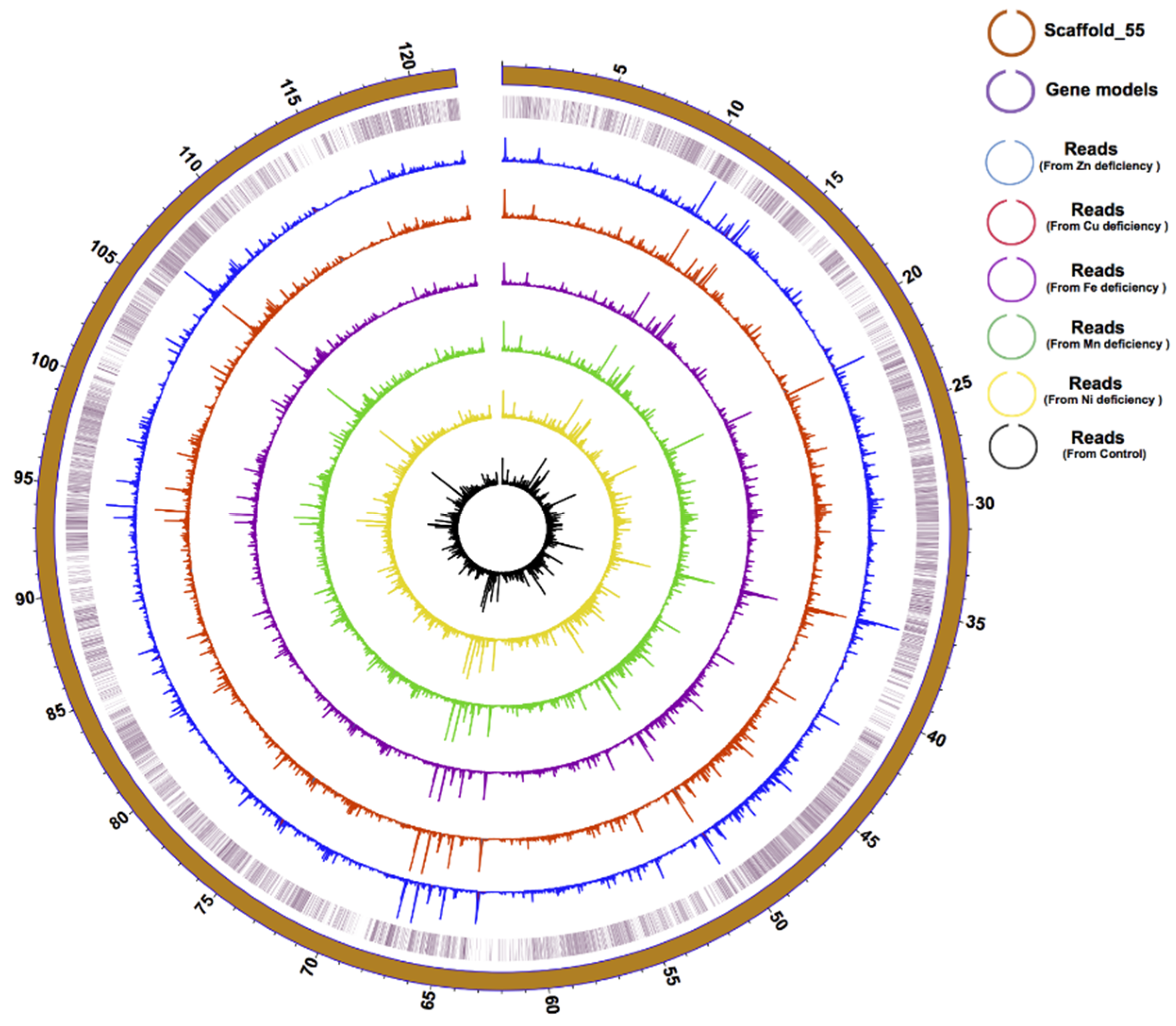
2.4. Genome Improvement and Gene Set Refinement
2.5. Core Gene Set Update
2.6. Validation of Reference Genes for Reverse Transcription Quantitative PCR (RT-qPCR)
3. Results and Discussion
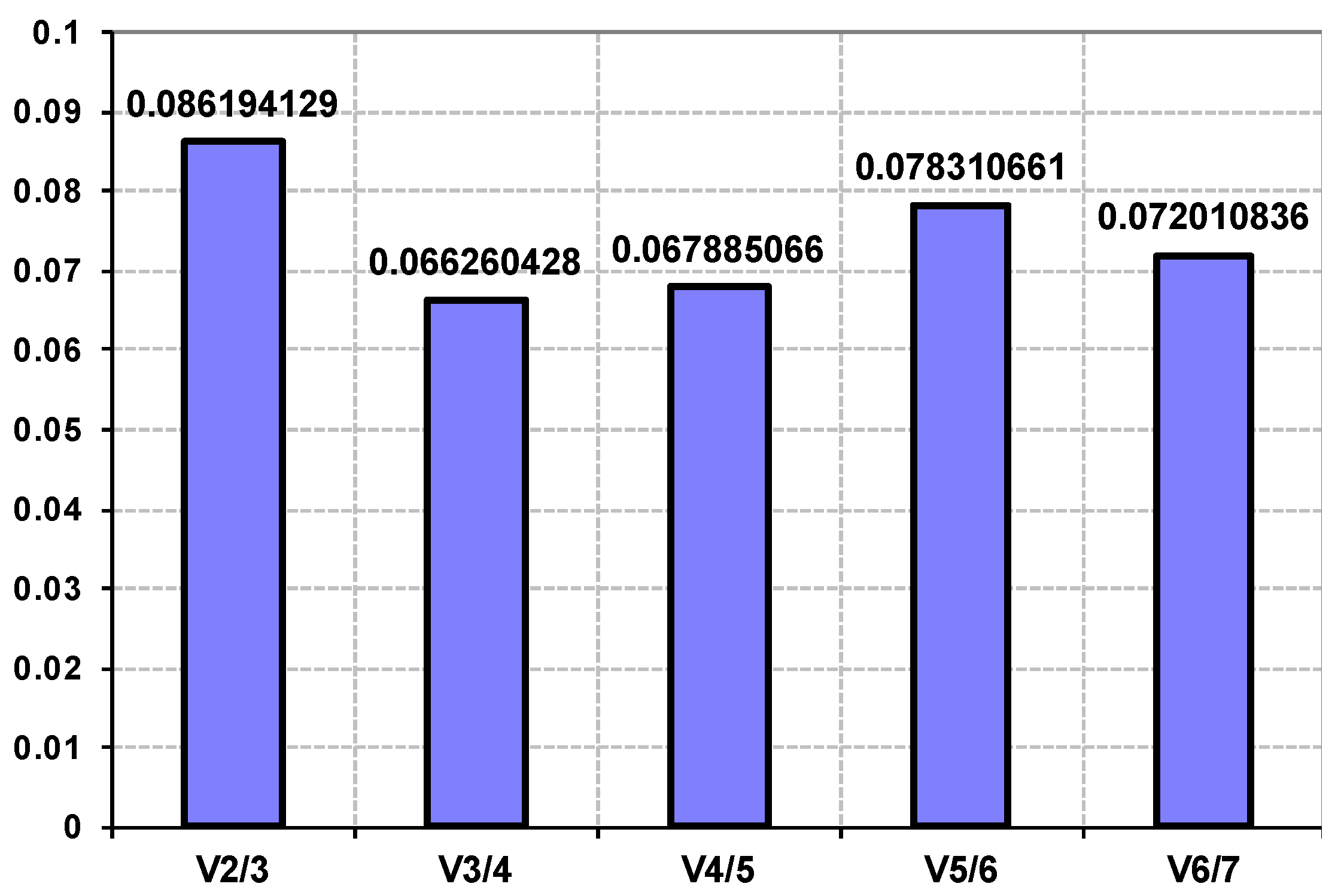
3.1. Genome Improvement and Gene Set Refinement
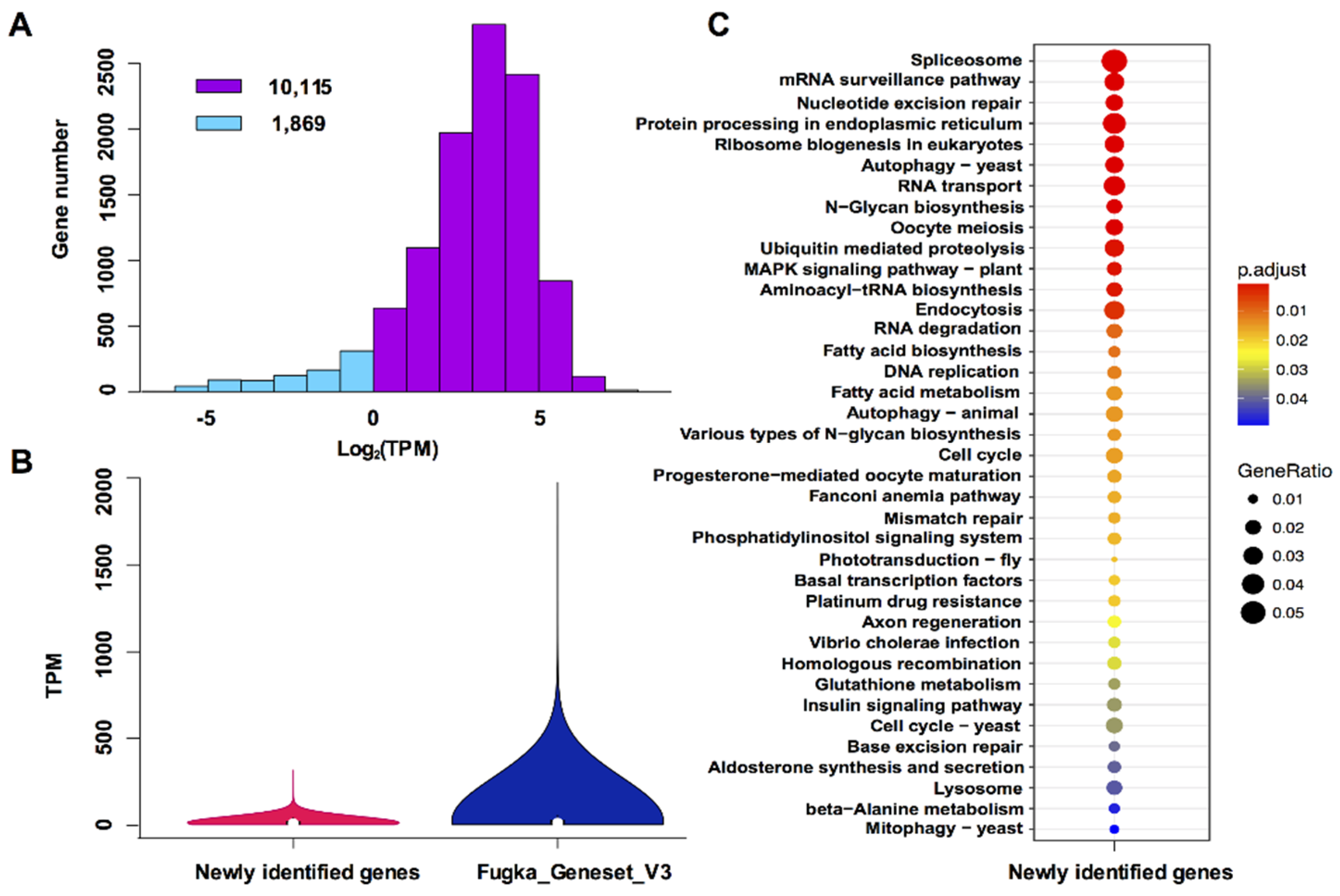
3.2. Characterization of Newly Identified Genes
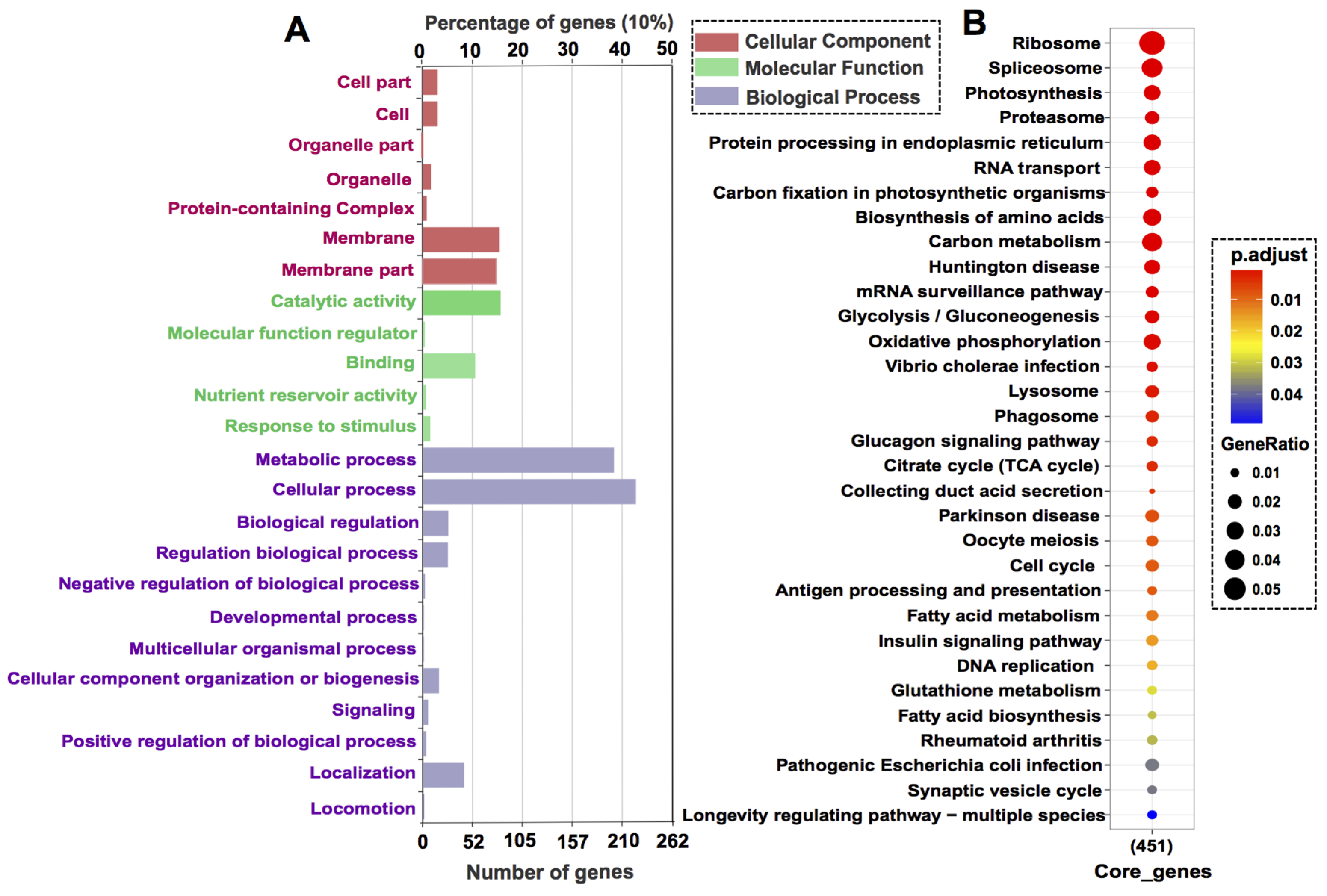
3.3. Core Gene Set Refinement
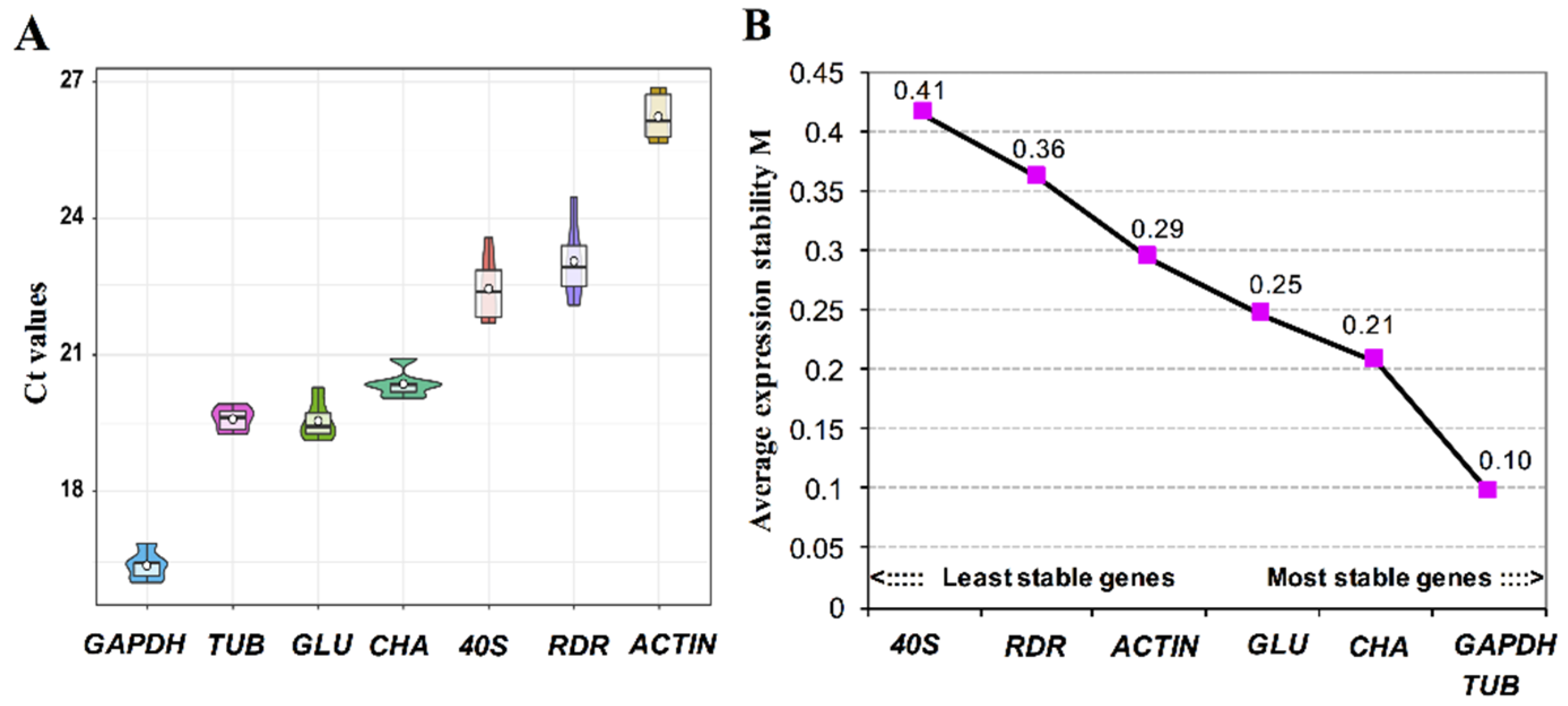
3.4. Validation of Reference Genes for Gene Expression Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

References
- Trench, R.K.; Blank, R.J. Symbiodinium microdriaticum ferudenthal, S. Goreauii sp. nov., S. kawagutii sp. nov. and S. pilosum sp. nov.: Gymnodinioid dinoflagellate symbionts of marine invertebrates 1. J. Phycol. 1987, 23, 469–481. [Google Scholar] [CrossRef]
- Muscatine, L.; Porter, J.W. Reef corals: Mutualistic symbioses adapted to nutrient-poor environments. Bioscience 1997, 27, 454–460. [Google Scholar] [CrossRef]
- Muller-Parker, G.; D’Elia, C.F.; Cook, C.B. Interactions between corals and their symbiotic algae. In Coral Reefs in the Anthropocene; Birkeland, C., Ed.; Springer: Dordrecht, The Netherlands, 2015; pp. 99–116. [Google Scholar]
- Hughes, T.P.; Kerry, J.T.; Álvarez-Noriega, M.; Álvarez-Romero, J.G.; Anderson, K.D.; Baird, A.H.; Babcock, R.C.; Beger, M.; Bellwood, D.R.; Berkelmans, R.; et al. Global warming and recurrent mass bleaching of corals. Nature 2017, 543, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Baird, A.H.; Bhagooli, R.; Ralph, P.J.; Takahashi, S. Coral bleaching: The role of the host. Trends Ecol. Evol. 2009, 24, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, D.A.; Petrou, K.; Gates, R.D. Coral bleaching from a single cell perspective. ISME J. 2018, 12, 1558–1567. [Google Scholar] [CrossRef]
