Developing Gut Microbiota Exerts Colonisation Resistance to Clostridium (syn. Clostridioides) difficile in Piglets


Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Sampling
2.2. DNA Extraction
2.3. Sequencing and Computational Data Analysis
2.4. qPCR and Sample Enrichment
3. Results
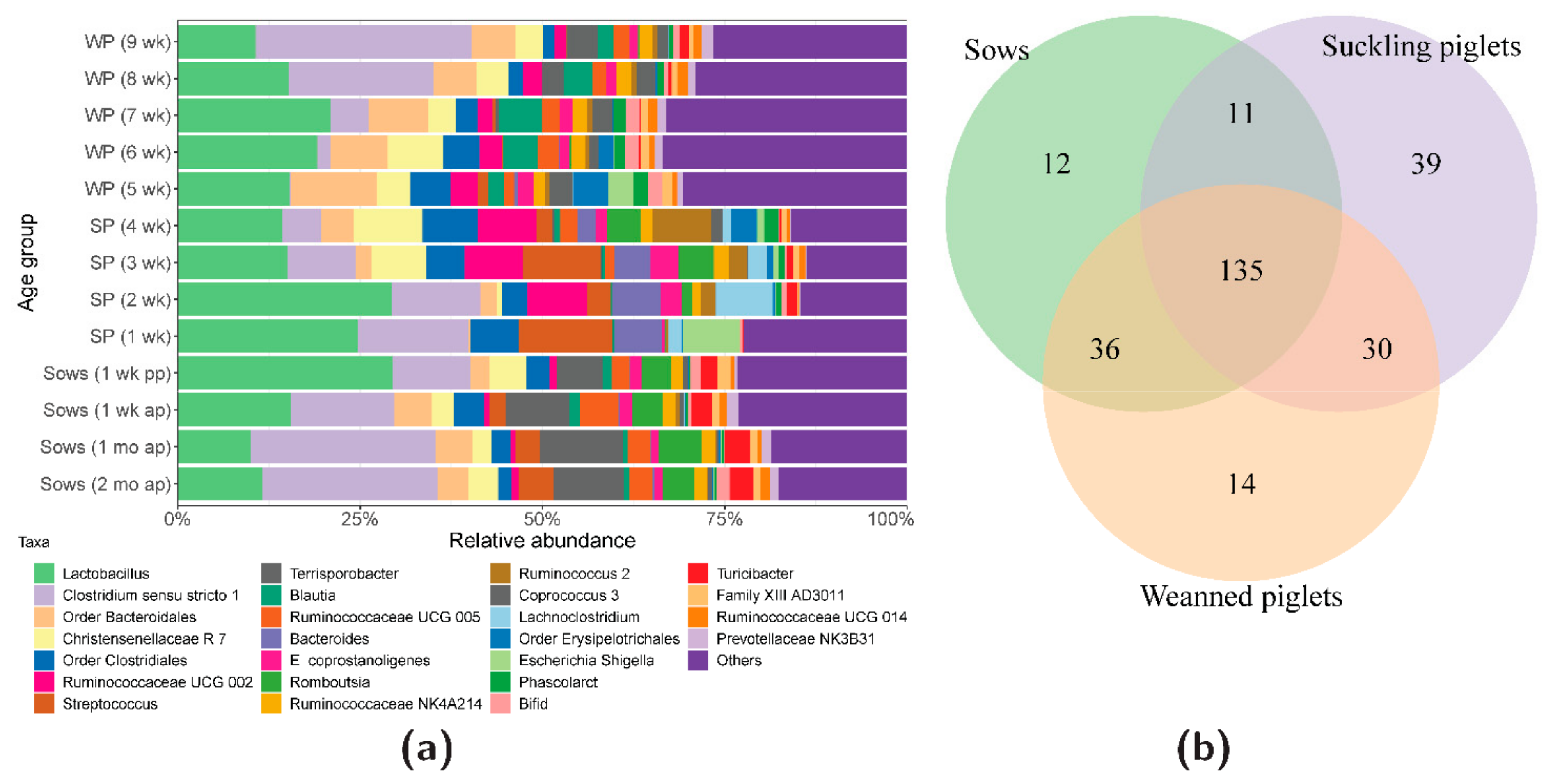
3.1. Bacterial Community Development
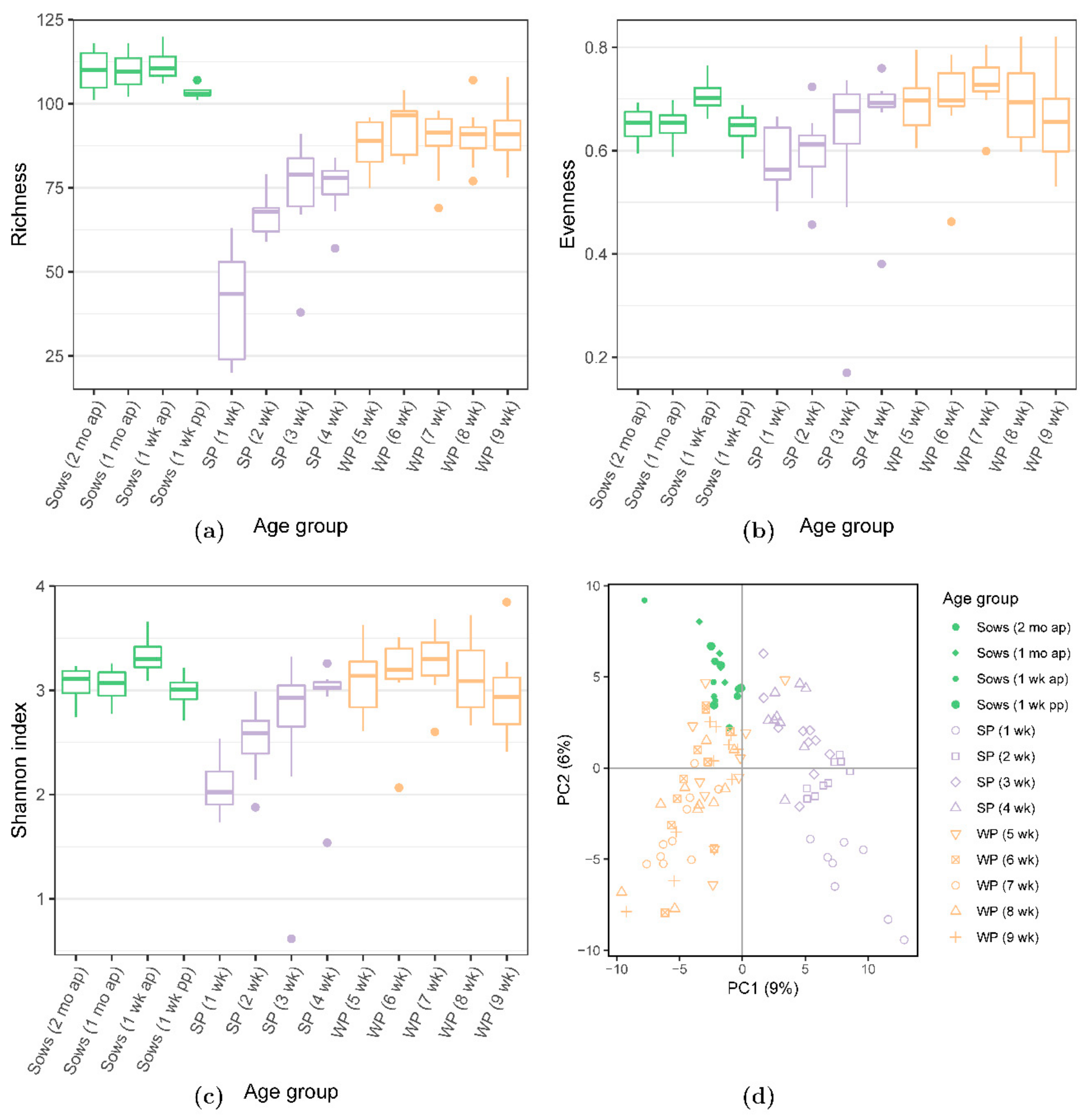
3.2. Bacterial Diversity Analyses
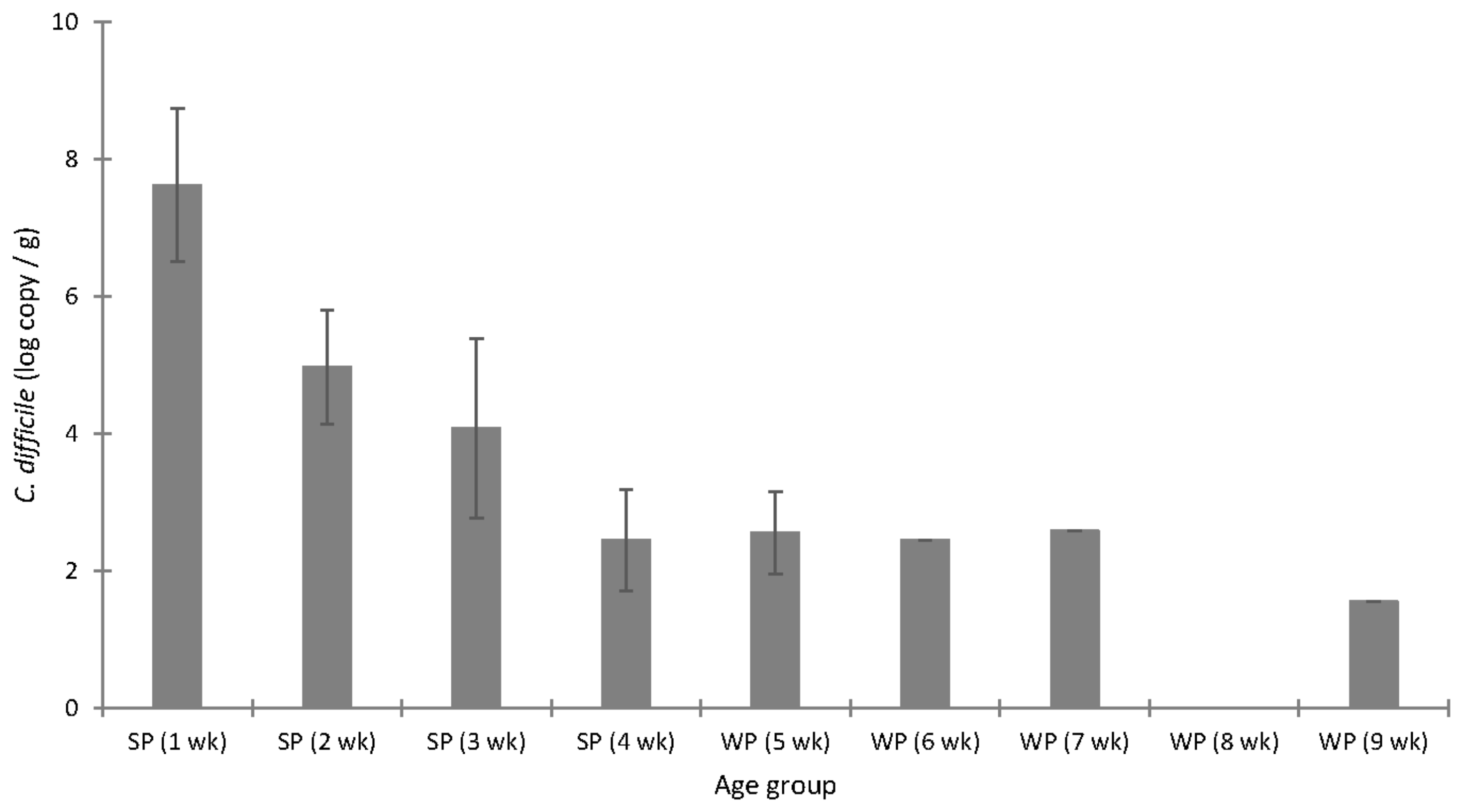
3.3. C. difficile Colonisation
3.4. Association of the Gut Microbiota and C. difficile in Suckling Piglets
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bian, G.; Ma, S.; Zhu, Z.; Su, Y.; Zoetendal, E.G.; Mackie, R.; Liu, J.; Mu, C.; Huang, R.; Smidt, H. Age, introduction of solid feed and weaning are more important determinants of gut bacterial succession in piglets than breed and nursing mother as revealed by a reciprocal cross-fostering model. Environ. Microbiol. 2016, 18, 1566–1577. [Google Scholar] [CrossRef] [PubMed]
- Grönlund, M.; Grzeskowiak, Ł.; Isolauri, E.; Salminen, S. Influence of mother’s intestinal microbiota on gut colonization in the infant. Gut Microbes 2011, 2, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Scharek-Tedin, L.; Kreuzer-Redmer, S.; Twardziok, S.O.; Siepert, B.; Klopfleisch, R.; Tedin, K.; Zentek, J.; Pieper, R. Probiotic Treatment Decreases the Number of CD14-Expressing Cells in Porcine Milk Which Correlates with Several Intestinal Immune Parameters in the Piglets. Front. Immunol. 2015, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Dou, S.; Gadonna-Widehem, P.; Rome, V.; Hamoudi, D.; Rhazi, L.; Lakhal, L.; Larcher, T.; Bahi-Jaber, N.; Pinon-Quintana, A.; Guyonvarch, A.; et al. Characterisation of Early-Life Fecal Microbiota in Susceptible and Healthy Pigs to Post-Weaning Diarrhoea. PLoS ONE 2017, 12, e0169851. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.L.; Wang, B.; Holmes, A.J. The immediate environment during postnatal development has long-term impact on gut community structure in pigs. ISME J. 2008, 2, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Pérez, S.; Blanco, J.L.; Bouza, E.; Alba, P.; Gibert, X.; Maldonado, J.; García, M.E. Prevalence of Clostridium difficile in diarrhoeic and non-diarrhoeic piglets. Vet. Microbiol. 2009, 137, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Grześkowiak, L.; Collado, M.C.; Mangani, C.; Maleta, K.; Laitinen, K.; Ashorn, P.; Isolauri, E.; Salminen, S.; Maleta, K. Distinct Gut Microbiota in Southeastern African and Northern European Infants. J. Pediatr. Gastroenterol. Nutr. 2012, 54, 812–816. [Google Scholar] [CrossRef]
- Rodriguez, C.; Taminiau, B.; Van Broeck, J.; Delmée, M.; Daube, G. Clostridium difficile in Food and Animals: A Comprehensive Review. Adv. Exp. Med. Biol. 2016, 932, 65–92. [Google Scholar] [PubMed]
- Weese, J.S.; Wakeford, T.; Reid-Smith, R.; Rousseau, J.; Friendship, R. Longitudinal investigation of Clostridium difficile shedding in piglets. Anaerobe 2010, 16, 501–504. [Google Scholar] [CrossRef]
- Grześkowiak, Ł.; Zentek, J.; Vahjen, W. Determination of the extent of Clostridium difficile colonisation and toxin accumulation in sows and neonatal piglets. Anaerobe 2016, 40, 5–9. [Google Scholar]
- Hopman, N.; Keessen, E.; Harmanus, C.; Sanders, I.; Van Leengoed, L.; Kuijper, E.; Lipman, L. Acquisition of Clostridium difficile by piglets. Vet. Microbiol. 2011, 149, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Keessen, E.; Donswijk, C.; Hol, S.; Hermanus, C.; Kuijper, E.; Lipman, L. Aerial dissemination of Clostridium difficile on a pig farm and its environment. Environ. Res. 2011, 111, 1027–1032. [Google Scholar] [CrossRef] [PubMed]
- Antharam, V.C.; Li, E.C.; Ishmael, A.; Sharma, A.; Mai, V.; Rand, K.H.; Wang, G.P. Intestinal Dysbiosis and Depletion of Butyrogenic Bacteria in Clostridium difficile Infection and Nosocomial Diarrhea. J. Clin. Microbiol. 2013, 51, 2884–2892. [Google Scholar] [CrossRef] [PubMed]
