Supercoil Levels in E. coli and Salmonella Chromosomes Are Regulated by the C-Terminal 35–38 Amino Acids of GyrA



Abstract
1. Introduction
2. Materials and Methods
2.1. Strains and Cloning
2.2. Growth Rate Measurements and Resolution Assays
2.3. Enzyme Purification
2.4. Biochemical Methods
2.5. Genetic Methods
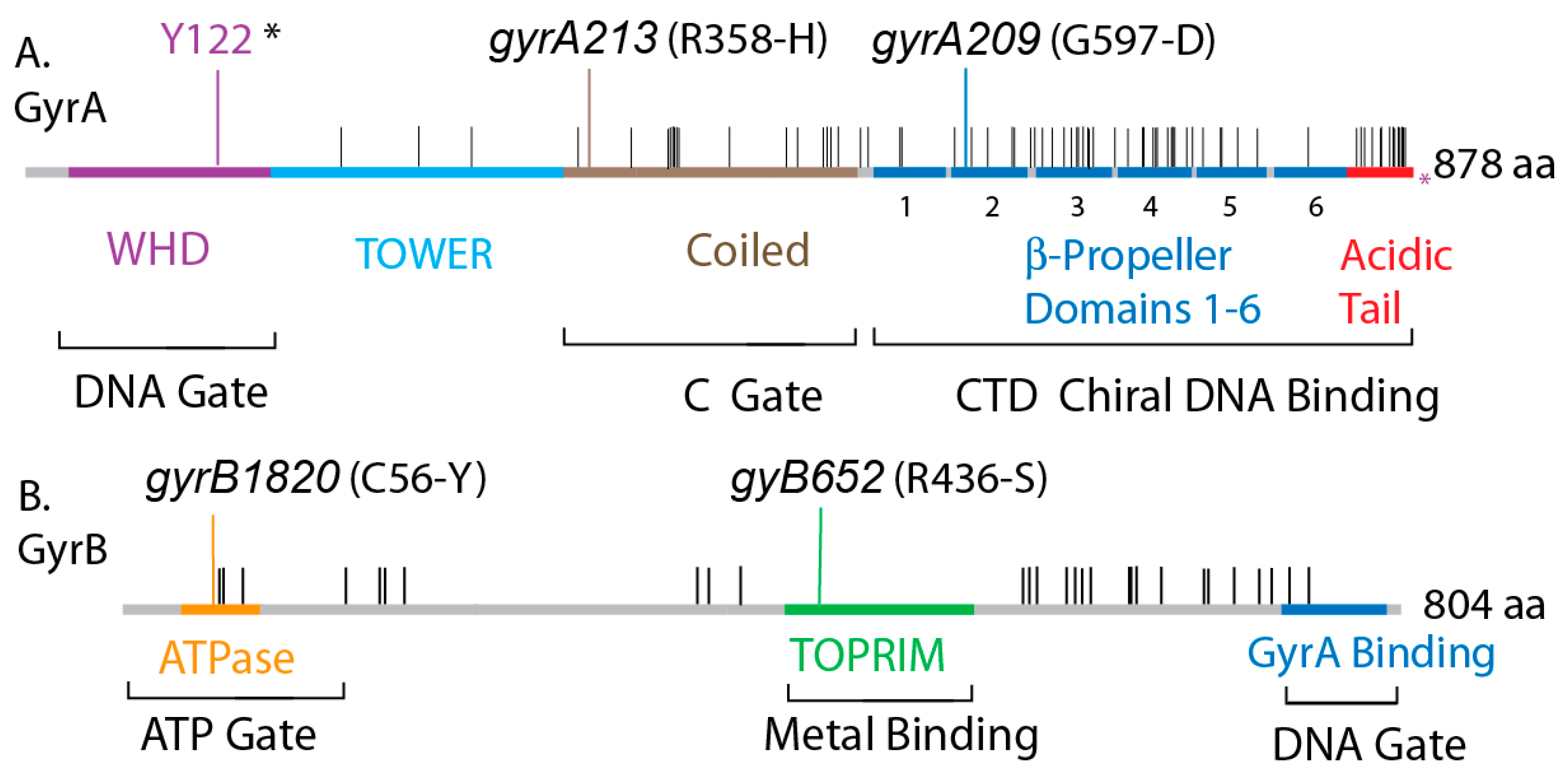
3. Results
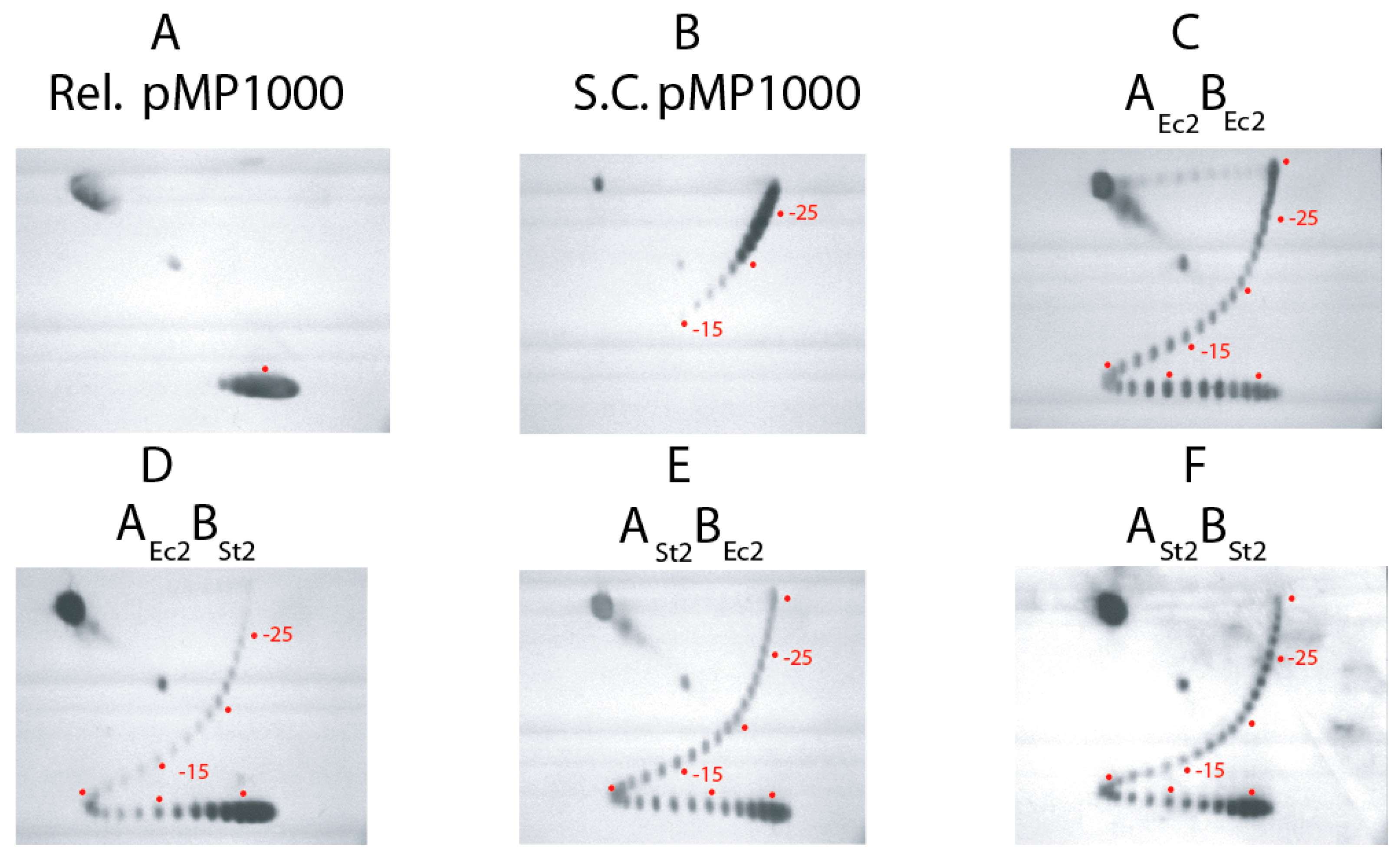
3.1. Supercoiling Assays of WT and Interspecies Forms of Gyrase
3.2. In Vivo Chromosome Supercoiling in Transgenic Salmonellae
3.3. Supercoiling in gyrBEc Transgenics
3.4. Salmonella GyrA + GyrB Double Transgenics
3.5. Transgenic Swaps of Salmonellae gyrBSt and gyrASt in E. coli
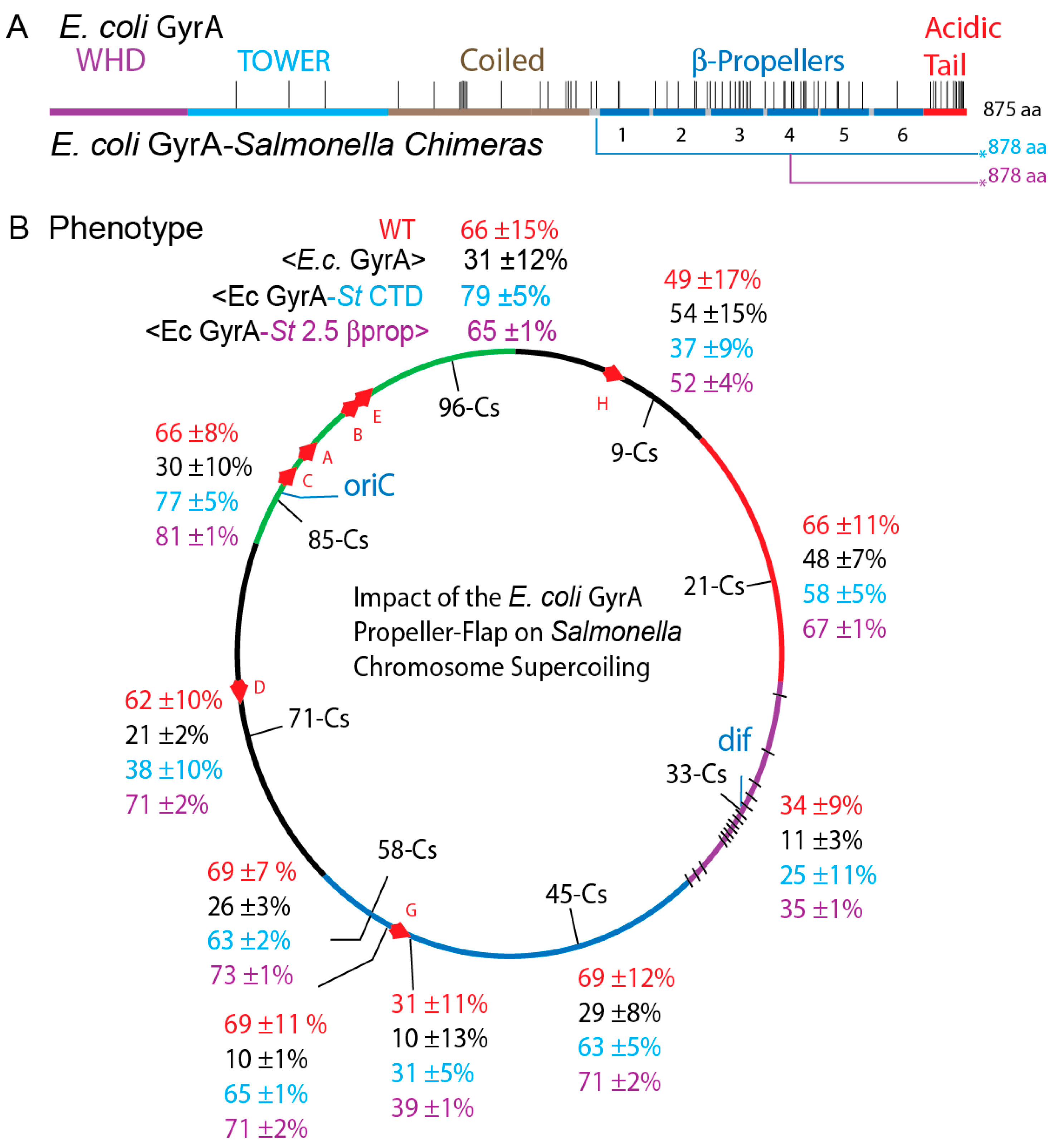
3.6. The GyrA CTD Controls Species Supercoiling
3.7. Transgenic Salmonella GyrA Chimeras in E. coli
3.8. Chromosome Supercoil Density in E. coli
4. Discussion
4.1. Evolution of Chromosomal Supercoil Control
4.2. Supercoil Domain Theory and Measurements
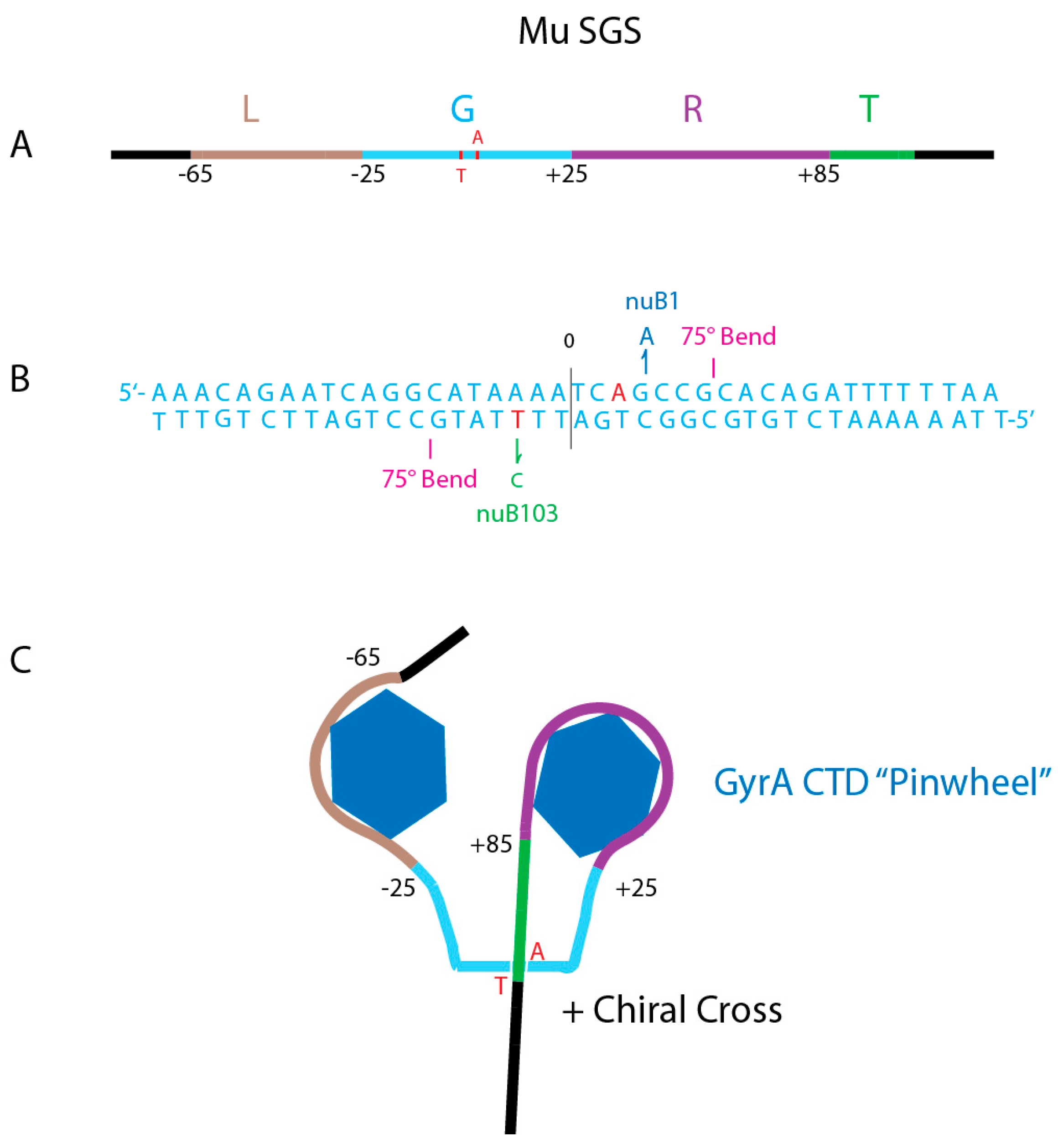
4.3. What Do Strong Gyrase Sites Look Like?
4.4. A Species-Specific Rheostat Regulates Supercoiling at Strong Binding Sites
4.5. Where Are the Strong Gyrase Sites in E. coli and Salmonella chromosomes?
4.6. Gyrase Supercoiling Mechanisms in Distantly Related Species
4.7. E. coli GyrA in Salmonella
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References and Note
- Martignoni, M.; Groothuis, G.M.; de Kanter, R. Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin. Drug Metab. Toxicol. 2006, 2, 875–894. [Google Scholar] [CrossRef] [PubMed]
- Higgins, N.P. Species-specific supercoil dynamics of the bacterial nucleoid. Biophys. Rev. 2016, 8, 113–121. [Google Scholar] [CrossRef]
- Champion, K.; Higgins, N.P. Growth rate toxicity phenotypes and homeostatic supercoil control differentiate Escherichia coli from Salmonella enterica serovar Typhimurium. J. Bacteriol. 2007, 189, 5839–5849. [Google Scholar] [CrossRef] [PubMed]
- Dorman, C.J.; Bhriain, N.N.; Higgins, C.F. DNA supercoiling and environmental regulation of virulence gene expression in Shigella flexneri. Nature 1990, 344, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Travers, A.; Muskhelishvili, G. DNA supercoiling—A global transcriptional regulator for enterobacterial growth? Nat. Rev. Microbiol. 2005, 3, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Higgins, N.P.; Deng, S.; Pang, Z.; Stein, R.; Champion, K.; Manna, D. Domain behavior and supercoil dynamics in bacterial chromosomes. In The Bacterial Chromosome; Higgins, N.P., Ed.; ASM Press: Washington, DC, USA, 2005; pp. 133–153. [Google Scholar]
