Food Preservatives Induce Proteobacteria Dysbiosis in Human-Microbiota Associated Nod2-Deficient Mice



Abstract
1. Introduction
1.1. Dysbiosis of Gut Microbiota
1.2. Food Additives and Gut Microbiota
2. Materials and Methods
2.1. Animals and Experimental Design
2.2. Rederivation of Germ-Free Mice, Mouse Diet, and Sterility Checks
2.3. Colonisation of Germ-Free Mice with Human Gut Microbiota and Experimental Diet
2.4. Antimicrobial Food Additives and Their Supplementation
2.5. Genomic DNA Extraction
2.6. PCR and Sequencing
2.7. Metagenomic Analysis
2.8. Statistics
3. Results
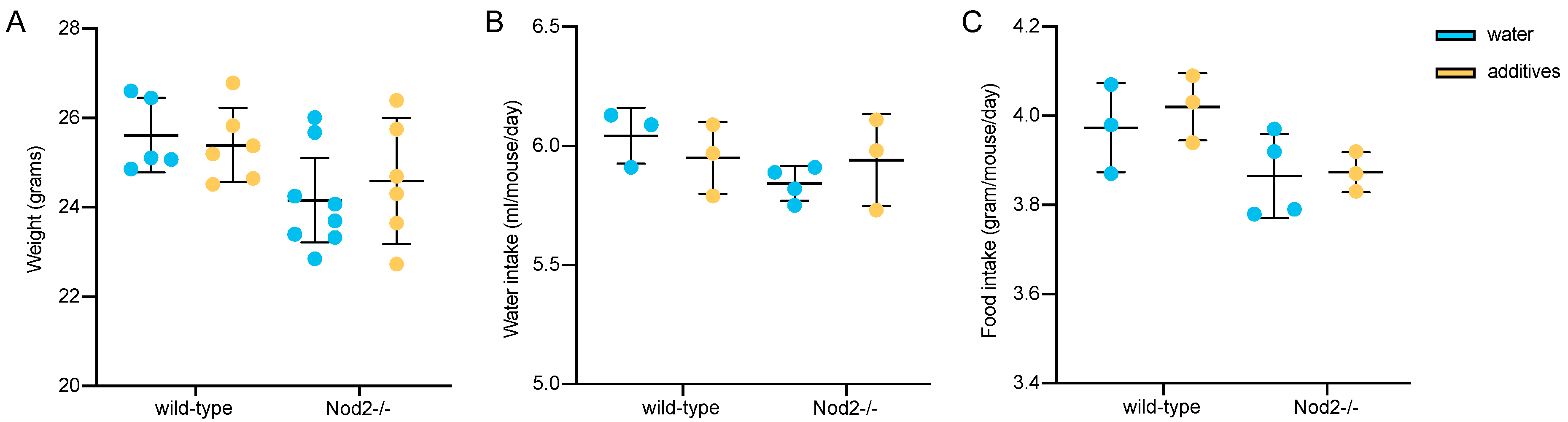
3.1. Body Weight, Water and Food Consumption
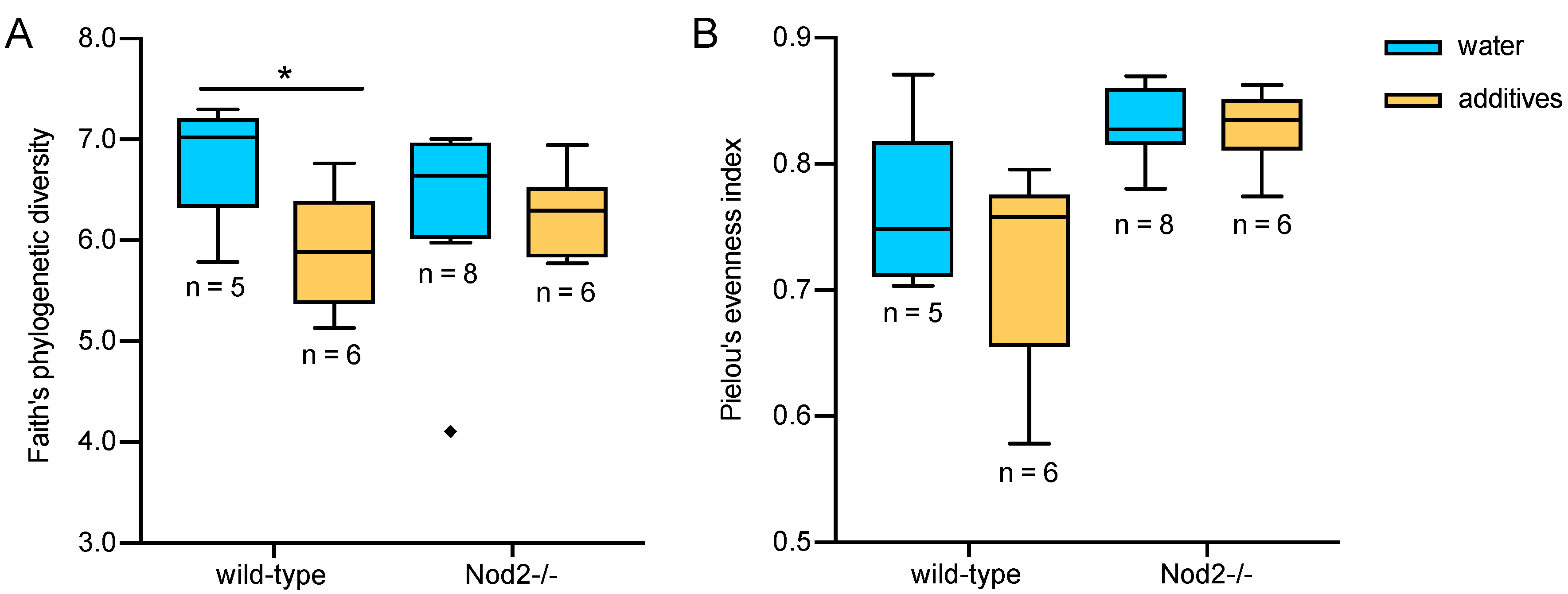
3.2. Alpha Diversity
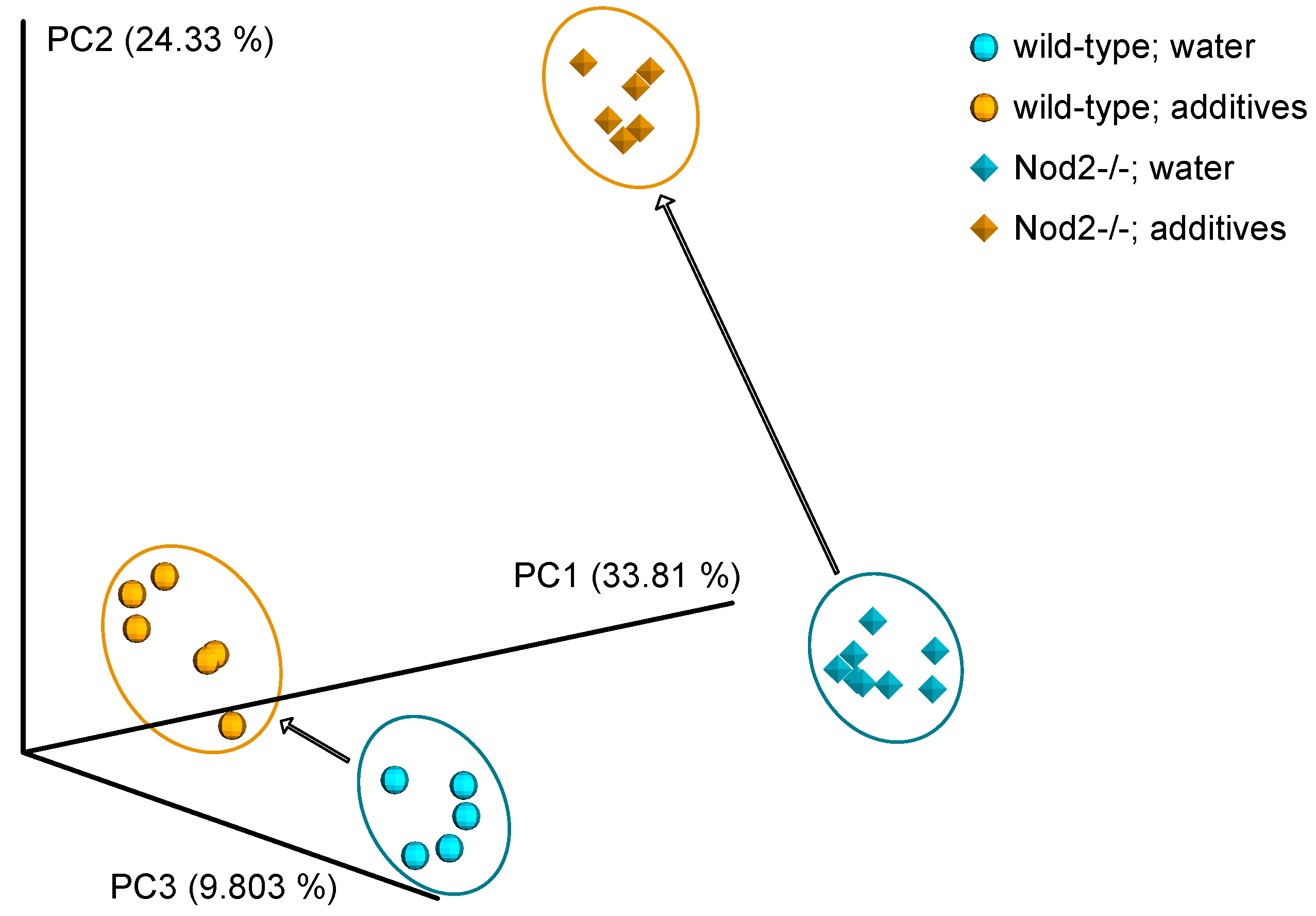
3.3. Beta Diversity
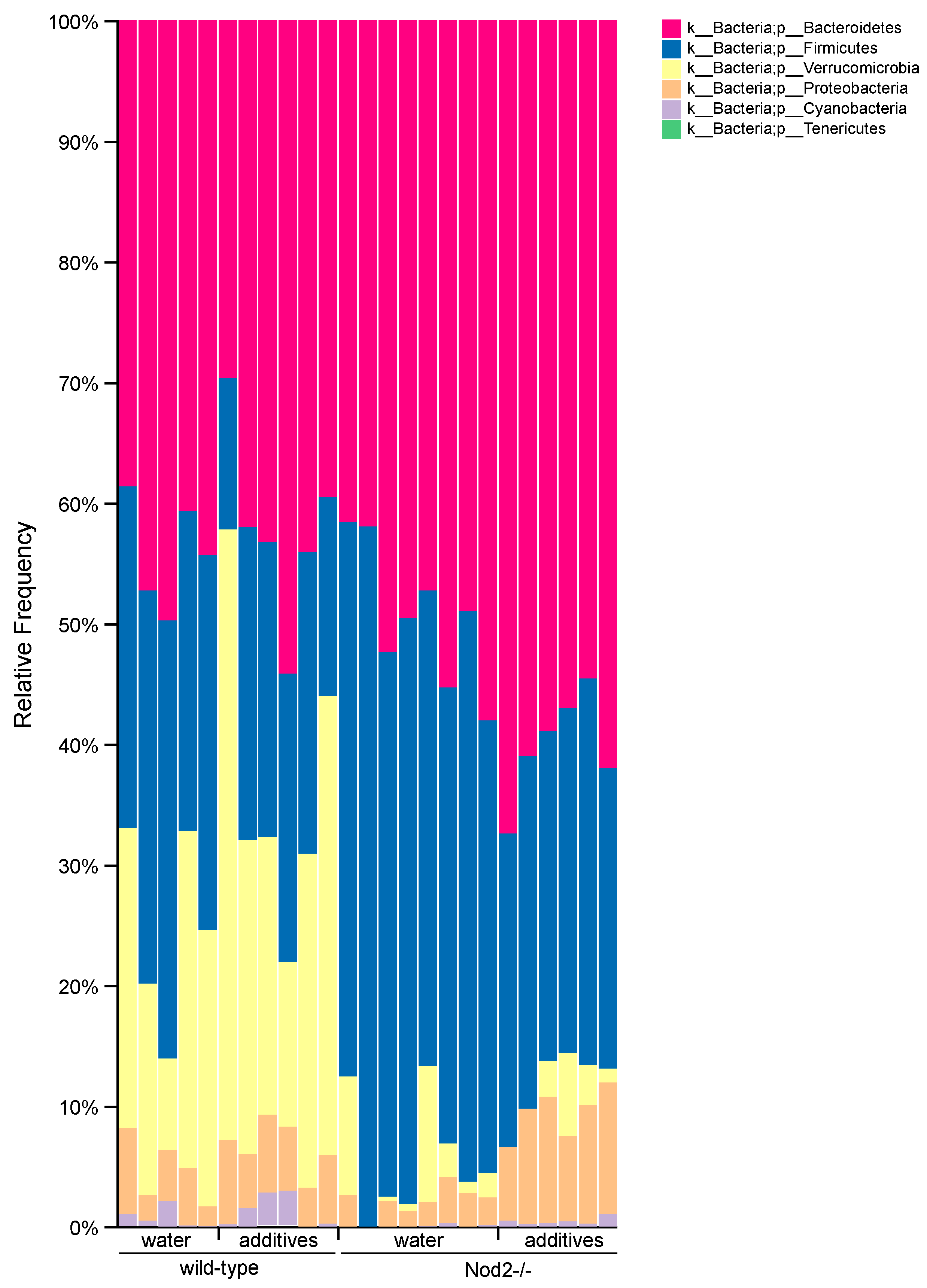
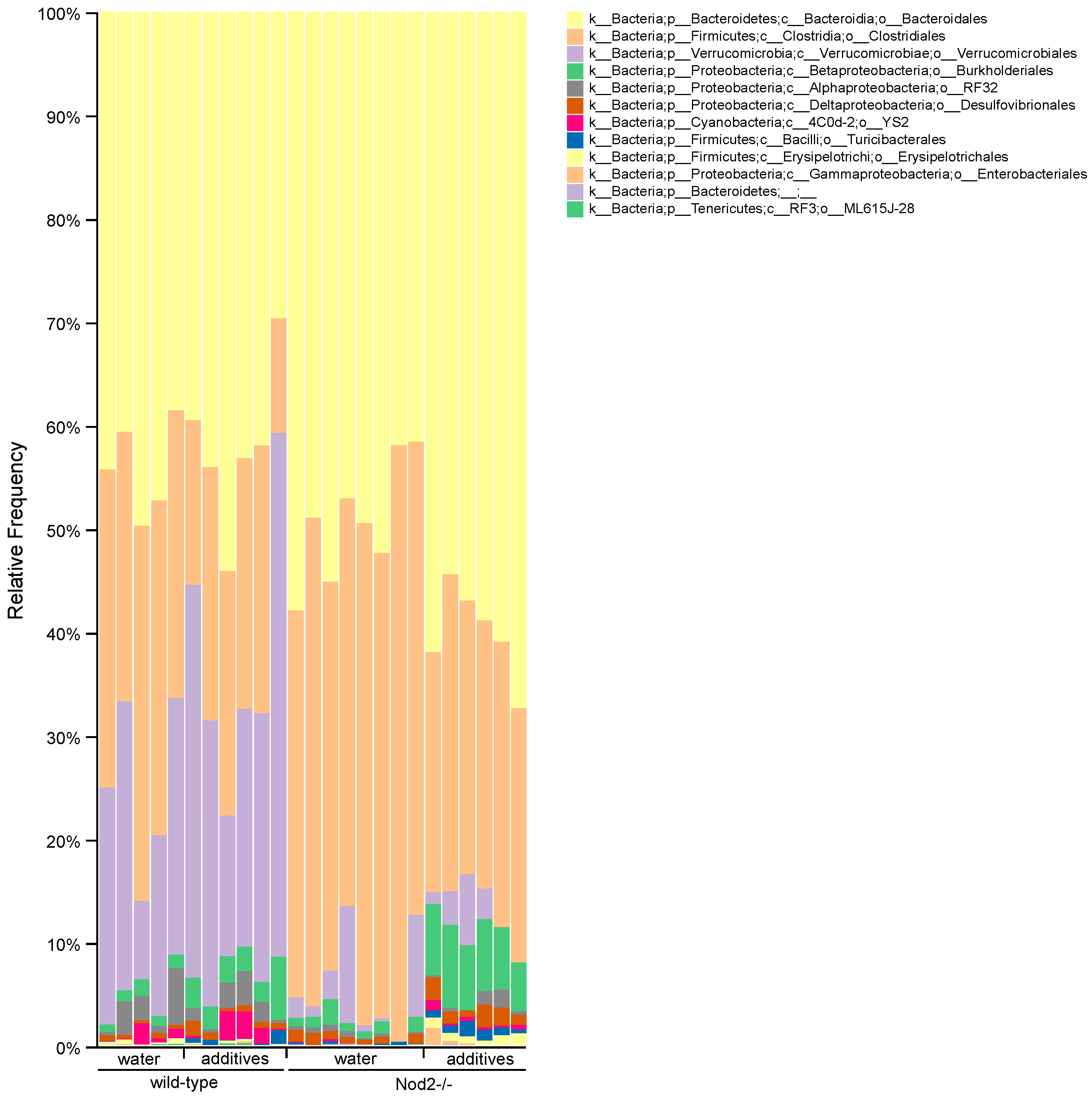
3.4. Taxonomic Analysis/Relative Abundance
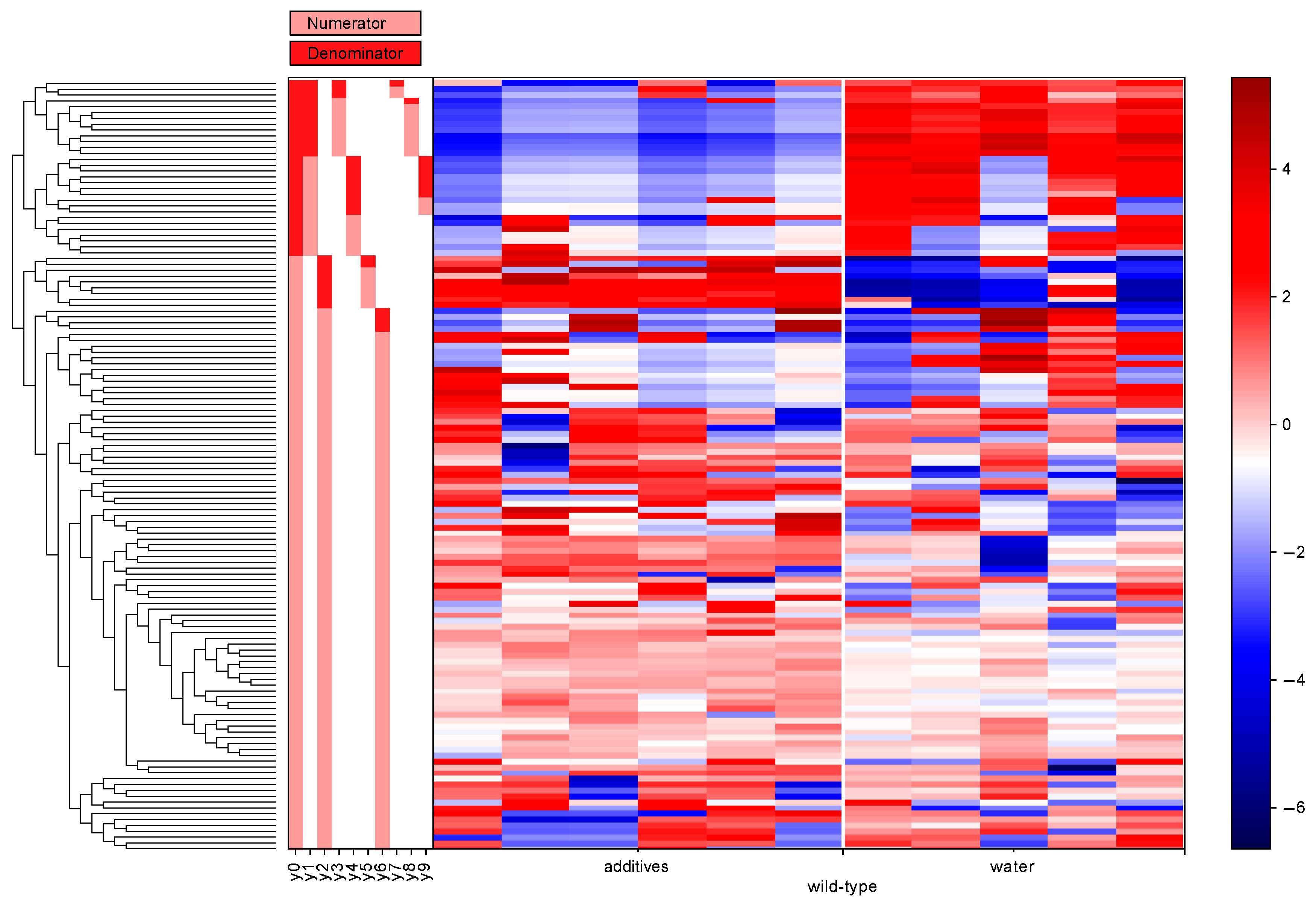
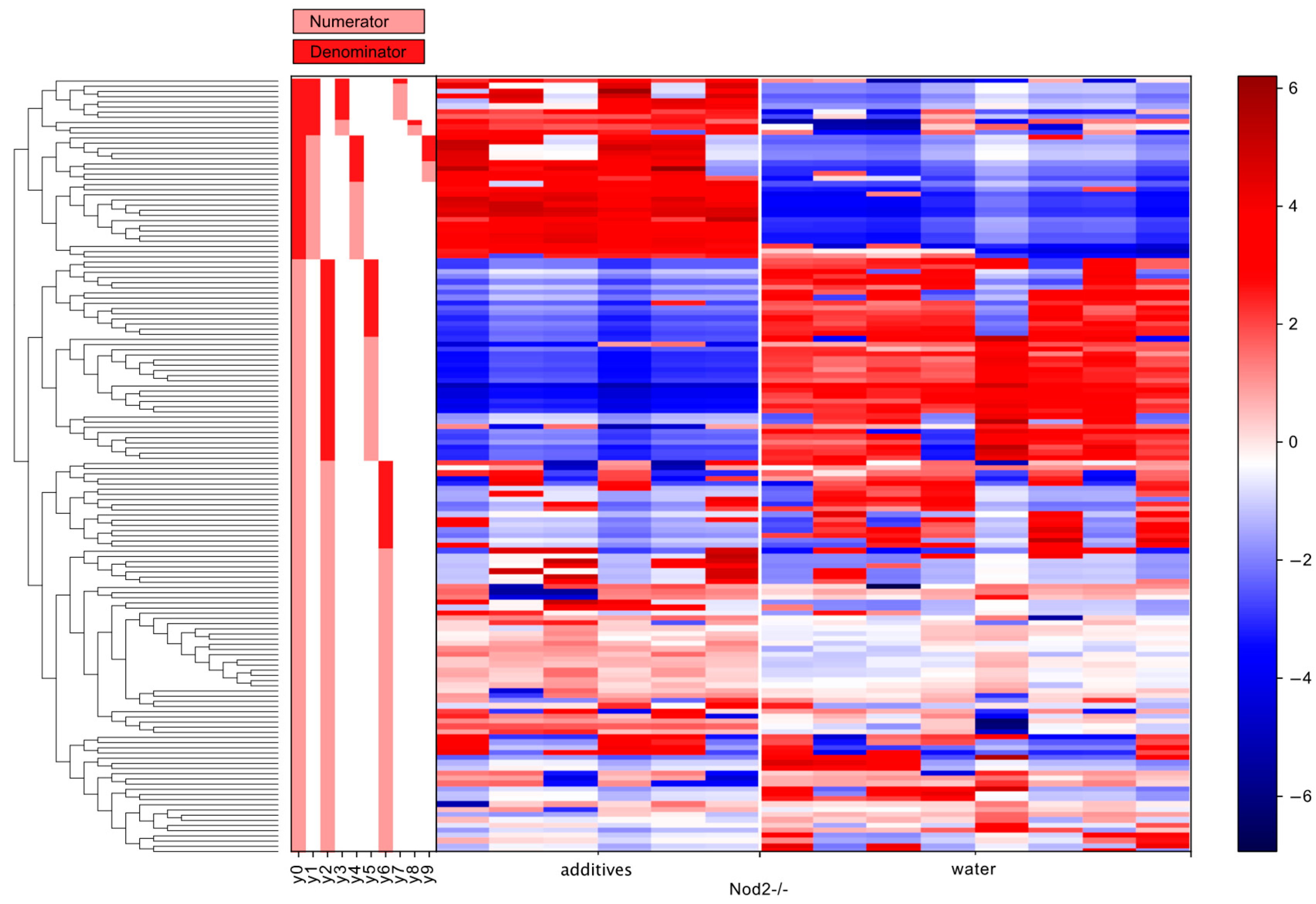