- Lajeunesse, T.C.; Parkinson, J.E.; Gabrielson, P.W.; Jeong, H.J.; Reimer, J.D.; Voolstra, C.R.; Santos, S.R. Systematic revision of Symbiodiniaceae highlights the antiquity and diversity of coral endosymbionts. Curr. Biol. 2018, 28, 2570–2580.e6. [Google Scholar] [CrossRef]
- Shoguchi, E.; Shinzato, C.; Kawashima, T.; Gyoja, F.; Mungpakdee, S.; Koyanagi, R.; Takeuchi, T.; Hisata, K.; Tanaka, M.; Fujiwara, M.; et al. Draft assembly of the Symbiodinium minutum nuclear genome reveals dinoflagellate gene structure. Curr. Biol. 2013, 23, 1399–1408. [Google Scholar] [CrossRef]
- Lin, S.; Cheng, S.; Song, B.; Zhong, X.; Lin, X.; Li, W.; Li, L.; Zhang, Y.; Zhang, H.; Ji, Z.; et al. The Symbiodinium kawagutii genome illuminates dinoflagellate gene expression and coral symbiosis. Science 2015, 350, 691–694. [Google Scholar] [CrossRef]
- Liu, H.; Stephens, T.G.; Gonzalezpech, R.A.; Beltran, V.H.; Lapeyre, B.; Bongaerts, P.; Bongaerts, P.; Cooke, I.; Aranda, M.; Bourne, D.G. Symbiodinium genomes reveal adaptive evolution of functions related to symbiosis. Commun. Biol. 2018, 1, 95. [Google Scholar] [CrossRef]
- Aranda, M.; Li, Y.; Liew, Y.J.; Baumgarten, S.; Simakov, O.; Wilson, M.C.; Piel, J.; Ashoor, H.; Bougouffa, S.; Bajic, V.B.; et al. Genomes of coral dinoflagellate symbionts highlight evolutionary adaptations conducive to a symbiotic lifestyle. Sci. Rep. 2016, 6, 39734. [Google Scholar] [CrossRef]
- Shoguchi, E.; Beedessee, G.; Tada, I.; Hisata, K.; Kawashima, T.; Takeuchi, T.; Arakaki, N.; Fujie, M.; Koyanagi, R.; Roy, M.C.; et al. Two divergent Symbiodinium genomes reveal conservation of a gene cluster for sunscreen biosynthesis and recently lost genes. BMC Genom. 2018, 19, 458. [Google Scholar] [CrossRef] [PubMed]
- Krueger, T.; Becker, S.; Pontasch, S.; Dove, S.; Hoeghguldberg, O.; Leggat, W.; Fisher, P.L.; Davy, S.K. Antioxidant plasticity and thermal sensitivity in four types of Symbiodinium sp. J. Phycol. 2014, 50, 1035–1047. [Google Scholar] [CrossRef] [PubMed]