- Zechiedrich, E.L.; Khodursky, A.B.; Bachellier, S.; Schneider, R.; Chen, D.; Lilley, D.M.; Cozzarelli, N.R. Roles of topoisomerases in maintaining steady-state DNA supercoiling in Escherichia coli. J. Biol. Chem. 2000, 275, 8103–8113. [Google Scholar] [CrossRef] [PubMed]
- Booker, B.M.; Deng, S.; Higgins, N.P. DNA topology of highly transcribed operons in Salmonella enterica serovar Typhimurium. Mol. Microbiol. 2010, 78, 1348–1364. [Google Scholar] [CrossRef]
- Rovinskiy, N.; Agbleke, A.A.; Chesnokova, O.; Pang, Z.; Higgins, N.P. Rates of gyrase supercoiling and transcription elongation control supercoil density in a bacterial chromosome. PLoS Genet. 2012, 8, e1002845. [Google Scholar] [CrossRef]
- Stupina, V.A.; Wang, J.C. Viability of Escherichia coli topA mutants lacking DNA topoisomerase I. Proc. Natl. Acad. Sci. USA 2005, 280, 355–360. [Google Scholar]
- Drolet, M.; Broccoli, S.; Rallu, F.; Hraiky, C.; Fortin, C.; Masse, E.; Baaklini, I. The problem of hypernegative supercoiling and R-loop formation in transcription. Front. Biosci. 2003, 8, D210–D221. [Google Scholar] [CrossRef]
- Higgins, N.P.; Booker, B.M.; Manna, D.M. Molecular Structure and Dynamics of Bacterial Nucleoids. In Bacterial Chromatin; Dorman, C.J., Dame, R.T., Eds.; Springer: New York, NY, USA, 2010; pp. 117–148. [Google Scholar]
- Das, A.; Liu, C.; Zeng, C.; Li, G.; Li, T.; Rosi, N.L.; Jin, R. Cyclopentanethiolato-protected Au36(SC5H9)24 nanocluster: Crystal structure and implications for the steric and electronic effects of ligand. J. Phys. Chem. A 2014, 118, 8264–8269. [Google Scholar] [CrossRef]
- Hayama, R.; Marians, K.J. Physical and functional interaction between the condensin MukB and the decatenase topoisomerase IV in Escherichia coli. Proc. Natl. Acad. Sci. USA 2010, 107, 18826–18831. [Google Scholar] [CrossRef]
- Hiasa, H.; Marians, K.J. Topoisomerase IV can support oriC DNA replication in vitro. J. Biol. Chem. 1994, 269, 16371–16375. [Google Scholar]