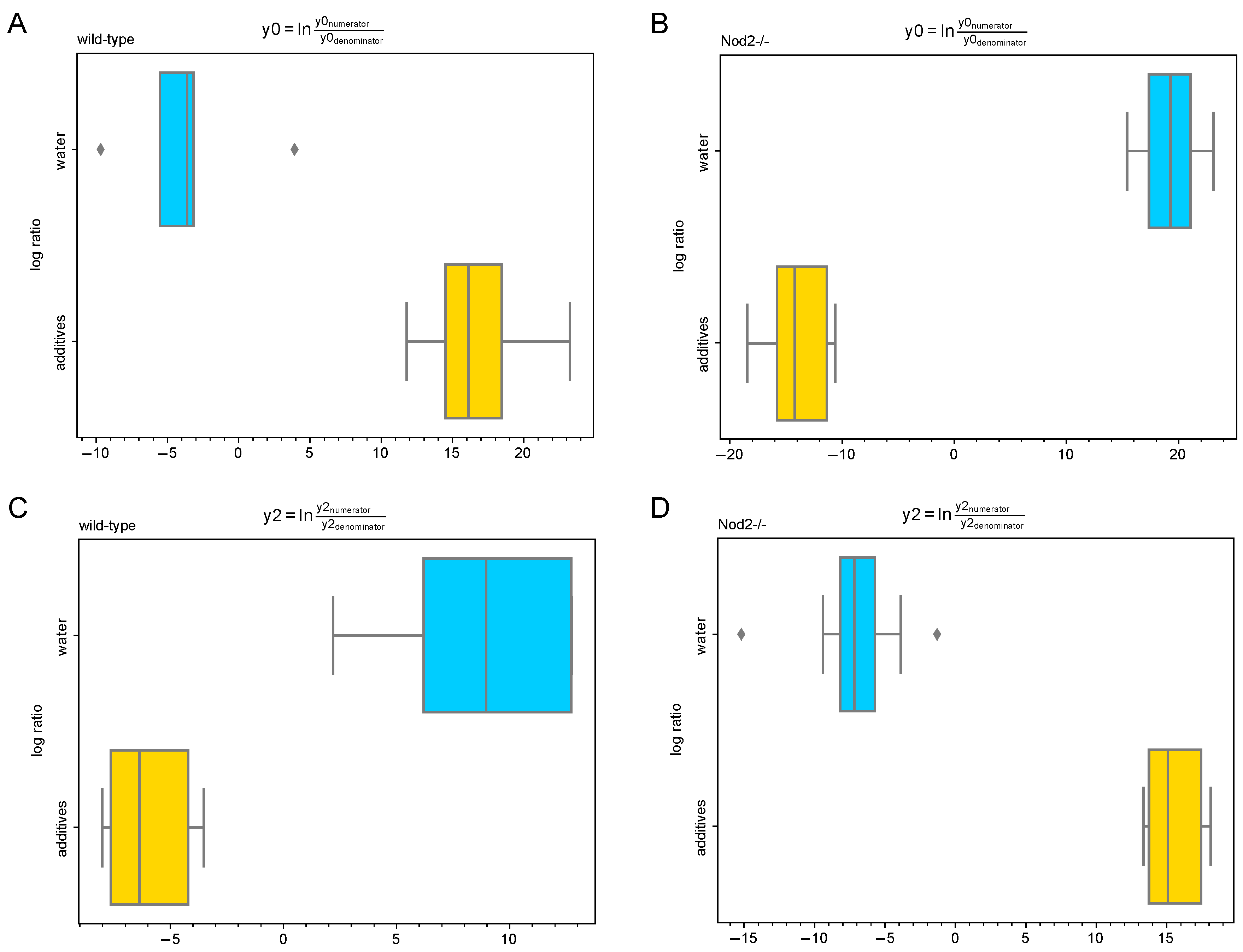
3.5. Differential Abundance Analysis (Gneiss Analysis)
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| IBD | inflammatory bowel disease |
| TLR | Toll-like receptor |
| EDTA | ethylenediaminetetraacetate |
| AMFA | antimicrobial food additives |
| PBS | reduced phosphate-buffered saline |
| ASV | amplicon sequence variants |
| OTU | operational taxonomic unit |
| FDR | false discovery rate |
| PCoA | principal coordinate analysis |
References
- Mosca, A.; Leclerc, M.; Hugot, J.P. Gut microbiota diversity and human diseases: Should we reintroduce key predators in our ecosystem? Front. Microbiol. 2016, 7, 455. [Google Scholar] [CrossRef] [PubMed]
- Walters, W.A.; Xu, Z.; Knight, R. Meta-analyses of human gut microbes associated with obesity and IBD. FEBS Lett. 2014, 588, 4223–4233. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Xu, Z.Z.; He, Y.; Yang, Y.; Liu, L.; Lin, Q.; Nie, Y.; Li, M.; Zhi, F.; Liu, S.; et al. Gut microbiota offers universal biomarkers across ethnicity in inflammatory bowel disease diagnosis and infliximab response prediction. mSystems 2018, 3, 1063. [Google Scholar] [CrossRef]