- Diazalmeyda, E.M.; Thome, P.E.; Hafidi, M.E.; Iglesiasprieto, R. Differential stability of photosynthetic membranes and fatty acid composition at elevated temperature in Symbiodinium. Coral Reefs 2011, 30, 217–225. [Google Scholar] [CrossRef]
- Rowan, R. Coral bleaching: Thermal adaptation in reef coral symbionts. Nature 2004, 430, 742. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.C.; Starger, C.J.; Mcclanahan, T.R.; Glynn, P.W. Coral reefs: Corals’ adaptive response to climate change. Nature 2004, 430, 741. [Google Scholar] [CrossRef] [PubMed]
- Larsen, P.E.; Trivedi, G.; Sreedasyam, A.; Lu, V.; Podila, G.K.; Collart, F.R. Using deep RNA sequencing for the structural annotation of the laccaria bicolor mycorrhizal transcriptome. PLoS ONE 2010, 5, e9780. [Google Scholar] [CrossRef] [PubMed]
- Filichkin, S.A.; Priest, H.D.; Givan, S.A.; Shen, R.; Bryant, U.W.; Fox, S.E.; Wong, W.-K.; Mockler, T.C. Genome-wide mapping of alternative splicing in Arabidopsis thaliana. Genome Res. 2009, 20, 45–58. [Google Scholar] [CrossRef]
- Guttman, M.; Garber, M.; Levin, J.Z.; Donaghey, J.; Robinson, J.; Adiconis, X.; Fan, L.; Koziol, M.J.; Gnirke, A.; Nusbaum, C.; et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat. Biotechnol. 2010, 28, 503–510. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Guillard, R.R.L.; Hargraves, P.E. Stichochrysis immobilis is a diatom, not a chrysophyte. Phycologia 1993, 32, 234–236. [Google Scholar] [CrossRef]
- Rodriguez, I.B.; Ho, T.-Y. Trace metal requirements and interactions in Symbiodinium kawagutii. Front. Microbiol. 2018, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Lin, X.; Yu, L.; Lin, S.; Rodriguez, I.B.; Ho, T.-Y. RNA-seq profiling of Fugacium kawagutii reveals strong responses in metabolic processes and symbiosis potential to deficiencies of iron and other trace metals. Sci. Total. Environ. 2019, 135767. [Google Scholar] [CrossRef]