- Cozzarelli, N.R.; Wang, J.C. DNA Topology and its Biological Effects; Cold Spring Harbor Press: Cold Spring Harbor, NY, USA, 1990; p. 480. [Google Scholar]
- Holmes, V.F.; Cozzarelli, N.R. Closing the ring: Links between SMC proteins and chromosome partitioning, condensation, and supercoiling. Proc. Natl. Acad. Sci. USA 2000, 97, 1322–1324. [Google Scholar] [CrossRef] [PubMed]
- Ogura, T.; Niki, H.; Mori, H.; Morita, M.; Hasegawa, M.; Ichinose, C.; Hiraga, S. Identification and characterization of gyrB mutants of Escherichic coli that are defective in partitioning of mini-F plasmids. J. Bacteriol. 1990, 172, 1562–1568. [Google Scholar] [CrossRef] [PubMed]
- Sternglanz, R.; DiNardo, S.; Voelkel, K.A.; Nishimura, Y.; Hirota, Y.; Becherer, K.; Zumstein, L.; Wang, J.C. Mutations in the gene coding for Escherichia coli DNA topoisomerase I affect transcription and transposition. Proc. Natl. Acad. Sci. USA 1981, 78, 2747–2751. [Google Scholar] [CrossRef] [PubMed]
- Peter, B.J.; Arsuaga, J.; Breier, A.M.; Khodursky, A.B.; Brown, P.O.; Cozzarelli, N.R. Genomic transcriptional response to loss of chromosomal supercoiling in Escherichia coli. Genome Biol. 2004, 5, R87. [Google Scholar] [CrossRef]
- Isberg, R.R.; Syvanen, M. DNA gyrase is a host factor required for transposition of Tn5. Cell 1982, 30, 9–18. [Google Scholar] [CrossRef]
- Ross, W.; Shore, S.H.; Howe, M.M. Mutants of Escherichia coli defective for replicative transposition of bacteriophage Mu. J. Bacteriol. 1986, 167, 905–919. [Google Scholar] [CrossRef]
- Pato, M.L.; Banerjee, M. The Mu strong gyrase-binding site promotes efficient synapsis of the prophage termini. Mol. Microbiol. 1996, 22, 283–292. [Google Scholar] [CrossRef]
- Stols, L.; Gu, M.; Dieckman, L.; Raffen, R.; Collart, F.R.; Donnelly, M.I. A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein Expr. Purif. 2002, 25, 8–15. [Google Scholar] [CrossRef]
- Dieckman, L.; Gu, M.; Stols, L.; Donnelly, M.I.; Collart, F.R. High throughput methods for gene cloning and expression. Protein Expr. Purif. 2002, 25, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.N.; Yang, X.; Hua, Z.C. Expression and purification of recombinant human annexin V in Escherichia coli. Prep. Biochem. Biotechnol. 2000, 30, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Higgins, N.P.; Peebles, C.L.; Sugino, A.; Cozzarelli, N.R. Purification of the subunits of Escherichia coli DNA gyrase and reconstitution of enzymatic activity. Proc. Natl. Acad. Sci. USA 1978, 75, 1773–1777. [Google Scholar] [CrossRef] [PubMed]