- Frank, D.N.; St Amand, A.L.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef] [PubMed]
- Garrett, W.S.; Lord, G.M.; Punit, S.; Lugo-Villarino, G.; Mazmanian, S.K.; Ito, S.; Glickman, J.N.; Glimcher, L.H. Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell 2007, 131, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Shin, N.-R.; Whon, T.W.; Bae, J.-W. Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef]
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the immune system. Nat. Rev. Immunol. 2017, 17, 219–232. [Google Scholar] [CrossRef]
- Kostic, A.D.; Gevers, D.; Siljander, H.; Vatanen, T.; Hyotylainen, T.; Hämäläinen, A.-M.; Peet, A.; Tillmann, V.; Pöhö, P.; Mattila, I.; et al. The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell Host Microbe 2015, 17, 260–273. [Google Scholar] [CrossRef]
- Scheperjans, F.; Aho, V.; Pereira, P.A.B.; Koskinen, K.; Paulin, L.; Pekkonen, E.; Haapaniemi, E.; Kaakkola, S.; Eerola-Rautio, J.; Pohja, M.; et al. Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov. Disord. 2015, 30, 350–358. [Google Scholar] [CrossRef]
- Schaubeck, M.; Clavel, T.; Calasan, J.; Lagkouvardos, I.; Haange, S.B.; Jehmlich, N.; Basic, M.; Dupont, A.; Hornef, M.; von Bergen, M.; et al. Dysbiotic gut microbiota causes transmissible Crohn’s disease-like ileitis independent of failure in antimicrobial defence. Gut 2016, 65, 225–237. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027131. [Google Scholar] [CrossRef] [PubMed]
- Chassaing, B.; Van de Wiele, T.; De Bodt, J.; Marzorati, M.; Gewirtz, A.T. Dietary emulsifiers directly alter human microbiota composition and gene expression ex vivo potentiating intestinal inflammation. Gut 2017, 66, 1414–1427. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Palacios, A.; Harding, A.; Menghini, P.; Himmelman, C.; Retuerto, M.; Nickerson, K.P.; Lam, M.; Croniger, C.M.; McLean, M.H.; Durum, S.K.; et al. The