- Zhang, H.; Hou, Y.; Miranda, L.; Campbell, D.A.; Sturm, N.R.; Gaasterland, T.; Lin, S. Spliced leader RNA trans-splicing in dinoflagellates. Proc. Natl. Acad. Sci. USA 2017, 104, 4618–4623. [Google Scholar] [CrossRef] [PubMed]
- Belton, J.M.; McCord, R.P.; Gibcus, J.H.; Naumova, N.; Zhan, Y.; Dekker, J. Hi-C: A comprehensive technique to capture the conformation of genomes. Methods 2012, 58, 268–276. [Google Scholar] [CrossRef]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef]
- Hackl, T.; Hedrich, R.; Schultz, J.; Förster, F. proovread: Large-scale high-accuracy PacBio correction through iterative short read consensus. Bioinformatics 2014, 30, 3004–3011. [Google Scholar] [CrossRef]
- Servant, N.; Varoquaux, N.; Lajoie, B.R.; Viara, E.; Chen, C.-J.; Vert, J.-P.; Heard, E.; Dekker, J.; Barillot, E. HiC-Pro: An optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015, 16, 259. [Google Scholar] [CrossRef]
- Durnd, N.C.; Shamim, M.S.; Machol, I.; Rao, S.S.P.; Huntley, M.H.; Lander, E.S.; Aiden, E.L. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Systems 2016, 3, 95–98. [Google Scholar] [CrossRef]
- Bonni, A.; Cohen, M.N.; Szabo, Q.; Fritsch, L.; Papadopoulos, G.L.; Lubling, Y.; Xu, L.; Lv, X.; Hugnot, J.-P.; Tanay, A. Faculty of 1000 evaluation for Multiscale 3D Genome Rewiring during Mouse Neural Development. Cell 2017, 171, 557–572. [Google Scholar]
- Perţea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Yu, L.; Zhang, H. Transcriptomic responses to thermal stress and varied 3 phosphorus conditions in Fugacium kawagutii. Microorganisms 2019, 7, 96. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Guo, C.; Zhang, Y.; Wang, C.; Lin, X.; Lin, S. Identification and expression analysis of an atypical alkaline phosphatase in Emiliania huxleyi. Front. Microbiol. 2018, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Boil. 2002, 3, research0034.1. [Google Scholar]
- Seligmann, H. An overlapping genetic code for frameshifted overlapping genes in Drosophila mitochondria: Antisense antitermination tRNAs UAR insert serine. J. Theor. Boil. 2012, 298, 51–76. [Google Scholar] [CrossRef]