- Higgins, N.P.; Cozzarelli, N.R. The binding of gyrase to DNA: Analysis by retention by nitrocellulose filters. Nucleic Acids Res. 1982, 10, 6833–6847. [Google Scholar] [CrossRef]
- Scheirer, K.E.; Higgins, N.P. The DNA cleavage reaction of DNA gyrase. Comparison of stable ternary complexes formed with enoxacin and CcdB protein. J. Biol. Chem. 1997, 272, 27202–27209. [Google Scholar] [CrossRef]
- Sharan, S.K.; Thomason, L.C.; Kuznetsov, S.G.; Court, D.L. Recombineering: A homologous recombination-based method of genetic engineering. Nat. Protoc. 2009, 4, 206–223. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Costantino, N.; Zhou, X.; Court, D.L. Identification and analysis of recombineering functions from Gram-negative and Gram-positive bacteria and their phages. Proc. Natl. Acad. Sci. USA 2008, 105, 1626–1631. [Google Scholar] [CrossRef]
- Pang, Z.; Chen, R.; Manna, D.; Higgins, N.P. A gyrase mutant with low activity disrupts supercoiling at the replication terminus. J. Bacteriol. 2005, 187, 7773–7783. [Google Scholar] [CrossRef]
- Tretter, E.M.; Berger, J.M. Mechanisms for defining supercoiling set point of DNA gyrase orthologs: I. A nonconserved acidic C-terminal tail modulates Escherichia coli gyrase activity. J. Biol. Chem. 2012, 287, 18636–18644. [Google Scholar] [CrossRef]
- Tretter, E.M.; Berger, J.M. Mechanisms for defining supercoiling set point of DNA gyrase orthologs: II. The shape of the GyrA subunit C-terminal domain (CTD) is not a sole determinant for controlling supercoiling efficiency. J. Biol. Chem. 2012, 287, 18645–18654. [Google Scholar] [CrossRef]
- Lanz, M.A.; Farhat, M.; Klostermeier, D. The acidic C-terminal tail of the GyrA subunit moderates the DNA supercoiling activity of Bacillus subtilis gyrase. J. Biol. Chem. 2014, 289, 12275–12285. [Google Scholar] [CrossRef]
- Pato, M.L.; Howe, M.M.; Higgins, N.P. A DNA gyrase-binding site at the center of the bacteriophage Mu genome is required for efficient replicative transposition. Proc. Natl. Acad. Sci. USA 1990, 87, 8716–8720. [Google Scholar] [CrossRef] [PubMed]