- Schubert, A.M.; Rogers, M.A.M.; Ring, C.; Mogle, J.; Petrosino, J.P.; Young, V.B.; Aronoff, D.M.; Schloss, P.D. Microbiome Data Distinguish Patients with Clostridium difficile Infection and Non-C. difficile-Associated Diarrhea from Healthy Controls. mBio 2014, 5, e01021-14. [Google Scholar] [CrossRef] [PubMed]
- Krůtová, M.; Zouharova, M.; Matejkova, J.; Tkadlec, J.; Krejci, J.; Faldyna, M.; Nyc, O.; Bernardy, J. The emergence of Clostridium difficile PCR ribotype 078 in piglets in the Czech Republic clusters with Clostridium difficile PCR ribotype 078 isolates from Germany, Japan and Taiwan. Int. J. Med. Microbiol. 2018, 308, 770–775. [Google Scholar] [CrossRef] [PubMed]
- DLG Verlag. GfE Empfehlungen Zur Energie-Und Nährstoffversorgung Von Schweinen; DLG Verlag: Frankfurt, Germany, 2006. [Google Scholar]
- Zentek, J.; Vahjen, W.; Grześkowiak, L. Physical Pre-Treatment Improves Efficient DNA Extraction and qPCR Sensitivity from Clostridium difficile Spores in Faecal Swine Specimens. Curr. Microbiol. 2016, 73, 727–731. [Google Scholar]
- Bushnell, B.; Rood, J.; Singer, E. BBMerge—Accurate paired shotgun read merging via overlap. PLoS ONE 2017, 12, e0185056. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. QIIME 2: Reproducible, interactive, scalable, and extensible microbiome data science. PeerJ Prepr. 2018, 6, e27295v2. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; A Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Holmes, S.P. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 2017, 11, 2639–2643. [Google Scholar] [CrossRef]
- Weiss, S.; Xu, Z.Z.; Peddada, S.; Amir, A.; Bittinger, K.; González, A.; Lozupone, C.; Zaneveld, J.R.; Vázquez-Baeza, Y.; Birmingham, A.; et al. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome 2017, 5, 27. [Google Scholar] [CrossRef] [PubMed]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Caporaso, J.G. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2014, 42, D643–D648. [Google Scholar] [CrossRef] [PubMed]
- RStudio Team. RStudio: Integrated Development for R; v0.98.1074; RStudio, Inc.: Boston, MA, USA, 2015. [Google Scholar]