artificial sweetener Splenda promotes gut Proteobacteria, dysbiosis, and myeloperoxidase reactivity in Crohn’s disease-like ileitis. Inflamm. Bowel. Dis. 2018, 24, 1005–1020. [Google Scholar] [CrossRef] [PubMed]
- Irwin, S.V.; Fisher, P.; Graham, E.; Malek, A.; Robidoux, A. Sulfites inhibit the growth of four species of beneficial gut bacteria at concentrations regarded as safe for food. PLoS ONE 2017, 12, e0186629. [Google Scholar] [CrossRef] [PubMed]
- You, X.; Einson, J.E.; Lopez-Pena, C.L.; Song, M.; Xiao, H.; McClements, D.J.; Sela, D.A. Food-grade cationic antimicrobial ε-polylysine transiently alters the gut microbial community and predicted metagenome function in CD-1 mice. NPJ Sci. Food 2017, 1, 8. [Google Scholar] [CrossRef] [PubMed]
- Hrncirova, L.; Hudcovic, T.; Sukova, E.; Machova, V.; Trckova, E.; Krejsek, J.; Hrncir, T. Human gut microbes are susceptible to antimicrobial food additives in vitro. Folia Microbiol. 2019, 1–12. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Knight, R.; Gordon, J.I. The effect of diet on the human gut microbiome: A metagenomic analysis in humanized gnotobiotic mice. Sci. Transl. Med. 2009, 1, 6ra14. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.; Al-Ghalith, G.A.; Alexander, H. QIIME 2: Reproducible, interactive, scalable, and extensible microbiome data science. PeerJ Prepr. 2018, 6, e27295v2. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.I.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Faith, D.P. Conservation evaluation and phylogenetic diversity. Biol. Conserv. 1992, 61, 1–10. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.J. Permutational Multivariate Analysis of Variance (PERMANOVA); John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2014; ISBN 9781118445112. [Google Scholar]
- Petnicki-Ocwieja, T.; Hrncir, T.; Liu, Y.-J.; Biswas, A.; Hudcovic, T.; Tlaskalova-Hogenova, H.; Kobayashi, K.S. Nod2 is required for the regulation of commensal microbiota in the intestine. Proc. Natl. Acad. Sci. USA 2009, 106, 15813–15818. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype | Balance of Interest | Family | Number of ASVs |