- EI Houmami, N.; Seligmann, H. Evolution of nucleotide punctuation marks: From structural to linear signals. Front. Genet. 2017, 8, 36. [Google Scholar] [CrossRef]
- Seligmann, H.; Warthi, G. Chimeric translation for mitochondrial peptides: Regular and expanded codons. Comput. Struct. Biotechnol. 2019, 17, 1195–1202. [Google Scholar] [CrossRef]
- Shinzato, C.; Shoguchi, E.; Kawashima, T.; Hamada, M.; Hisata, K.; Tanaka, M.; Fujie, M.; Fujiwara, M.; Koyanagi, R.; Ikuta, T.; et al. Using the Acropora digitifera genome to understand coral responses to environmental change. Nature 2011, 476, 320–323. [Google Scholar] [CrossRef]
- Banaszak, A.T.; Lajeunesse, T.C.; Trench, R.K. The synthesis of mycosporine-like amino acids (MAAs) by cultured, symbiotic dinoflagellates. J. Exp. Mar. Boil. Ecol. 2000, 249, 219–233. [Google Scholar] [CrossRef]
- Banaszak, A.T.; Santos, M.G.B.; Lajeunesse, T.C.; Lesser, M.P. The distribution of mycosporine-like amino acids (MAAs) and the phylogenetic identity of symbiotic dinoflagellates in cnidarian hosts from the Mexican Caribbean. J. Exp. Mar. Boil. Ecol. 2006, 337, 131–146. [Google Scholar] [CrossRef]
- Davy, S.K.; Allemand, D.; Weis, V.M. Cell biology of cnidarian-dinoflagellate symbiosis. Microbiol. Mol. Boil. Rev. 2012, 76, 229–261. [Google Scholar] [CrossRef] [PubMed]
- Lajeunesse, T.C.; Pettay, D.T.; Sampayo, E.M.; Phongsuwan, N.; Brown, B.; Obura, D.O.; Hoegh-Guldberg, O.; Fitt, W.K. Long-standing environmental conditions, geographic isolation and host-symbiont specificity influence the relative ecological dominance and genetic diversification of coral endosymbionts in the genus Symbiodinium. J. Biogeogr. 2010, 37, 785–800. [Google Scholar] [CrossRef]
- Thornhill, D.J.; Lewis, A.M.; Wham, D.C.; LaJeunesse, T.C. Host-specialist lineages dominate the adaptive radiation of reef coral endosymbionts. Evolution 2014, 68, 352–367. [Google Scholar] [CrossRef] [PubMed]