- Oram, M.; Travers, A.A.; Howells, A.J.; Maxwell, A.; Pato, M.L. Dissection of the bacteriophage Mu strong gyrase site (SGS): Significance of the SGS right arm in Mu biology and DNA gyrase mechanism. J. Bacteriol. 2006, 188, 619–632. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Schoeffler, A.J.; Berger, J.M.; Bryant, Z. ATP binding controls distinct structural transitions of Escherichia coli DNA gyrase in complex with DNA. Nat. Struct. Mol. Biol. 2012, 19, 538–546. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Depew, R.E.; Wang, J.C. Conformational fluctuations of DNA helix. Proc. Natl. Acad. Sci. USA 1975, 72, 4275–4279. [Google Scholar] [CrossRef] [PubMed]
- Vologodskii, A.V.; Cozzarelli, N.R. Conformational and thermodynamic properties of supercoiled DNA. Annu. Rev. Biophys. Biomol. Struct. 1994, 23, 609–643. [Google Scholar] [CrossRef]
- Wendorff, T.J.; Schmidt, B.H.; Heslop, P.; Austin, C.A.; Berger, J.M. The structure of DNA-bound human topoisomerase II alpha: Conformational mechanisms for coordinating inter-subunit interactions with DNA cleavage. J. Mol. Biol. 2012, 424, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Pei, J.; Grishin, N.V. C-terminal domain of gyrase A is predicted to have a beta-propeller structure. Proteins 2002, 47, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Corbett, K.D.; Shultzaberger, R.K.; Berger, J.M. The C-terminal domain of DNA gyrase A adopts a DNA-bending beta-pinwheel fold. Proc. Natl. Acad. Sci. USA 2004, 101, 7293–7298. [Google Scholar] [CrossRef]
- Ochman, H.; Groisman, E.A. The origin and evolution of species differences in Escherichia coli and Salmonella typhimurium. EXS 1994, 69, 479–493. [Google Scholar] [PubMed]
- Menzel, R.; Gellert, M. Regulation of the genes for E. coli DNA gyrase: Homeostatic control of DNA supercoiling. Cell 1983, 34, 105–113. [Google Scholar] [CrossRef]
- Gmuender, H.; Kuratli, K.; Di Padova, K.; Gray, C.P.; Keck, W.; Evers, S. Gene expression changes triggered by exposure of Haemophilus influenzae to novobiocin or ciprofloxacin: Combined transcription and translation analysis. Genome Res. 2010, 11, 28–42. [Google Scholar] [CrossRef]