- Berthold, M.R.; Cebron, N.; Dill, F.; Gabriel, T.R.; Kötter, T.; Meinl, T.; Ohl, P.; Thiel, K.; Wiswedel, B. KNIME—The Konstanz Information Miner: Version 2.0 and Beyond. SIGKDD Explor. Newsl. 2009, 11, 26–31. [Google Scholar] [CrossRef]
- Chen, X.; Xu, J.; Ren, E.; Su, Y.; Zhu, W. Co-occurrence of early gut colonization in neonatal piglets with microbiota in the maternal and surrounding delivery environments. Anaerobe 2018, 49, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Frese, S.A.; Parker, K.; Calvert, C.C.; Mills, D.A. Diet shapes the gut microbiome of pigs during nursing and weaning. Microbiome 2015, 3, 1237439. [Google Scholar] [CrossRef]
- Pluske, J.R.; Turpin, D.L.; Kim, J.C. Gastrointestinal tract (gut) health in the young pig. Anim. Nutr. 2018, 4, 187–196. [Google Scholar] [CrossRef]
- Bandelj, P.; Harmanus, C.; Blagus, R.; Cotman, M.; Kuijper, E.J.; Ocepek, M.; Vengust, M. Quantification of Clostridioides (Clostridium) difficile in feces of calves of different age and determination of predominant Clostridioides difficile ribotype 033 relatedness and transmission between family dairy farms using multilocus variable-number tandem-repeat analysis. BMC Vet. Res. 2018, 14, 298. [Google Scholar]
- Buffie, C.G.; Bucci, V.; Stein, R.R.; McKenney, P.T.; Ling, L.; Gobourne, A.; No, D.; Liu, H.; Kinnebrew, M.; Viale, A.; et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 2015, 517, 205–208. [Google Scholar] [CrossRef]
- Grześkowiak, Ł.M.; Pieper, R.; Huynh, H.A.; Cutting, S.M.; Vahjen, W.; Zentek, J. Impact of early-life events on the susceptibility to Clostridium difficile colonisation and infection in the offspring of the pig. Gut Microbes 2018, 10, 251–259. [Google Scholar]
- Nagaro, K.J.; Phillips, S.T.; Cheknis, A.K.; Sambol, S.P.; Zukowski, W.E.; Johnson, S.; Gerding, D.N. Nontoxigenic Clostridium difficile Protects Hamsters against Challenge with Historic and Epidemic Strains of Toxigenic BI/NAP1/027 C. difficile. Antimicrob. Agents Chemother. 2013, 57, 5266–5270. [Google Scholar] [CrossRef] [PubMed]