- Levin, R.A.; Beltran, V.H.; Hill, R.; Kjelleberg, S.; Mcdougald, D.; Steinberg, P.D. Sex, scavengers, and chaperones: Transcriptome secrets of divergent Symbiodinium thermal tolerances. Mol. Biol. Evol. 2016, 33, 2201–2215. [Google Scholar] [CrossRef]
- Kaur, J.; Sebastian, J.; Siddiqi, I. The Arabidopsis-mei2-like genes play a role in meiosis and vegetative growth in Arabidopsis. Plant Cell 2006, 18, 545–559. [Google Scholar] [CrossRef]
- Lin, S. Genomic understanding of dinoflagellates. Res. Microbiol. 2011, 162, 551–569. [Google Scholar] [CrossRef]
- Takahashi, S.; Yoshioka-Nishimura, M.; Nanba, D.; Badger, M.R. Thermal acclimation of the symbiotic alga Symbiodinium spp. alleviates photobleaching under heat stress. Plant Physiol. 2013, 161, 477–485. [Google Scholar] [CrossRef]
- Rosic, N.N.; Pernice, M.; Rodriguez-Lanetty, M.; Hoegh-Guldberg, O. Validation of housekeeping genes for gene expression studies in Symbiodinium exposed to thermal and light stress. Mar. Biotechnol. 2011, 13, 355–365. [Google Scholar] [CrossRef]
- Krueger, T.; Fisher, P.L.; Becker, S.; Pontasch, S.; Dove, S.; Hoegh-Guldberg, O.; Leggat, W.; Davy, S.K. Transcriptomic characterization of the enzymatic antioxidants FeSOD, MnSOD, APX and KatG in the dinoflagellate genus Symbiodinium. BMC Evol. Boil. 2015, 15, 48. [Google Scholar] [CrossRef]
- Liu, C.; Wu, G.; Huang, X.; Liu, S.; Cong, B. Validation of housekeeping genes for gene expression studies in an ice alga Chlamydomonas during freezing acclimation. Extremophiles 2012, 16, 419–425. [Google Scholar] [CrossRef]
- De Jonge, H.J.M.; Fehrmann, R.S.N.; De Bont, E.S.J.M.; Hofstra, R.M.W.; Gerbens, F.; Kamps, W.A.; De Vries, E.G.E.; Van Der Zee, A.G.J.; Meerman, G.J.T.; Ter Elst, A. Evidence based selection of housekeeping genes. PLoS ONE 2007, 2, e898. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Zhang, H.; Lin, S. Tandem repeats, high copy number and remarkable diel expression rhythm of form II RuBisCO in Prorocentrum donghaiense (Dinophyceae). PLoS ONE 2013, 8, e71232. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Lin, S.; Huang, L.; Lu, W.; Li, M.; Liu, S. Suppression subtraction hybridization analysis revealed regulation of some cell cycle and toxin genes in Alexandrium catenella by phosphate limitation. Harmful Algae 2014, 39, 26–39. [Google Scholar] [CrossRef]
- Li, M.; Shi, X.; Guo, C.; Lin, S. Phosphorus deficiency inhibits cell division but not growth in the Dinoflagellate Amphidinium carterae. Front. Microbiol. 2016, 7, 559. [Google Scholar] [CrossRef]
- Mou, S.; Zhang, X.; Miao, J.; Zheng, Z.; Xu, D.; Ye, N. Reference genes for gene expression normalization in Chlamydomonas sp. ICE-L by quantitative real-time RT-PCR. J. Plant Biochem. Biotechnol. 2015, 24, 276–282. [Google Scholar] [CrossRef]
- Expósito-Rodríguez, M.; Borges, A.A.; Borges-Pérez, A.A.; Pérez, J. Selection of internal control genes for quantitative real-time RT-PCR studies during tomato development process. BMC Plant Biol. 2008, 8, 131. [Google Scholar] [CrossRef]
- Løvdal, T.; Lillo, C. Reference gene selection for quantitative real-time PCR normalization in tomato subjected to nitrogen, cold, and light stress. Anal. Biochem. 2009, 387, 238–242. [Google Scholar] [CrossRef]
- Ji, N.; Li, L.; Lin, L.; Lin, S. Screening for suitable reference genes for quantitative real-time PCR in Heterosigma akashiwo (Raphidophyceae). PLoS ONE 2015, 10, e0132183. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Assembled Genome Size (M) | % Genome Assembled | Gene No. | Scaffold N50 (kbp) | Gene Average Length (+ Intron) | Gene Supported by EST *(%) | Reference |
|---|---|---|---|---|---|---|---|
| B. minutum | 616 | 41.07 | 41,925 | 126.2 | 11,959 | 77.20 | [8] |
| S. microadriacticum | 808 | 73.45/57.71 | 49,109 | 573.5 | 12,898 | 76.30 | [11] |
| C. goreaui | 1030 | 85.55 | 35,913 | 98.9 | 6967 | 67.02 | [10] |
| Symbiodinium spp. | 767 | NA | 69,018 | 133.4 | 8834 | 67.50 | [12] |