|---|---|---|---|
| wild-type | y0 denominator | Lachnospiraceae | 13 |
| Ruminococcaceae | 7 | ||
| Bacteroidaceae | 6 | ||
| Veillonellaceae | 1 | ||
| Clostridiaceae | 1 | ||
| Peptococcaceae | 1 | ||
| Alcaligenaceae | 1 | ||
| y2 denominator | Bacteroidaceae | 4 | |
| Lachnospiraceae | 2 | ||
| Turicibacteraceae | 1 | ||
| Ruminococcaceae | 1 | ||
| Alcaligenaceae | 1 | ||
| Nod2-deficient | y0 denominator | Lachnospiraceae | 12 |
| Bacteroidaceae | 7 | ||
| Ruminococcaceae | 4 | ||
| Desulfovibrionaceae | 2 | ||
| Rikenellaceae | 2 | ||
| Erysipelotrichaceae | 2 | ||
| Clostridiales (order) | 2 | ||
| Porphyromonadaceae | 1 | ||
| Enterobacteriaceae | 1 | ||
| Turicibacteraceae | 1 | ||
| y2 denominator | Lachnospiraceae | 20 | |
| Ruminococcaceae | 8 | ||
| Clostridiales (order) | 7 | ||
| Bacteroidaceae | 2 | ||