- Songer, J.G.; Jones, R.; Anderson, M.A.; Barbara, A.J.; Post, K.W.; Trinh, H.T. Prevention of porcine Clostridium difficile-associated disease by competitive exclusion with nontoxigenic organisms. Veter. Microbiol. 2007, 124, 358–361. [Google Scholar] [CrossRef] [PubMed]
- May, T.; Mackie, R.I.; Fahey, G.C.; Cremin, J.C.; Garleb, K.A. Effect of Fiber Source on Short-Chain Fatty Acid Production and on the Growth and Toxin Production by Clostridium difficile. Scand. J. Gastroenterol. 1994, 29, 916–922. [Google Scholar] [CrossRef] [PubMed]
- Grześkowiak, Ł.; Martínez-Vallespín, B.; Dadi, T.H.; Radloff, J.; Amasheh, S.; Heinsen, F.A.; Franke, A.; Reinert, K.; Vahjen, W.; Zentek, J.; et al. Formula feeding predisposes neonatal piglets to Clostridium difficile gut infection. J. Infect. Dis. 2018, 217, 1442–1452. [Google Scholar] [CrossRef] [PubMed]
- Elliott, B.; Chang, B.J.; Golledge, C.L.; Riley, T.V. Clostridium difficile-associated diarrhoea. Intern. Med. J. 2007, 37, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Kyne, L.; Warny, M.; Qamar, A.; Kelly, C.P. Association between antibody response to toxin A and protection against recurrent Clostridium difficile diarrhoea. Lancet 2001, 357, 189–193. [Google Scholar] [CrossRef]
- Steele, J.; Mukherjee, J.; Parry, N.; Tzipori, S. Antibody against TcdB, but not TcdA, prevents development of gastrointestinal and systemic Clostridium difficile disease. J. Infect. Dis. 2013, 207, 323–330. [Google Scholar] [CrossRef]
- Nerandzic, M.M.; Donskey, C.J. Effective and reduced-cost modified selective medium for isolation of Clostridium difficile. J. Clin. Microbiol. 2009, 47, 397–400. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Age (Week) | % of Positive Samples | |
|---|---|---|
| Before Enrichment | After Enrichment | |
| 1 | 100 | 100 |
| 2 | 100 | 100 |
| 3 | 90 | 90 |
| 4 | 56 | 100 |
| 5 | 30 | 80 |
| 6 | 10 | 60 |
| 7 | 10 | 60 |
| 8 | 0 | 10 |
| 9 | 11 | 56 |