| Cladocopium spp. | 705 | NA | 65,850 | 248.9 | 8192 | 62.50 | [12] |
| F. kawagutii | 935 | 79.24 | 36,850 | 381 | 3788 | 72.82 | [9] |
| 1050 | 88.98 | 26,609 | 268.8 | 6507 | 64.40 | [10] | |
| 937 | 79.41 | 45,192 | 13,533.5 | 7242 | 90.09 | This study |
| Project | Methodology | Growth Condition | Clean Read Data | Mapping Rate (Genome, Geneset) | ||
|---|---|---|---|---|---|---|
| V1 * | V2 ** | V3 *** | ||||
| Transcriptome1 # | BGI RNA-seq (DGE) (SE 50) | Normal Low Cu, Zn, Fe, Mn, Ni | 329 M | 75%, 33% | 69%, 33% | 69%, 54% |
| Transcriptome2 # | Illumina NGS (PE150) | Mix of 1# | 10 Gbp | 88%, 25% | 87%, 29% | 87%, 50% |
| Transcriptome3 # | Pacbio Sequel (full-length) | Mix of 1# | 15 Gbp | 99%, NA | 99%, NA | 99%, NA |
| BUSCO | V1 | V2 | V3 |
|---|---|---|---|
| Complete | 68 | 126 | 141 |
| Complete and single-copy | 56 | 112 | 130 |
| Complete and duplicated | 12 | 14 | 11 |
| Fragmented | 57 | 67 | 92 |
| Missing | 304 | 236 | 196 |
| Total detected (%) | 29% | 45% | 55% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, T.; Yu, L.; Song, B.; Song, Y.; Li, L.; Lin, X.; Lin, S. Genome Improvement and Core Gene Set Refinement of Fugacium kawagutii. Microorganisms 2020, 8, 102. https://doi.org/10.3390/microorganisms8010102
Li T, Yu L, Song B, Song Y, Li L, Lin X, Lin S. Genome Improvement and Core Gene Set Refinement of Fugacium kawagutii. Microorganisms. 2020; 8(1):102. https://doi.org/10.3390/microorganisms8010102
Chicago/Turabian StyleLi, Tangcheng, Liying Yu, Bo Song, Yue Song, Ling Li, Xin Lin, and Senjie Lin. 2020. "Genome Improvement and Core Gene Set Refinement of Fugacium kawagutii" Microorganisms 8, no. 1: 102. https://doi.org/10.3390/microorganisms8010102
APA StyleLi, T., Yu, L., Song, B., Song, Y., Li, L., Lin, X., & Lin, S. (2020). Genome Improvement and Core Gene Set Refinement of Fugacium kawagutii. Microorganisms, 8(1), 102. https://doi.org/10.3390/microorganisms8010102