- Marrer, E.; Satoh, A.T.; Johnson, M.M.; Piddock, L.J.V.; Page, M.B.P. Global Transcriptome Analysis of the Responses of a Fluoroquinolone-Resistant Streptococcus pneumoniae Mutant and Its Parent to Ciprofloxacin. Antimicrob. Agents Chemother. 2006, 50, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Cirz, R.T.; O’Neil, B.M.; Hammond, J.A.; Head, S.R.; Romesberg, F.E. Defining the Pseudomonas aeruginosa SOS Response and Its Role in the Global Response to the Antibiotic Ciprofloxacin. J. Bacteriol. 2006, 188, 7101–7110. [Google Scholar] [CrossRef]
- Jensen, P.R.; van der Weijden, C.C.; Jensen, L.B.; Westerhoff, H.V.; Snoep, J.L. Extensive regulation compromises the extent to which DNA gyrase controls DNA supercoiling and growth rate of Escherichia coli. Eur. J. Biochem. 1999, 226, 865–877. [Google Scholar] [CrossRef]
- Snoep, J.L.; van der Weijden, C.C.; Andersen, H.W.; Westerhoff, H.V.; Jensen, P.R. DNA supercoiling in Escherichia coli is under tight and subtle homeostatic control, involving gene-expression and metabolic regulation of both topoisomerase I and DNA gyrase. Eur. J. Biochem. 2002, 269, 1662–1669. [Google Scholar] [CrossRef]
- Hsieh, L.-S.; Rouviere-Yaniv, J.; Drlica, K. Bacterial DNA supercoiling and ATP/ADP ratio: changes associated with salt shock. J. Bacteriol. 1991, 173, 3914–3917. [Google Scholar] [CrossRef]
- Hsieh, L.S.; Burger, R.M.; Drlica, K. Bacterial DNA supercoiling and ATP/ADP. Changes associated with a transition to anaerobic growth. J. Mol. Biol. 1991, 219, 443–450. [Google Scholar] [CrossRef]
- Reyes-Dominguez, Y.; Contreras-Ferrat, G.; Ramirez-Santos, J.; Membrillo-Hernandez, J.; Gomez-Eichelmann, M.C. Plasmid DNA supercoiling and gyrase activity in Escherichia coli wild-type and rpoS stationary-phase cells. J. Bacteriol. 2003, 185, 1097–1100. [Google Scholar] [CrossRef]
- Sinden, R.R.; Pettijohn, D.E. Chromosomes in living Escherichia coli cells are segregated into domains of supercoiling. Proc. Natl. Acad. Sci. USA 1981, 78, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Pettijohn, D.E. The nucleoid. In Escherichia coli and Salmonella, 2nd ed.; Neidhardt, F.C., Ed.; ASM Press: Washington, DC, USA, 1996; Volume 1, pp. 158–166. [Google Scholar]
- In retrospect, the Sinden and Pettijohn estimate of 50 domains with 100 kb dimension was correct for cells treated with X-rays and then held in salt solution for recovery. There would be no transcription in these cells. When Salmonella cells enter stationary phase, which has a low transcription rate, the domain size expands to 100 kb.