| Rikenellaceae | 2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hrncirova, L.; Machova, V.; Trckova, E.; Krejsek, J.; Hrncir, T. Food Preservatives Induce Proteobacteria Dysbiosis in Human-Microbiota Associated Nod2-Deficient Mice. Microorganisms 2019, 7, 383. https://doi.org/10.3390/microorganisms7100383
Hrncirova L, Machova V, Trckova E, Krejsek J, Hrncir T. Food Preservatives Induce Proteobacteria Dysbiosis in Human-Microbiota Associated Nod2-Deficient Mice. Microorganisms. 2019; 7(10):383. https://doi.org/10.3390/microorganisms7100383
Chicago/Turabian StyleHrncirova, Lucia, Vladimira Machova, Eva Trckova, Jan Krejsek, and Tomas Hrncir. 2019. "Food Preservatives Induce Proteobacteria Dysbiosis in Human-Microbiota Associated Nod2-Deficient Mice" Microorganisms 7, no. 10: 383. https://doi.org/10.3390/microorganisms7100383
APA StyleHrncirova, L., Machova, V., Trckova, E., Krejsek, J., & Hrncir, T. (2019). Food Preservatives Induce Proteobacteria Dysbiosis in Human-Microbiota Associated Nod2-Deficient Mice. Microorganisms, 7(10), 383. https://doi.org/10.3390/microorganisms7100383