| Taxon | Pearson’s r (n = 31) | p |
|---|---|---|
| Ruminococcus 2 spp. | −0.5960 | 0.0004 |
| Christensenellaceae R7 | −0.5936 | 0.0004 |
| Romboutsia spp. | −0.5936 | 0.0004 |
| Marvinbryantia spp. | −0.5669 | 0.0009 |
| Ruminococcaceae UCG 002 | −0.5566 | 0.0012 |
| Fournierella spp. | −0.5488 | 0.0014 |
| Ruminococcaceae UCG 010 | −0.5296 | 0.0022 |
| Terrisporobacter spp. | −0.5286 | 0.0023 |
| Coprococcus 3 spp. | −0.5213 | 0.0026 |
| Hydrogenoanaerobacterium spp. | −0.5193 | 0.0028 |
| Prevotellaceae UCG 004 | −0.5188 | 0.0028 |
| Corynebacterium 1 spp. | −0.5163 | 0.0029 |
| Globicatella spp. | 0.5025 | 0.0040 |
| Pasteurella spp. | 0.5257 | 0.0024 |
| Clostridioides spp. | 0.5381 | 0.0018 |
| Clostridium sensu stricto 2 spp. | 0.5838 | 0.0006 |
| Peptostreptococcus spp. | 0.6064 | 0.0003 |
| Enterococcus spp. | 0.6272 | 0.0002 |
| Staphylococcus spp. | 0.6428 | 9.6402 × 10−5 |
| Moraxella spp. | 0.6447 | 9.0628 × 10−5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grześkowiak, Ł.; Dadi, T.H.; Zentek, J.; Vahjen, W. Developing Gut Microbiota Exerts Colonisation Resistance to Clostridium (syn. Clostridioides) difficile in Piglets. Microorganisms 2019, 7, 218. https://doi.org/10.3390/microorganisms7080218
Grześkowiak Ł, Dadi TH, Zentek J, Vahjen W. Developing Gut Microbiota Exerts Colonisation Resistance to Clostridium (syn. Clostridioides) difficile in Piglets. Microorganisms. 2019; 7(8):218. https://doi.org/10.3390/microorganisms7080218
Chicago/Turabian StyleGrześkowiak, Łukasz, Temesgen Hailemariam Dadi, Jürgen Zentek, and Wilfried Vahjen. 2019. "Developing Gut Microbiota Exerts Colonisation Resistance to Clostridium (syn. Clostridioides) difficile in Piglets" Microorganisms 7, no. 8: 218. https://doi.org/10.3390/microorganisms7080218
APA StyleGrześkowiak, Ł., Dadi, T. H., Zentek, J., & Vahjen, W. (2019). Developing Gut Microbiota Exerts Colonisation Resistance to Clostridium (syn. Clostridioides) difficile in Piglets. Microorganisms, 7(8), 218. https://doi.org/10.3390/microorganisms7080218