- Higgins, N.P.; Yang, X.; Fu, Q.; Roth, J.R. Surveying a supercoil domain by using the gd resolution system in Salmonella typhimurium. J. Bacteriol. 1996, 178, 2825–2835. [Google Scholar] [CrossRef] [PubMed]
- Stein, R.; Deng, S.; Higgins, N.P. Measuring chromosome dynamics on different timescales using resolvases with varying half-lives. Mol. Microbiol. 2005, 56, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Postow, L.; Hardy, C.D.; Arsuaga, J.; Cozzarelli, N.R. Topological domain structure of the Escherichia coli chromosome. Genes Dev. 2004, 18, 1766–1779. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.F.; Wang, J.C. Supercoiling of the DNA template during transcription. Proc. Natl. Acad. Sci. USA 1987, 84, 7024–7027. [Google Scholar] [CrossRef]
- Deng, S.; Stein, R.A.; Higgins, N.P. Transcription-induced barriers to supercoil diffusion in the Salmonella typhimurium chromosome. Proc. Natl. Acad. Sci. USA 2004, 101, 3398–3403. [Google Scholar] [CrossRef]
- Higgins, N.P. RNA polymerase: Chromosome domain boundary maker and regulator of supercoil density. Curr. Opin. Microbiol. 2014, 22, 138–143. [Google Scholar] [CrossRef]
- Taniguchi, Y.; Choi, P.J.; Li, G.W.; Chen, H.; Babu, M.; Hearn, J.; Emili, A.; Xie, X.S. Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science 2010, 329, 533–538. [Google Scholar] [CrossRef]
- Chong, S.; Chen, C.; Ge, H.; Xie, X.S. Mechanism of transcriptional bursting in bacteria. Cell 2014, 158, 314–326. [Google Scholar] [CrossRef]
- Higgins, N.P.; Manlapaz-Ramos, P.; Gandhi, R.T.; Olivera, B.M. Bacteriophage Mu: A transposing replicon. Cell 1983, 33, 623–628. [Google Scholar] [CrossRef]
- Yoshida, R.K.; Miller, J.K.; Miller, H.I.; Friedman, K.I.; Howe, M.M. Isolation and mapping of Mu nu mutants which grow in him mutants of E. coli. Virology 1982, 120, 269–272. [Google Scholar] [CrossRef]
- Pato, M.L. Central location of the Mu strong gyrase binding site is obligatory for optimal rates of replicative transposition. Proc. Natl. Acad. Sci. USA 1994, 91, 7056–7060. [Google Scholar] [CrossRef] [PubMed]
- Pato, M.L.; Karlok, M.; Wall, C.; Higgins, N.P. Characterization of Mu prophage lacking the central strong gyrase binding site: Localization of the block in replication. J. Bacteriol. 1995, 177, 5937–5942. [Google Scholar] [CrossRef]
- Pato, M.L.; Banerjee, M. Genetic analysis of the strong gyrase site (SGS) of bacteriophage Mu: Localization of determinants required for promoting Mu replication. Mol. Microbiol. 2000, 37, 800–810. [Google Scholar] [CrossRef] [PubMed]
- Oram, M.; Howells, A.J.; Maxwell, A.; Pato, M.L. A biochemical analysis of the interaction of DNA gyrase with the bacteriophage Mu, pSC101 and pBR322 strong gyrase sites: The role of DNA sequence in modulating gyrase supercoiling and biological activity. Mol. Microbiol. 2003, 50, 333–347. [Google Scholar] [CrossRef]
- Kirkegaard, K.; Wang, J.C. Mapping the topography of DNA wrapped around gyrase by nucleolytic and chemical probing of complexes of unique DNA sequences. Cell 1981, 23, 721–729. [Google Scholar] [CrossRef]
- Rice, P.A.; Yang, S.; Mizuuchi, K.; Nash, H.A. Crystal structure of an IHF-DNA complex: A protein-induced DNA U-turn. Cell 1996, 87, 1295–1306. [Google Scholar] [CrossRef]
- Swinger, K.K.; Lemberg, K.M.; Zhang, Y.; Rice, P.A. Flexible DNA bending in HU-DNA cocrystal structures. EMBO J. 2003, 22, 3749–3760. [Google Scholar] [CrossRef]
- Dong, K.C.; Berger, J.M. Structural basis for gate-DNA recognition and bending by type IIA topoisomerases. Nature 2007, 450, 1201–1205. [Google Scholar] [CrossRef]
- Brown, P.O.; Cozzarelli, N.R. A sign inversion mechanism for enzymatic supercoiling of DNA. Science 1979, 206, 1081–1083. [Google Scholar] [CrossRef]
- Segal, E.; Fondufe-Mittendorf, Y.; Chen, L.; Thastrom, A.; Field, Y.; Moore, I.K.; Wang, J.P.; Widom, J. A genomic code for nucleosome positioning. Nature 2006, 442, 772–778. [Google Scholar] [CrossRef] [PubMed]
- Sugino, A.; Higgins, N.P.; Brown, P.O.; Peebles, C.L.; Cozzarelli, N.R. Energy coupling in DNA gyrase and the mechanism of action of novobiocin. Proc. Natl. Acad. Sci. USA 1978, 74, 4838–4842. [Google Scholar] [CrossRef]
- Yang, Y.; Ames, G.F. DNA gyrase binds to the family of prokaryotic repetitive extragenic palindromic sequences. Proc. Natl. Acad. Sci. USA 1988, 85, 8850–8854. [Google Scholar] [CrossRef] [PubMed]
- Bachellier, S.; Clement, J.M.; Hofnung, M.; Gilson, E. Bacterial interspersed mosaic elements (BIMEs) are a major source of sequence polymorphism in Escherichia coli intergenic regions including specific associations with a new insertion sequence. Genetics 1997, 145, 551–562. [Google Scholar] [PubMed]
- Bachellier, S.; Perrin, D.; Hofnung, M.; Gilson, E. Bacterial interspersed mosaic elements (BIMEs) are present in the genome of Klebsiella. Mol. Microbiol. 1993, 7, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Espeli, O.; Boccard, F. In vivo cleavage of Escherichia coli BIME-2 repeats by DNA gyrase: Genetic characterization of the target and identification of the cut site. Mol. Microbiol. 1997, 26, 767–777. [Google Scholar] [CrossRef]
- Espeli, O.; Moulin, L.; Boccard, F. Transcription attenuation associated with bacterial repetitive extragenic BIME elements. J. Mol. Biol. 2001, 314, 375–386. [Google Scholar] [CrossRef]
- Sutormin, D.; Rubanova, N.; Logacheva, M.; Ghilarov, D.; Severinov, K. Single-nucleotide-resolution mapping of DNA gyrase cleavage sites across the Escherichia coli genome. Nucleic Acids Res. 2018. [Google Scholar] [CrossRef]
- Lother, H.; Lurz, R.; Orr, E. DNA binding and antigenic specifications of DNA gyrase. Nucleic Acids Res. 1984, 12, 901–914. [Google Scholar] [CrossRef]
- Moore, C.L.; Klevan, L.; Wang, J.C.; Griffith, J.D. Gyrase. DNA complexes visualized as looped structures by electron microscopy. J. Biol. Chem. 1983, 258, 4612–4617. [Google Scholar]
- Ruthenburg, A.J.; Graybosch, D.M.; Huetsch, J.C.; Verdine, G.L. A superhelical spiral in the Escherichia coli DNA gyrase A C-terminal domain imparts unidirectional supercoiling bias. J. Biol. Chem. 2005, 280, 26177–26184. [Google Scholar] [CrossRef]
- Sugino, A.; Cozzarelli, N.R. The intrinsic ATPase of DNA gyrase. J. Biol. Chem. 1980, 255, 6299–6306. [Google Scholar]
- Johnson, R.C.; Johnson, L.M.; Schmidt, J.W.; Gardner, J.F. The major nucleoid proteins in the structure and function of the E. coli chromosome. In The Bacterial Chromosome; Higgins, N.P., Ed.; ASM Press: Washington, DC, USA, 2005; pp. 65–132. [Google Scholar]
- Jaworski, A.; Higgins, N.P.; Wells, R.D.; Zacharias, W. Topoisomerase mutants and physiological conditions control supercoiling and Z-DNA formation in vivo. J. Biol. Chem. 1991, 266, 2576–2581. [Google Scholar]
- Dame, R.T.; Noom, M.C.; Wuite, G.J. Bacterial chromatin organization by H-NS protein unravelled using dual DNA manipulation. Nature 2006, 444, 387–390. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, H.; Kenney, L.J.; Yan, J. A divalent switch drives H-NS/DNA-binding conformations between stiffening and bridging modes. Genes Dev. 2010, 24, 339–344. [Google Scholar] [CrossRef]
- Dilweg, I.W.; Dame, R.T. Post-translational modification of nucleoid-associated proteins: An extra layer of functional modulation in bacteria? Biochem. Soc. Trans. 2018, 46, 1381–1392. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | GyrA | GyrB | Doubling Time (min) on LB 30 °C |
|---|---|---|---|
| S.Tm NH6303 | S. Tm | S. Tm | 40 ± 1 |
| S. Tm NH6304 | S. Tm | E. coli | 43 ± 1 |
| S.Tm NH6281 | E. coli | S.Tm | 43 ± 1 |
| S.Tm NH6292 | E. coli | E. coli | 44 ±1 |
| S.Tm NH6390 | E. coli <1/2 CTD S.Tm> | S.Tm | 40 ± 2 |
| S.Tm NN6392 | E. coli <CTD S.Tm> | S. Tm | 40 ± 1 |
| S.Tm NH6391 | E. coli <CTD S.Tm> | E. coli | 39 ± 1 |
| E. coli NH1013 | E. coli | E. coli | 39 ± 1 |
| E. coli NH6386 | S. Tm | E. coli | 60 ± 3 |
| E. coli NH6451 | S. Tm <CTD E. coli> | E. coli | 41 ± 1 |
| E. coli NH6453 | S. Tm <32 aa E. coli Tail> | E. coli | 37 ± 1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rovinskiy, N.S.; Agbleke, A.A.; Chesnokova, O.N.; Higgins, N.P. Supercoil Levels in E. coli and Salmonella Chromosomes Are Regulated by the C-Terminal 35–38 Amino Acids of GyrA. Microorganisms 2019, 7, 81. https://doi.org/10.3390/microorganisms7030081
Rovinskiy NS, Agbleke AA, Chesnokova ON, Higgins NP. Supercoil Levels in E. coli and Salmonella Chromosomes Are Regulated by the C-Terminal 35–38 Amino Acids of GyrA. Microorganisms. 2019; 7(3):81. https://doi.org/10.3390/microorganisms7030081
Chicago/Turabian StyleRovinskiy, Nikolay S., Andrews A. Agbleke, Olga N. Chesnokova, and N. Patrick Higgins. 2019. "Supercoil Levels in E. coli and Salmonella Chromosomes Are Regulated by the C-Terminal 35–38 Amino Acids of GyrA" Microorganisms 7, no. 3: 81. https://doi.org/10.3390/microorganisms7030081
APA StyleRovinskiy, N. S., Agbleke, A. A., Chesnokova, O. N., & Higgins, N. P. (2019). Supercoil Levels in E. coli and Salmonella Chromosomes Are Regulated by the C-Terminal 35–38 Amino Acids of GyrA. Microorganisms, 7(3), 81. https://doi.org/10.3390/microorganisms7030081